

Abstract
Regulatory T (Treg) cells are essential in preventing the host from developing certain autoimmune diseases and limiting excessive immune responses against pathogens. The normal function of most Treg cells requires sustained expression of functional FOXP3, a member of the FOXP family transcription factors. FOXP3 is distinct from other subfamily members because of its unique proline rich amino (N)-terminal domain. Mutations in this region are occasionally identified in certain patients with X-linked autoimmunity–allergic dysregulation syndrome (XLAAD) and similar mutations also increase susceptibility of autoimmune diseases in rodent models. Previous analyses of the FOXP3 N- domain revealed a role in nuclear import, interaction with other transcription factors, and as sites of specific post-translational modifications of FOXP3 that contribute to FOXP3 stability.
Introduction
Regulatory or suppressor T cells represent a small portion of the CD4+ T cell pool but mediate an important role in maintaining the balance between inflammation and immune tolerance (Ziegler, 2006). Proliferation of CD4+ T effector cells can be suppressed by Treg cells so that these cells do not attack self antigens or respond excessively to external antigens (Shevach, 2009; Thornton and Shevach, 1998). A key transcription factor regulating the development and function of Treg cells was identified as a member of the FOXP family, FOXP3 (FOXP3 in human, Foxp3 in mice; hereafter referred to as FOXP3 unless specified). Mutations of Foxp3 account for the scurfy phenotype in mice and the XLAAD syndrome in human (Brunkow et al., 2001; Chatila et al., 2000). These mutations are dominantly located at three domains of FOXP3, the forkhead domain responsible for DNA binding, the zinc finger and leucine zipper region responsible for higher order clustering of FOXP3, and less frequently in the N-terminal domain (Bennett et al., 2001; Lopes et al., 2006; Wildin et al., 2002). The contributions of the large N-terminal region of FOXP3 are not well understood to the function of this important transcriptional molecule.
Recent studies showed that disruption of a small region of the Foxp3 N-terminal domain caused by an unappreciated cloning alteration that occurred when GFP was fused to the N-terminal domain of Foxp3 created a hypomorphic species (Bettini et al., 2012; Darce et al., 2012; Fontenot et al., 2005), These hypomorphic forms of FOXP3 render rodents more susceptible to diabetes but more resistant to antibody mediated arthritis, indicating the functional and complex role of the Foxp3 N-terminal domain in mediating different functional outcomes of Treg cells.
This review will summarize recent insights into the structural, molecular and functional contributions of the FOXP3 N-terminal domain, which is defined as the protein sequence from 1 to 197 aa of human FOXP3 including the exon 2 encoding region and the proline-rich domain (Figure 1).
Figure 1.
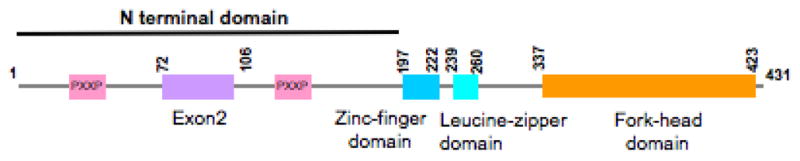
Schematic representation of structural domains in human FOXP3.
FOXP3 and Treg
FOXP3 was identified through a positional cloning approach to isolate the gene responsible for X-linked autoimmunity–allergic dysregulation syndrome (XLAAD, also known as immunedysregulation polyendocrinopathy enteropathy, X-linked (IPEX)) in human, and scurfy disease in mice (Brunkow et al., 2001; Chatila et al., 2000). A 2-bp insertion within the Foxp3 exon 8 leads to a truncated gene product lacking the C-terminal DNA binding domain in mice with scurfy phenotype. It was soon discovered that Foxp3 is specifically and stably expressed in CD4+CD25+ Treg cells of both rodents and human and plays a dominant role in maintaining self-tolerance through regulating Treg function (Fontenot et al., 2003; Hori et al., 2003; Khattri et al., 2003).
