

Abstract
Inflammatory macrophages have been implicated in hepatotoxicity induced by the analgesic, acetaminophen (APAP). In these studies we characterized the phenotype of macrophages accumulating in the liver following APAP intoxication and evaluated the role of galectin-3 (Gal-3) in macrophage activation. Administration of APAP (300 mg/kg, i.p.) to wild type mice resulted in the appearance of two distinct subpopulations of CD11b+ cells in the liver, which expressed high or low levels of the monocyte/macrophage activation marker Ly6C. Whereas CD11b+/Ly6Chi macrophages exhibited a classically activated proinflammatory phenotype characterized by increased expression of TNF-α, inducible nitric oxide synthase (iNOS), and CCR2, CD11b+/Ly6Clo macrophages were alternatively activated, expressing high levels of the anti-inflammatory cytokine, IL-10. APAP intoxication was also associated with an accumulation of Gal-3+ macrophages in the liver; the majority of these cells were Ly6Chi. APAP-induced increases in CD11b+/Ly6Chi macrophages were significantly reduced in Gal-3−/− mice. This was evident 72 h post-APAP and was correlated with reduced expression of the classical macrophage activation markers, iNOS, IL-12, and TNF-α, as well as the proinflammatory chemokines, CCL2 and CCL3, and chemokine receptors CCR1, and CCR2. Conversely, numbers of CD11b+/Ly6Clo macrophages increased in livers of APAP-treated Gal-3−/− mice. This was associated with increased expression of the alternative macrophage activation markers Ym1 and Fizz1, increased liver repair and reduced hepatotoxicity. These data demonstrate that both classically and alternatively activated macrophages accumulate in the liver following APAP intoxication; moreover, Gal-3 plays a role in promoting a persistent proinflammatory macrophage phenotype.
Introduction
Liver injury caused by overdose of the analgesic acetaminophen (APAP) is the major cause of acute liver failure in the United States (1). APAP intoxication is characterized by centrilobular hepatocellular necrosis, which is initiated by covalent binding of the reactive APAP metabolite, N-acetyl-parabenzoquinoneimine (NAPQI), to critical protein targets in the liver (2). Evidence suggests that activated macrophages contribute to the pathogenic response to APAP. However, the role of these cells in APAP hepatotoxicity depends on their origin, the timing of their appearance in the liver, and the inflammatory mediators they encounter, which control their phenotype and function. Based on studies using macrophage inhibitors and transgenic mice, two subpopulations of macrophages have been identified in the liver after APAP intoxication that play distinct roles in hepatotoxicity: classically activated proinflammatory/cytotoxic macrophages, and alternatively activated anti-inflammatory/wound repair macrophages (3-7). It appears that the outcome of tissue injury depends on which macrophage subpopulation predominates. Thus, hepatotoxicity results from exaggerated or persistent responses of classically activated macrophages, whereas hepatoprotection is associated with increases in numbers of alternatively activated macrophages (reviewed in 8 and 9). The mechanisms regulating classical and alternative macrophage activation in the liver after APAP intoxication have not been established.
Gal-3 is a β-galactoside binding lectin secreted by macrophages in response to LPS, TNF-α, or IFN-γ (10, 11). Gal-3 acts in an autocrine and paracrine manner to promote macrophage release of proinflammatory mediators, including TNF-α, IL-12, CCL3, and CCL4, as well as reactive nitrogen species generated via inducible nitric oxide synthase (iNOS) (10-13). Loss of Gal-3 has been reported to result in reduced susceptibility to antigen-induced arthritis, renal ischemia-reperfusion injury, hypoxic-ischemic brain injury, and concanavalin A-induced hepatotoxicity, pathologies associated with exaggerated proinflammatory mediator activity (14-17). These findings led us to hypothesize that Gal-3 plays a role in promoting classical macrophage activation and inflammatory mediator production in the liver following APAP intoxication. This is supported by our findings of reduced hepatotoxicity and inflammatory mediator production in response to APAP in mice lacking Gal-3 (18). In the present studies, we extended these observations and characterized the role of Gal-3 in regulating the phenotype of hepatic macrophage subpopulations accumulating in the liver during APAP-induced hepatotoxicity. Results from these studies provide additional support for a contribution of Gal-3 to promoting inflammation in the liver following APAP intoxication.