Foxp3−/− bone marrow stem cells failed to give rise to Treg cells in lethally irraditated rodent recipients, reflecting a critical role of Foxp3 in the development of Treg cells (Fontenot et al., 2003). As Foxp3 is required for the lineage commitment of Treg cells (Gavin et al., 2007), dysfunction of Foxp3 has multiple effects on the Treg cells. Expression of a nonfunctional Foxp3-EGFP fusion protein results in heightened apoptosis as well as a survival disadvantage of Treg cells (Lin et al., 2007). Several distinct mutations in Foxp3 also abolish the suppressive function of Treg cells (Khattri et al., 2003).
Ectopic expression of Foxp3 inhibits cytokine production, up-regulates expression of cell surface molecules related to Treg function, and confers naïve CD4+ T cells with suppressor function (Fontenot et al., 2003; Hori et al., 2003). Foxp3 transduced CD4+ T cells are able to prevent mice from developing autoimmune diseases such as IBD, colitis, and prevent mortality in CTLA4−/− mice (Hori et al., 2003; Khattri et al., 2003). FOXP3 serves as a key regulator in maintaining the normal function of Treg cells, which is critical for the prevention of autoimmune diseases.
Domain structure of FOXP3
FOXP3 is a multi-domain protein which contains three major domains: a N-terminal domain responsible for transcriptional repression, a zinc finger and leucine zipper domain (ZL domain) centrally located which facilitates the formation of FOXP3 homo-dimer or tetramers, or association with other transcription factors like FOXP1, and a highly conserved forkhead domain located at the C-terminal responsible for DNA binding (Figure 1) (Zhou et al., 2008b). The crystal structure of both the zinc finger and leucine zipper domain was resolved by our laboratory (Song et al., 2012) and the DNA binding domain (Bandukwala et al., 2011) has also been determined. The forkhead domain in the C-terminal is essential for the suppressive function of FOXP3 (Bandukwala et al., 2011). The coiled coil structure in the leucine zipper region mediates the loose or transient dimerization of FOXP3, and its conformation can be modulated by post-translational modifications under physiological conditions, but disturbed or impaired irreversibly by disease-causing mutations. Dimerization through the coiled coil motif is essential for FOXP3 to function as a transcriptional regulator in Tregs. As shown in figure 2, the anti-parallel conformation of FOXP3 dimer may render both the N-terminal TIP60 binding repressor domain and the C-terminal AML-1/Runx binding linker region spatially adjacent to the zinc finger and leucine zipper motif, which then facilitates the coordinated function of a variety of regulators recruited into the regulatory complex.
Figure 2.
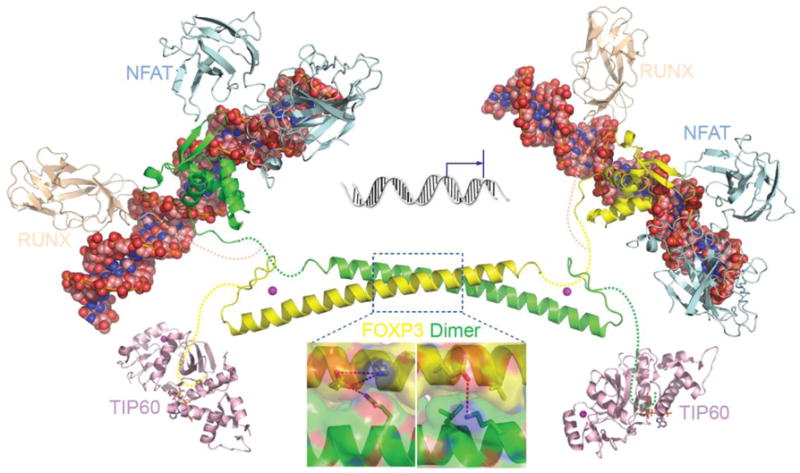
Structure model of FOXP3 mediated regulatory complex. The overall architecture of the intact FOXP3 associated DNA-protein regulatory complex are constructed by using the crystal structure of FOXP2 forkhead domain complexed with DNA and NFAT (Wu et al., 2006), and the crystal structure of the FOXP3 coiled coil motif reported (Song et al., 2012). To regulate gene transcription, the repressor unit from one FOXP3 molecule cooperates with the DNA binding unit from another FOXP3 molecule.
FOXP3 differs from other FOXP family members in the N-terminal region, which is proline rich and has little similarity to the glutamine rich N-terminal regions of FOXP1, FOXP2 and FOXP4 (Li and Greene, 2007). The crystal structure of the FOXP3 N-terminal region is still unavailable, possibly because this region could be intrinsically disordered although we have been successful in developing small crystals of an N terminal region that extends to amino acid 262. Partner binding and/or posttranslational modification may also stabilize this region and help to define the structural features of the FOXP3 N-terminal domain.
Functional importance of the FOXP3 N-terminal domain
FOXP3 possesses a proline rich N-terminal domain rather than the glutamine rich N-terminal domain that has been found in FOXP1, FOXP2 and FOXP4. Therefore, the N-terminal domain of FOXP3 is expected to play a critical role in mediating the specific regulatory effect of FOXP3 on Treg cells.
An N-terminal truncated Foxp3 (1–150 aa deleted) is unable to repress IL-2 production when transduced into CD4+ T cells (Bettelli et al., 2005). FOXP3 also enhances HIV-1 LTR activity and deletion of FOXP3 N-terminal 190 aa abrogated the Foxp3 mediated LTR enhancement (Holmes et al., 2007). A Gal4-FOXP3 fusion protein has been used to study domain requirements of FOXP3 in the inhibition of the expression of firefly luciferase reporter gene driven by the Gal4-binding site. A construct containing the first 198 aa of FOXP3 at the N terminus mediates transcription repression equivalent to forkhead domain deleted FOXP3, suggesting that the N-terminal domain of FOXP3 is both required and is sufficient to convey certain features of transcription repression activity of FOXP3 (Lopes et al., 2006). CD4+ T cells transduced with an N-terminal deleted Foxp3 construct failed to repress IL-4 production and the effect of the transduced CD4+ T cells on preventing IBD in mouse models was also abrogated by N-terminal deletion of Foxp3 (Zeng et al., 2011).
Mutations in the FOXP3 N-terminal domain have been identified in IPEX patients (Lopes et al., 2006; van der Vliet and Nieuwenhuis, 2007). Despite having little functional activity, N-terminal deletion does not appreciably affect binding to DNA, a feature that must be taken into account when considering functionalities.
A Foxp3gfp knockin mutant, which contains an in-frame insertion of GFP between amino acid 5 and 6 of Foxp3, was broadly used in Foxp3gfp reporter mice to track Treg cells by many investigators. However, this well accepted methodology was recently challenged by two groups who observed the accelerated incidence of the diabetes coupled with the reduced number of Treg cells, suggesting that this Foxp3gfp knockin mutant leads to reduced Foxp3 function and may indeed exert effects on the function of Treg cells (Bettini et al., 2012); (Darce et al., 2012). Although Treg cells expressing Foxp3 with an in-frame GFP insertion is as effective as wild type Treg cells in suppressing certain T effector cell proliferation properties in vitro, they are far less functional in vivo and are unable to suppress the T helper 1 (Th1) cell-driven pathology in Foxp3−/− mice. Compared with wildtype Foxp3 expressing cells, there was a ~50% reduction in the efficiency of TGF-β-induced Treg (iTreg) cell conversion from Tconv cells in the Foxp3gfp NOD knockin mice (Bettini et al., 2012).
Surprisingly Foxp3 with this GFP insertion may confer Treg cells an increased capability to suppress Th17 cell differentiation and T cell help to B cells. While the GFP-Foxp3 mice have greater susceptibility to diabetes on the NOD background, a pathology which involves autoreactive Th1 cell-like T cells, the protection from arthritis in the K/BxN model may reflect an effect on IL-4 and IL-17 production (Darce et al., 2012). The GFP insertion disrupts the interaction of Foxp3 with some proteins known to interact with the N-terminal domain (described in detail later in this review).