Materials and Methods
Animals
Male specific pathogen-free C57Bl/6J wild type and Gal-3−/− mice (8-12 weeks old) were obtained from the Jackson Laboratory (Bar Harbor, ME). Gal-3−/− mice were backcrossed to a C57Bl/6 background for more than 10 generations. Mice were housed in microisolation cages and allowed free access to food and water. All animals received humane care in compliance with the institution’s guidelines, as outlined in the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health. Mice were fasted overnight prior to i.p administration of APAP (300 mg/kg) or PBS control. Mice were euthanized 24-72 h later with nembutal (200 mg/kg). Liver samples (100 mg aliquots) were collected and stored at −20°C in RNAlater until RNA isolation. The remaining tissue was snap frozen in liquid nitrogen.
Hepatic nonparenchymal cell isolation
Nonparenchymal cells were isolated from the liver as previously described, with some modifications (19). The liver was perfused through the portal vein with warm Ca2+/Mg2+-free Hank’s balanced salt solution (pH 7.3) containing 25 mM HEPES and 0.5 mM EGTA, followed by Leibowitz L-15 medium containing HEPES, 0.2 U/ml Liberase 3 Blendzyme, and 0.5 mg/ml protease type XIV. The liver was excised, disaggregated, and incubated with 2 mg/ml protease type XIV for 15 min at 37°C. The resulting cell suspension was filtered through a 220 μm nylon mesh. Hepatocytes were separated from nonparenchymal cells by four successive washes (50×g, 3 min). Supernatants containing nonparenchymal cells were centrifuged (300×g, 7 min), and the cells purified by density gradient centrifugation using Optiprep medium (Sigma-Aldrich, St Louis, MO). Viability was assessed by trypan blue dye exclusion and was >95%.
Flow cytometry/cell sorting
Cells were analyzed immediately following isolation. Non-specific binding was blocked by incubation of the cells with anti-mouse-FcRII/III antibody (BD Biosciences, Franklin Lakes, NJ) for 5 min at 4°C. This was followed by 30 min incubation with FITC-conjugated anti-CD11b and PE-conjugated anti-Ly6C antibodies or isotype controls (BioLegend, San Diego, CA). Cells were then fixed in 3% paraformaldehyde, permeabilized in buffer containing 0.1% saponin, 0.1% sodium azide, and 1% fetal bovine serum in PBS, and stained with anti-Gal-3 antibody or goat IgG (R&D Systems, Minneapolis, MN), followed by isotype-specific AlexaFluor633-conjugated secondary antibody (Molecular Probes, Carlsbad, CA). Cells were analyzed using an FC500 flow cytometer (Beckman Coulter, Brea, CA). For sorting, cells were incubated with anti-mouse-FcRII/III antibody, followed by FITC-conjugated anti-CD45, AlexaFluor647-conjugated CD11b, and PE-conjugated anti-Ly6C antibodies (BioLegend, San Diego, CA) for 30 min. DAPI was added to the cell suspension immediately before analysis to exclude dead cells. Cells were sorted into DAPI−/CD45+/CD11b+/Ly6Chi and DAPI−/CD45+/CD11b+/Ly6Clo subpopulations using a Beckman Coulter MoFlo XDP Cell Sorter (Brea, CA), and immediately processed for RNA isolation.
Histology and immunohistochemistry
Livers were collected and 5 mm samples of the left lobes immediately fixed overnight at 4°C in 3% paraformaldehyde/2% sucrose. Tissue was washed three times in PBS containing 2% sucrose, and then transferred to 50% ethanol. After embedding in paraffin, 5 μm sections were prepared. For immunohistochemistry, sections were rehydrated and stained with antibody to PCNA (1:800, Abcam, Cambridge, MA), Ym1 (1:450, StemCell, Vancouver, BC) or IgG control (ProSci, Poway, CA). Binding was visualized using a Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA). Three to five random sections of each liver were examined.
Immunofluorescence
Liver samples (5 mm) were snap frozen in liquid nitrogen-cooled isopentane and embedded in OCT medium (Sakura Finetek, Torrance, CA). Six μm sections were prepared and fixed in 90% acetone/10% methanol. For double immunofluorescence, a sequential staining procedure was used (20). Sections were stained with anti-Ly6C antibody (1:50, AbD Serotec, Kidlington, UK), followed by isotype-specific AlexaFluor488-conjugated secondary antibody (Molecular Probes, Carlsbad, CA). After blocking with 5% rat serum, sections were stained with FITC-conjugated anti-F4/80 antibody (1:50, AbD Serotec, Kidlington, UK), followed by anti-FITC AlexaFluor488-conjugated secondary antibody. Images were acquired using a Leica SP5 confocal microscope (Leica Microsystems, Wetzlar, Germany). Identical laser power, gain and offset settings were used for all analyses.
Western blotting
Liver samples (30 mg) were lysed in buffer containing 20 mM HEPES, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1.5 mM MgCl2, 1 mM diethylene-triamine pentaaacetic acid (DTPA), 1 mM phenylmethylsulfonylfluoride, 10 mM sodium pyrophosphate, 50 mM sodium fluoride, 2 mM sodium orthovanadate, and protease inhibitor cocktail. Protein concentrations were measured using the Bradford Assay (Bio-Rad, Hercules, CA). Proteins were separated on Tris-glycine polyacrylamide gels (Bio-Rad, Hercules, CA) and transferred onto nitrocellulose membranes. Non-specific binding was blocked by incubation of the blots for 1 h at room temperature with buffer containing 5% non-fat milk, 10 mM Tris-base, 200 mM sodium chloride, and 0.1% polysorbate 20. Membranes were then incubated overnight at 4°C with anti-Gal-3 (1:2000) or anti-actin (1:1000) primary antibodies, followed by incubation with isotype-specific HRP-conjugated secondary antibodies (1:10,000) for 1 h at room temperature. Binding was visualized using an ECL Plus chemiluminescence kit (GE Healthcare, Piscataway, NJ).
Real-time PCR
Total RNA was isolated from liver samples using an RNeasy Mini kit, and from sorted monocytes/macrophages using an RNeasy Micro kit (Qiagen, Valencia, CA). RNA purity and concentration were measured using a Nanodrop spectrophotometer (Thermo Fisher Scientific, Wilmington, DE). RNA was converted into cDNA using a High Capacity cDNA Reverse Transcription kit according to manufacturer’s directions (Applied Biosystems, Foster City, CA). Standard curves were generated using serial dilutions from pooled randomly selected cDNA samples. Real-time PCR was performed using SYBR Green PCR Master Mix (Applied Biosystems) on a 7900HT thermocycler (Applied Biosystems). All PCR primer pairs were generated using Primer Express 2.0 (Applied Biosystems), and synthesized by Integrated DNA Technologies (Coralville, IA). For each sample, gene expression changes was normalized relative to 18s RNA. Data are expressed as fold change relative to control. Forward and reverse primer sequences were: TNF-α, AGGGATGAGAAGTTCCCAAATG and TGTGAGGGTCTGGGCCATA; iNOS, GGCAGCCTGTGAGACCTTTG and TGAAGCGTTTCGGGATCTG; IL-12, CCTGGAGCACTC CCCATTC and TGCGCTGGATTCGAACAA; CCL2, TTGAATGTGAAGTTGACCCGTAA and GCTTGAGGTTGTGGAAAAG; CCL3, TCTTCTCAGCGCCATATGGA and TCCGGCTGTAGGAGAAGCA; CCL4, AGGGTTCTCAGCACCAATGG and CCGGGAGGTGTAAGAGAAACAG; CX3CL1, GCACAGGATGCAGGGCTTAC and TGTCAGCCGCCTCAAAACT; CCR1, CTGAGGGCCCGAACTGTTAC and GGCTAGGGCCCAGGTGAT; CCR5, TGATAAGCTGCAAAAAGCTGAAGA and GTCAGAGATGGCCAGGTTGAG; CCR2, TCCACGGCATACTATCAACATCTC and GGCCCCTTCATCAAGCTCTT; CX3CR1, TCGGTCTGGTGGGAAATCTG and GGCTTCCGGCTGTTGGT; Gal-1, CGGACGCCAAGAGCTTTGT and TGAAGTGTAGGCACAGGTTGTTG; found in inflammatory zone-1 (Fizz-1), CAGCTGATGGTCCCAGTGAA and TTCCTTGACCTTATTCTCCACGAT; Ym1, TCTGGTGAAGGAAATGCGTAAA and GCAGCCTTGGAATGTCTTTCTC; 18sRNA, CGGCTACCACATCCAAGGAA and GCTGGAATTACCGCGGCT.