Regulating FOXP3 activity through N-terminal domain
FOXP3 is a dominant transcriptional regulator in Treg cells and functions by activating or inactivating different sets of genes (Katoh et al., 2011; Rudra et al., 2012). The ability of FOXP3 to bind DNA is essential for its physiological activity. This feature is supported by the fact that many mutations found in IPEX patient are located in the forkhead domain of FOXP3 and those mutant species cannot bind to DNA effectively. It is reported that the N-terminal domain of FOXP3 alone is sufficient to repress reporter gene expression as effectively as full length FOXP3 when fused to Gal4 (Lopes et al., 2006), while deletion of the entire N-terminal domain only results in a minimal decrease in the DNA binding ability of Foxp3 (Zeng et al., 2011). We propose that the N-terminal domain does not regulate DNA binding of FOXP3 directly, but functions through a trans-repression mechanism by interacting with other repressors.
Functional motifs in the N-terminal domain
Post-translational cleavage of a precursor protein sometimes is required to yield an activated protein with physiological functions, a process that is catalyzed by the family of proprotein convertases (Artenstein and Opal, 2011). There are at least 9 members in the proprotein convertases family, seven of which cleave protein at basic motif [R/K]–Xn–[R/K]↓, where X indicates any amino acids, n indicates the number of space residues and “↓” indicates the cleavage site (Seidah and Chretien, 1999). Analysis of Foxp3 sequence has revealed two proprotein convertase cleavage motifs, one located at the N-terminal domain (48RXXR51), and the other located at the C-terminal forkhead domain (414RXXR417) (de Zoeten et al., 2009). The cleaved form of Foxp3 is only found in the chromatin fraction of activated Treg cells and cleavage at both N- and C-terminal of Foxp3 is dependent on the intact RXXR motifs.
Compared to full length Foxp3, the cleaved form of Foxp3 mediates different effects in regulating distinct patterns of gene expression when transduced to CD4+ T, with a decreased ability to up-regulate CTLA-4 expression and an increased ability to up-regulate IL-10 expression. Cleavage at the N-terminal of Foxp3 seems to have a less effect in activating Treg cells, since CD4+ T cells expressing the N-terminal cleaved Foxp3 inhibits T effector cell proliferation comparable to full length Foxp3, while CD4+ T cells expressing the C-terminal cleaved Foxp3 has a stronger effect on inhibiting effector T cell proliferation.
Foxp3 is constitutively localized in the nucleus in Treg cells (Fontenot et al., 2005), but the mechanism regulating Foxp3 localization in the nucleus is poorly defined. The N-terminal cleaved Foxp3 (1–51 aa deleted) has reduced localization to the nucleus, indicating a role of the Foxp3 N-terminal domain in regulating subcellular localization of Foxp3 (Hancock and Ozkaynak, 2009). However, the classical nuclear localization sequence (NLS) that is rich in basic residues and consists either one or two stretches of basic amino acids is not found in this region (Kalderon et al., 1984; Robbins et al., 1991). Alternatively, FOXP3 may also interact with other nuclear factors or chaperones that help the nuclear transport of FOXP3.
Interaction of N-terminal domain with other transcription factors
Formation of a macromolecular complex with various transcription factors is an important process for FOXP3 to regulate the expression of a variety of genes relevant to Treg function (Rudra et al., 2012; Zhou et al., 2008b). The N-terminal of FOXP3 is involved in the interaction with several transcription factors, which are related to the suppressive transcriptional activity of FOXP3.
One of the key features of Treg cells is to maintain the anergic status in response to stimulation, which is due to the impaired DNA-binding activity of AP-1 in Treg cells (Lee et al., 2008). The N-terminal region of Foxp3 contributes to this feature by interacting with phosphorylated and active c-Jun, therefore altering c-Jun subnuclear localization and inhibiting AP-1 promoter binding of c-Jun. Another key feature of Treg cells, the suppression of CD4+ T cells proliferation, is also maintained through the interaction of Foxp3 N-terminal domain with Eos, a zinc-finger transcription factor of the Ikaros family (Pan et al., 2009). Foxp3 and Eos, together with C-terminal binding protein-1 (CtBP1), form a suppression complex for epigenetically modified promoter regions (e.g. increased histone methylation (MeH3K9) and decreased histone acetylation (H3Ac and H4Ac)) and inhibit gene expressions. Disruption of the interaction of Foxp3 and Eos leads to the failure of Treg to suppress proliferation of T effector cells and to prevent the development of colitis in the mouse model. However, N-terminal of Foxp3 alone is not sufficient to confer Treg cells suppressive function, and interaction of NFAT and Foxp3 C-terminal forkhead domain is also required for the suppressive function of Treg cells (Lee et al., 2008; Wu et al., 2006). Therefore, different from the anergic status of Treg cells, the suppressive function of Treg cells requires the cooperative effect of both the N-terminal and C-terminal of Foxp3.
Different from mice Treg cells, there are two FOXP3 isoforms expressed in human Treg cells, FOXP3a and FOXP3b that lacks Exon 2 (Allan et al., 2005; Yagi et al., 2004). These two FOXP3 isoforms have the same capability to inhibit cytokine production and suppress the proliferation of effector T cells (Aarts-Riemens et al., 2008; Allan et al., 2005; Smith et al., 2006). However, the two FOXP3 isoforms do have functional difference in that only FOXP3a but not FOXP3b is able to induce the expression of PIM-2 kinase, which confers rapamycin resistance in Treg cells (Basu et al., 2008).
Exon 2 of Foxp3 is responsible for the interaction with RORα and RORγt, transcriptional factors that activate the expression of genes required for Th17 differentiation, such as IL-17 and IL-22. Interaction of Foxp3 with RORα and RORγt inhibits the expression of these genes (Ichiyama et al., 2008; Zhou et al., 2008a) (Du et al., 2008). HIF-1α binds to the C terminus of Foxp3 and targets Foxp3 for degradation during Th17 development (Dang et al., 2011). As mentioned above, the differentiation of Th2 and Th17 differentiation can also be suppressed by the GFP insertion at the N-terminal domain of Foxp3, which blocked HIF-1α and increased IRF4 interactions with Foxp3 (Darce et al., 2012). Taken together, these interactions indicate the importance of Foxp3 N-terminal domain in balancing the differentiation between Treg cells and other type cells such as Th17.
The N-terminal of Foxp3 is also reported to interact with the C-terminal of c-Rel, a member of the NF-κB family that is responsible for the up-regulation of a variety of pro-inflammatory cytokines. The interaction between Foxp3 and c-Rel may also contribute to the nucleus shuttling of Foxp3, as forkhead domain deleted Foxp3, which is mainly located at cytoplasm, were predominantly found in the nucleus in the presence of ectopically expressed c-Rel (Bettelli et al., 2005; Loizou et al., 2011).
In addition to the interaction with numerous transcriptional factors, FOXP3 also interacts with some post-translational enzymes that regulate the activity of FOXP3 at post-translational level (Xiao et al., 2010; Zhang et al., 2012). Among these enzymes, histone acetyltransferase TIP60 and histone deacetylase HDAC7 interacts with the N-terminal of FOXP3 and increases its repressive transcriptional activity (Li et al., 2007). TIP60 promotes the acetylation of FOXP3 and the acetyltransferase activity of TIP60 is required for the enhanced repressive transcriptional activity of FOXP3, but TIP60 may also play a role in regulating FOXP3 activity independent of its HAT activity. Binding of TIP60 with FOXP3 may regulate the oligomerization status of FOXP3 and therefore its DNA binding, for it is reported before that FOXP3 N-terminal domain may form high-order clustering. Besides, TIP60 may also recruit additional factors such as HDAC7 required for transcriptional repression/activation. A summary of the functions and binding partners of FOXP3 N-terminal domain is indicated in Figure 3.
Figure 3.