Statistical analysis
Experiments were repeated two to three times. Data were analyzed using the Student’s t test or one-way ANOVA followed by Dunn’s post hoc analysis. A p value of < 0.05 was considered statistically significant.
Results
Distinct macrophage subpopulations accumulate in the liver following APAP intoxication
In our initial series of studies we used techniques in flow cytometry/cell sorting to assess the phenotype of macrophages accumulating in the liver after APAP intoxication. CD11b is the alpha chain of the Mac-1 integrin expressed on myeloid cells (21). APAP administration resulted in a time-related increase in CD11b+ cells in the liver (Fig. 1, panels A and D). This was evident within 24 h and persisted for at least 72 h after APAP. To characterize these cells, we analyzed their expression of Ly6C, a surface antigen present at high levels on proinflammatory monocytes/macrophages (22). In livers of both control and APAP-treated mice, two distinct subpopulations of CD11b+ cells were identified based on their expression of Ly6C. These consisted of Ly6Clo cells and Ly6Chi cells (Fig. 1, panel A). In wild type mice, the majority of CD11b+ cells expressed low levels of Ly6C. Following APAP administration, time-related increases in both Ly6Clo and Ly6Chi subpopulations were observed. Time-related increases in cells expressing Ly6C were also observed in histological sections following APAP administration (Fig. 2). These cells were mainly noted in centrilobular regions of the liver and were distinct from F4/80+ resident Kupffer cells. In contrast to Ly6C+ cells, F4/80+ macrophages decreased in areas surrounding the central veins following APAP administration, a response which persisted for 48 h; subsequently, they began to reappear.
Fig. 1. Characterization of macrophage subpopulations in the liver following APAP intoxication.
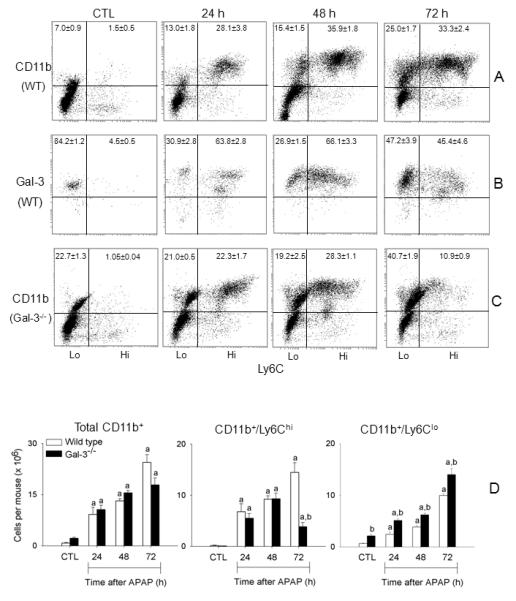
Liver nonparenchymal cells were isolated 24-72 h after treatment of wild type mice (panels A and B) or Gal-3−/− mice (panel C) with APAP or PBS control (CTL). Cells were stained with antibodies to CD11b, Ly6C and Gal-3, or appropriate isotypic controls as described in the Materials and Methods, and then analyzed by flow cytometry. Panel A. Expression of CD11b and Ly6C by liver nonparenchymal cells from wild type (WT) mice. Panel B. Expression of Ly6C and Gal-3 by CD11b+ nonparenchymal cells from WT mice. Panel C. Expression of CD11b and Ly6C by liver nonparenchymal cells from Gal-3−/− mice. The percentages (mean ± SE) of each cell population are indicated. One representative plot from 4-12 mice is shown. Panel D. The number of CD11b+ cells, CD11b+/Ly6Chi cells, and CD11b+/Ly6Clo cells was calculated from the percentage-positive cells relative to the total number of liver nonparenchymal cells recovered. Bars represent the mean ± SE (n=4-12 mice). aSignificantly different (p<0.05) from CTL. bSignificantly different (p<0.05) from wild type mice.