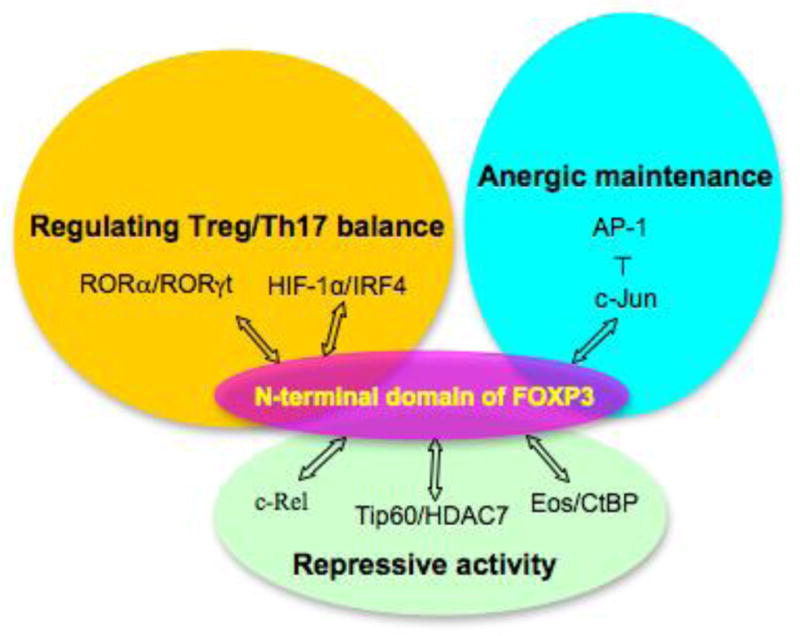
Summary of the functions and binding partners of FOXP3 N-terminal domain described in the text.
Post-translational modification of N-terminal domain
So far, among the various post-translational modifications of Foxp3, acetylation is the best understood modification in Foxp3. Multiple sites in Foxp3 are acetylated, and one of them, K31, is located in the N-terminal domain of Foxp3. One effect of acetylation is to counteract with ubiquitination, therefore increasing Foxp3 stability by preventing proteosome-dependent degradation (van Loosdregt et al., 2011; van Loosdregt et al., 2010). In accordance with this, acetylation of K31 by p300, or substitution of K31 with R, greatly increases the stability of Foxp3, indicating that K31 is also an ubiquitination site (Kwon et al., 2012).
In addition to acetylation and ubiquitination, mass spectrum analysis also shows that there are phosphorylation sites in the N-terminal domain of Foxp3. One potential kinase responsible for Foxp3 phosphorylation has been identified already in our lab, but the role of phosphorylation in regulating Foxp3 activity is still under investigation (Guoping Deng and Mark I. Greene, unpublished data). Possible crosstalk between acetylation, ubiquitination and phosphorylation of FOXP3, possibly in response to different extra-cellular stimulations, may exist to regulate FOXP3 activity and stability required for Treg cells development and maintenance.
Conclusion
FOXP3 has a unique N-terminal domain that has no sequence similarity with other members of the FOXP family. This N-terminal domain plays an essential role in the suppressive activity of FOXP3 by regulating the nuclear importation of FOXP3, the interaction with other transcription factor, and the stability of FOXP3 through post-translational modifications. More importantly, specific functions of Treg cells can be regulated through the differential interaction of FOXP3 N-terminal domain with other transcription factors.
The interaction of FOXP3 with other transcription factors could be regulated through post-translational modifications of FOXP3. Acetylation of FOXP3 K249/251 in the zinc finger region has been shown to destabilize FOXP3 homodimer and heterodimer with FOXP1, therefore decreasing the DNA binding ability of FOXP3 (Song et al., 2012). Since N-terminal domain of FOXP3 also subjects to post-translational modifications, it will be interesting to investigate whether post-translational modification would affect the interaction of N-terminal domain with other transcription factors, therefore conferring Treg cells different functions. It may be possible to modulate the specific function of Treg cells for the treatment of specific autoimmune diseases by regulating the interaction of FOXP3 N-terminal domain with other interacting proteins.
Acknowledgments
This work was supported by the grant from the National Institutes of Health to M.I.G. (PO1 AI073489-03).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aarts-Riemens T, et al. Forced overexpression of either of the two common human Foxp3 isoforms can induce regulatory T cells from CD4(+)CD25(−) cells. European journal of immunology. 2008;38:1381–90. doi: 10.1002/eji.200737590. [DOI] [PubMed] [Google Scholar]
- Allan SE, et al. The role of 2 FOXP3 isoforms in the generation of human CD4+ Tregs. The Journal of clinical investigation. 2005;115:3276–84. doi: 10.1172/JCI24685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artenstein AW, Opal SM. Proprotein convertases in health and disease. The New England journal of medicine. 2011;365:2507–18. doi: 10.1056/NEJMra1106700. [DOI] [PubMed] [Google Scholar]
- Bandukwala HS, et al. Structure of a domain-swapped FOXP3 dimer on DNA and its function in regulatory T cells. Immunity. 2011;34:479–91. doi: 10.1016/j.immuni.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S, et al. Cutting edge: Foxp3-mediated induction of pim 2 allows human T regulatory cells to preferentially expand in rapamycin. Journal of immunology. 2008;180:5794–8. doi: 10.4049/jimmunol.180.9.5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett CL, et al. A rare polyadenylation signal mutation of the FOXP3 gene (AAUAAA-->AAUGAA) leads to the IPEX syndrome. Immunogenetics. 2001;53:435–9. doi: 10.1007/s002510100358. [DOI] [PubMed] [Google Scholar]
- Bettelli E, et al. Foxp3 interacts with nuclear factor of activated T cells and NF-kappa B to repress cytokine gene expression and effector functions of T helper cells. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:5138–43. doi: 10.1073/pnas.0501675102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettini ML, et al. Loss of epigenetic modification driven by the Foxp3 transcription factor leads to regulatory T cell insufficiency. Immunity. 2012;36:717–30. doi: 10.1016/j.immuni.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunkow ME, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nature genetics. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- Chatila TA, et al. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. The Journal of clinical investigation. 2000;106:R75–81. doi: 10.1172/JCI11679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang EV, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146:772–84. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darce J, et al. An N-terminal mutation of the Foxp3 transcription factor alleviates arthritis but exacerbates diabetes. Immunity. 2012;36:731–41. doi: 10.1016/j.immuni.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Zoeten EF, et al. Foxp3 processing by proprotein convertases and control of regulatory T cell function. The Journal of biological chemistry. 2009;284:5709–16. doi: 10.1074/jbc.M807322200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, et al. Isoform-specific inhibition of ROR alpha-mediated transcriptional activation by human FOXP3. Journal of immunology. 2008;180:4785–92. doi: 10.4049/jimmunol.180.7.4785. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, et al. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nature immunology. 2003;4:330–6. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, et al. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–41. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Gavin MA, et al. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007;445:771–5. doi: 10.1038/nature05543. [DOI] [PubMed] [Google Scholar]
- Hancock WW, Ozkaynak E. Three distinct domains contribute to nuclear transport of murine Foxp3. PloS one. 2009;4:e7890. doi: 10.1371/journal.pone.0007890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes D, et al. FoxP3 enhances HIV-1 gene expression by modulating NFkappaB occupancy at the long terminal repeat in human T cells. The Journal of biological chemistry. 2007;282:15973–80. doi: 10.1074/jbc.M702051200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori S, et al. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–61. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- Ichiyama K, et al. Foxp3 inhibits RORgammat-mediated IL-17A mRNA transcription through direct interaction with RORgammat. The Journal of biological chemistry. 2008;283:17003–8. doi: 10.1074/jbc.M801286200. [DOI] [PubMed] [Google Scholar]
- Kalderon D, et al. Sequence requirements for nuclear location of simian virus 40 large-T antigen. Nature. 1984;311:33–8. doi: 10.1038/311033a0. [DOI] [PubMed] [Google Scholar]