Fig. 2. Effects of APAP on liver macrophage expression of Ly6C and F4/80.
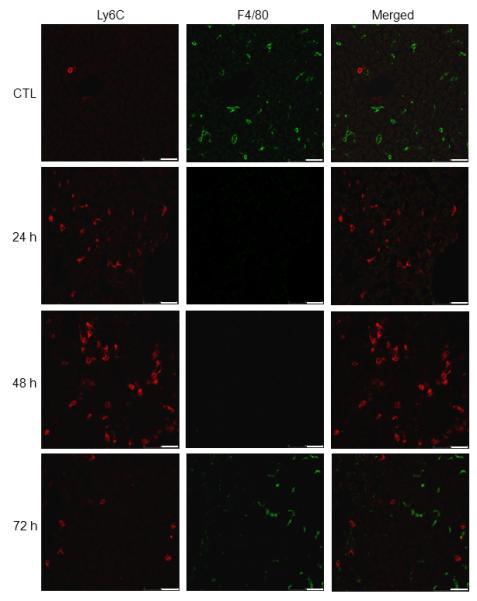
Liver sections, prepared 24-72 h after treatment of wild type mice with APAP or PBS control (CTL), were sequentially stained with anti-Ly6C and anti-F4/80 antibodies, as described in the Materials and Methods. Scale bar, 25 μm. One representative section from 3-4 mice is shown.
To further characterize the Ly6Clo and Ly6Chi macrophage subpopulations responding to APAP, the cells were sorted and examined microscopically. Giemsa staining confirmed that both subpopulations consisted of mononuclear phagocytes (Fig. 3, upper panel). Ly6Clo cells were enlarged relative to Ly6Chi cells, and more irregularly shaped. Ly6Clo cells also contained highly vacuolated cytoplasm, and displayed an increased cytoplasmic:nuclear ratio. RT-PCR analysis of the sorted cells revealed that Ly6Chi cells expressed higher mRNA levels of the proinflammatory proteins, TNF-α and iNOS, and the chemokine receptor CCR2, when compared to Ly6Clo cells (Fig. 3, lower panel). In contrast, mRNA expression of the anti-inflammatory cytokine IL-10 was reduced in Ly6Chi cells when compared to Ly6Clo cells.
Fig. 3. Characterization of Ly6Chi and Ly6Clo macrophages accumulating in the liver following APAP intoxication.
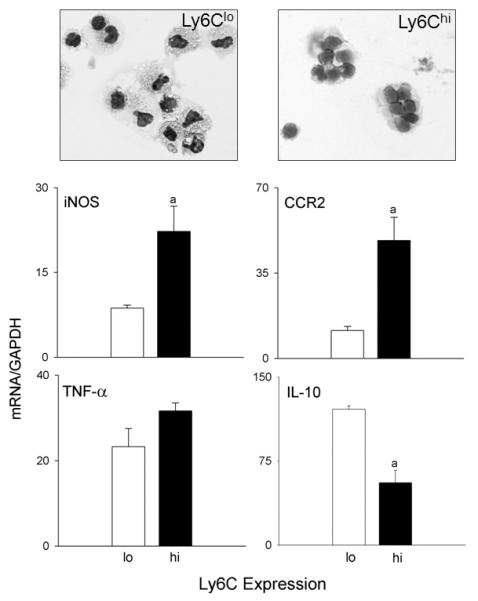
Liver nonparenchymal cells, isolated 48 h after treatment of wild type mice with APAP, were stained with anti-CD11b and anti-Ly6C antibodies, as described in the Materials and Methods. CD11b+ cells were sorted based on expression of Ly6C into CD11b+/Ly6Chi and CD11b+/Ly6Clo subpopulations. Upper panel. Cytospin preparations of sorted cells were stained with Giemsa. Lower panel. mRNA expression of sorted cells was assessed by RT-PCR. Data were normalized relative to GAPDH; bars represent mean ± SE (n=4-6).
Role of Gal-3 in macrophage activation in the liver following APAP intoxication
Consistent with our earlier studies (18), we found that APAP intoxication was associated with a time-dependent increase in Gal-3 protein expression in the liver, which was evident within 24 h and persisted for at least 72 h (Fig. 4, upper and middle panel). Immunostaining of the liver showed that Gal-3 was expressed by macrophages infiltrating into necrotic areas of the liver (Fig. 4, lower panel and 18). Flow cytometric analyses revealed that the majority of the Gal-3+ macrophages infiltrating into the liver 24-48 h after APAP expressed high levels of Ly6C (Fig. 1, panel B). In contrast, in control mice, most Gal-3-positive macrophages were Ly6Clo. APAP administration resulted in a decrease in the percentage of Gal-3+/LyClo cells at 24 h and 48 h; by 72 h, the percentage of these cells began to increase (Fig. 1, panel B).
Fig. 4. Effects of APAP intoxication on Gal-3 expression in the liver.
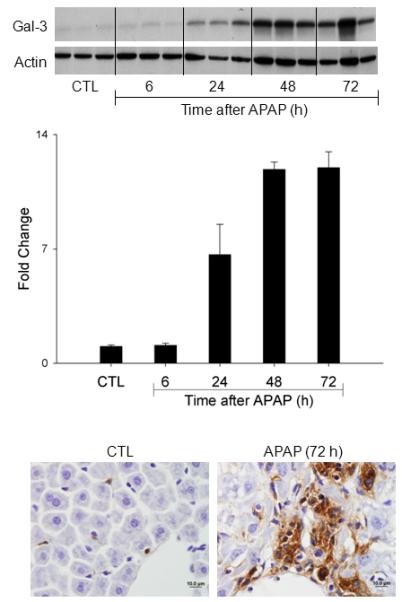
Livers were collected 6-72 h after treatment of wild type mice with APAP or PBS control (CTL). Upper panel. Gal-3 expression was analyzed by western blotting. Actin was used as the loading control. Each lane represents a different animal. Middle panel. Densitometric analysis was performed using ImageJ. Each bar represents the mean ± SE (n = 3 mice). Lower panel. Livers were collected 72 h after treatment of wild type mice with APAP or control. Gal-3 expression was analyzed by immunohistochemistry. One representative section from 6 mice is shown. Original magnification, 100x.