- Katoh H, et al. FOXP3 orchestrates H4K16 acetylation and H3K4 trimethylation for activation of multiple genes by recruiting MOF and causing displacement of PLU-1. Molecular cell. 2011;44:770–84. doi: 10.1016/j.molcel.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khattri R, et al. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nature immunology. 2003;4:337–42. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- Kwon HS, et al. Three novel acetylation sites in the Foxp3 transcription factor regulate the suppressive activity of regulatory T cells. Journal of immunology. 2012;188:2712–21. doi: 10.4049/jimmunol.1100903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SM, et al. FoxP3 maintains Treg unresponsiveness by selectively inhibiting the promoter DNA-binding activity of AP-1. Blood. 2008;111:3599–606. doi: 10.1182/blood-2007-09-115014. [DOI] [PubMed] [Google Scholar]
- Li B, Greene MI. FOXP3 actively represses transcription by recruiting the HAT/HDAC complex. Cell cycle. 2007;6:1432–6. [PubMed] [Google Scholar]
- Li B, et al. FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:4571–6. doi: 10.1073/pnas.0700298104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, et al. Regulatory T cell development in the absence of functional Foxp3. Nature immunology. 2007;8:359–68. doi: 10.1038/ni1445. [DOI] [PubMed] [Google Scholar]
- Loizou L, et al. Foxp3 interacts with c-Rel to mediate NF-kappaB repression. PloS one. 2011;6:e18670. doi: 10.1371/journal.pone.0018670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes JE, et al. Analysis of FOXP3 reveals multiple domains required for its function as a transcriptional repressor. Journal of immunology. 2006;177:3133–42. doi: 10.4049/jimmunol.177.5.3133. [DOI] [PubMed] [Google Scholar]
- Pan F, et al. Eos mediates Foxp3-dependent gene silencing in CD4+ regulatory T cells. Science. 2009;325:1142–6. doi: 10.1126/science.1176077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins J, et al. Two interdependent basic domains in nucleoplasmin nuclear targeting sequence: identification of a class of bipartite nuclear targeting sequence. Cell. 1991;64:615–23. doi: 10.1016/0092-8674(91)90245-t. [DOI] [PubMed] [Google Scholar]
- Rudra D, et al. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nature immunology. 2012 doi: 10.1038/ni.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidah NG, Chretien M. Proprotein and prohormone convertases: a family of subtilases generating diverse bioactive polypeptides. Brain research. 1999;848:45–62. doi: 10.1016/s0006-8993(99)01909-5. [DOI] [PubMed] [Google Scholar]
- Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30:636–45. doi: 10.1016/j.immuni.2009.04.010. [DOI] [PubMed] [Google Scholar]
- Smith EL, et al. Splice variants of human FOXP3 are functional inhibitors of human CD4+ T-cell activation. Immunology. 2006;119:203–11. doi: 10.1111/j.1365-2567.2006.02425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, et al. Structural and Biological Features of FOXP3 Dimerization Relevant to Regulatory T Cell Function. Cell reports. 2012;1:665–75. doi: 10.1016/j.celrep.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. The Journal of experimental medicine. 1998;188:287–96. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vliet HJ, Nieuwenhuis EE. IPEX as a result of mutations in FOXP3. Clinical & developmental immunology. 2007;2007:89017. doi: 10.1155/2007/89017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loosdregt J, et al. Rapid temporal control of Foxp3 protein degradation by sirtuin-1. PloS one. 2011;6:e19047. doi: 10.1371/journal.pone.0019047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loosdregt J, et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood. 2010;115:965–74. doi: 10.1182/blood-2009-02-207118. [DOI] [PubMed] [Google Scholar]
- Wildin RS, et al. Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. Journal of medical genetics. 2002;39:537–45. doi: 10.1136/jmg.39.8.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–87. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- Xiao Y, et al. Histone acetyltransferase mediated regulation of FOXP3 acetylation and Treg function. Current opinion in immunology. 2010;22:583–91. doi: 10.1016/j.coi.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi H, et al. Crucial role of FOXP3 in the development and function of human CD25+CD4+ regulatory T cells. International immunology. 2004;16:1643–56. doi: 10.1093/intimm/dxh165. [DOI] [PubMed] [Google Scholar]
- Zeng WP, et al. Domain requirements for the diverse immune regulatory functions of foxp3. Molecular immunology. 2011;48:1932–9. doi: 10.1016/j.molimm.2011.05.023. [DOI] [PubMed] [Google Scholar]
- Zhang H, et al. Immune regulation by histone deacetylases: a focus on the alteration of FOXP3 activity. Immunology and cell biology. 2012;90:95–100. doi: 10.1038/icb.2011.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008a;453:236–40. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, et al. FOXP3 and its partners: structural and biochemical insights into the regulation of FOXP3 activity. Immunologic research. 2008b;42:19–28. doi: 10.1007/s12026-008-8029-x. [DOI] [PubMed] [Google Scholar]
- Ziegler SF. FOXP3: of mice and men. Annual review of immunology. 2006;24:209–26. doi: 10.1146/annurev.immunol.24.021605.090547. [DOI] [PubMed] [Google Scholar]