We previously reported reduced injury and inflammation in the liver following APAP intoxication in mice lacking Gal-3 (18). In further studies we determined if this was associated with alterations in the macrophage subpopulations that appeared in the liver in response to APAP. Whereas loss of Gal-3 had no effect on the total number of CD11b+ cells in the liver following APAP intoxication, a significant decrease in CD11b+/Ly6Chi macrophages was observed (Fig. 1, panels C and D). This was associated with an increase in CD11b+/Ly6Clo macrophages. Increased numbers of CD11b+/Ly6Clo macrophages were also observed in PBS control-treated Gal-3−/− mice. Changes in liver macrophage subpopulations in APAP-treated Gal-3−/− mice were correlated with decreases in APAP-induced expression of the proinflammatory proteins IL-12, iNOS, and TNF-á, which was evident at 24 h and 48 h for IL-12, and at 48 h and 72 h for iNOS and TNF-á, as well as the chemokines CCL2 and CCL3, and the chemokine receptors CCR1 and CCR2, effects most prominent after 72 h (Figs. 5 and 6). Delayed CCL4 expression was also noted. Conversely, increases in expression of CX3CL1/CX3CR1 were observed in Gal-3−/− mice relative to wild type mice at 24 h post-APAP; however, by 72 h, CX3CR1 expression was reduced in these mice (Fig. 6). APAP-induced expression of CCR5 was not altered by loss of Gal-3 (Fig. 6). mRNA expression of Ym1 and Fizz-1, markers of alternatively activated anti-inflammatory/wound repair macrophages (23), was also increased in Gal-3−/− mice, when compared to wild type mice, after APAP administration (Figs. 5 and 7). Interestingly, whereas in control mice Ym1 protein was expressed in hepatic sinusoidal endothelial cells, after APAP administration Ym1 was upregulated in macrophages (Fig. 7). This was first evident 48 h post-APAP, and was correlated with decreased Ym1 expression in endothelial cells. Loss of Gal-3 was associated with a more rapid and abundant increase in Ym1-positive macrophages in the liver, which was apparent within 24 h, and remained elevated for at least 72 h. In contrast, expression of the anti-inflammatory lectin, Gal-1, which increased in wild type mice 48 h and 72 h after APAP administration, was not significantly altered by loss of Gal-3 (Fig. 5). Alterations in liver macrophage subpopulations and inflammatory mediator expression in Gal-3−/− mice were also associated with increased repair of APAP-induced injury. Thus, significant increases in hepatocyte proliferation, as measured by the number of PCNA-positive cells and mitotic index, were observed in the livers of Gal-3−/− mice when compared to wild type mice (Figs. 8 and 9).
Fig. 5. Effects of APAP intoxication on expression of markers of classical and alternative macrophage activation.
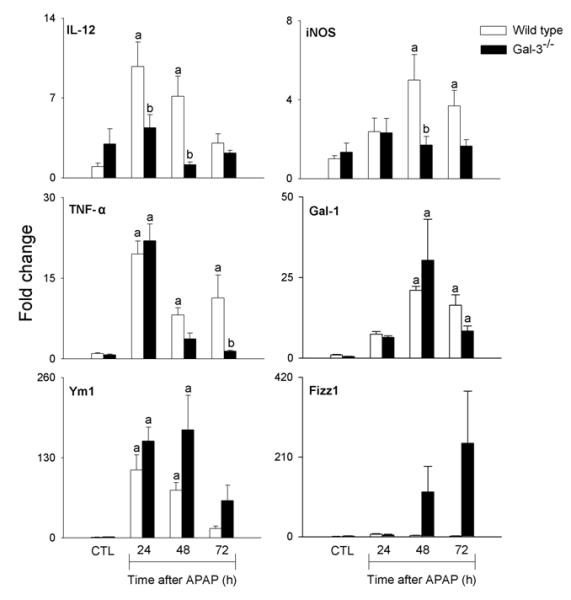
mRNA was prepared from liver samples 24-72 h after treatment of wild type and Gal-3−/− mice with APAP or PBS control (CTL), and analyzed by RT-PCR. Data were normalized to 18s RNA and presented as fold change relative to control. Each bar represents the mean ± SE (n=3-8 mice). aSignificantly different (p<0.05) from CTL. bSignificantly different (p<0.05) from wild type mice.
Fig. 6. Effects of APAP intoxication on expression of chemokines and chemokine receptors.
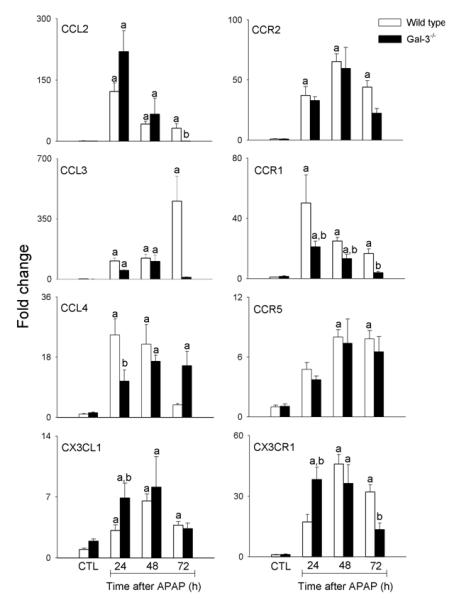
mRNA was prepared from liver samples collected 24-72 h after treatment of wild type and Gal-3−/− mice with APAP or PBS control (CTL), and analyzed by RT-PCR. Data were normalized to 18s RNA and presented as fold change relative to PBS control. Each bar represents the mean ± SE (n=3-8 mice). aSignificantly different (p<0.05) from CTL. bSignificantly different (p<0.05) from wild type mice.
Fig. 7. Effects of APAP intoxication on Ym1 expression.
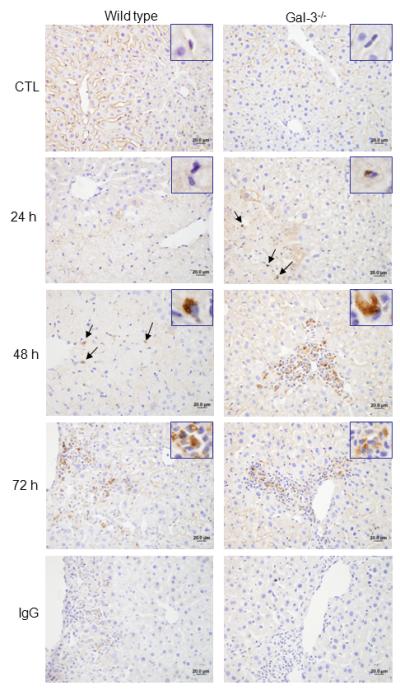
Liver sections, prepared 24-72 h after treatment of wild type and Gal-3−/− mice with APAP or PBS control (CTL), were stained with anti-Ym1 antibody or IgG control. Binding was visualized using a Vectastain Elite ABC kit, with 3,3′-diaminobenzidine as substrate. One representative section from 3 mice is shown. Original magnification, 40x. Insets, 100x.
Fig. 8. Effects of APAP administration on PCNA expression.
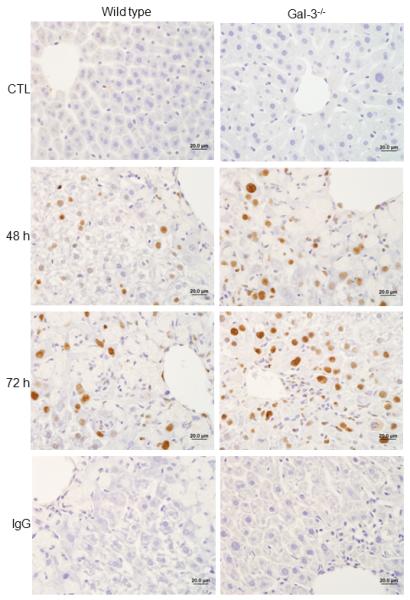
Liver sections, prepared 48-72 h after treatment of wild type and Gal-3−/− mice with APAP or PBS control, were stained with anti-PCNA antibody or IgG control. One representative section from 4-6 mice is shown. Original magnification, 63x.
Fig. 9. Effects of loss of Gal-3 on liver repair following APAP intoxication.
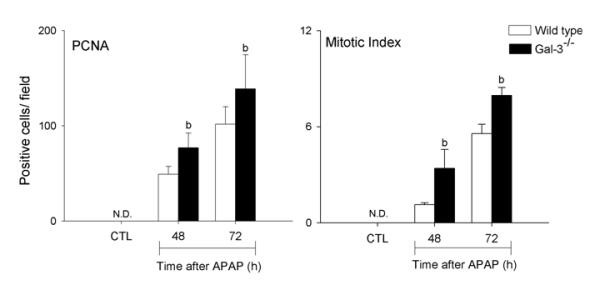
Liver sections were prepared 48-72 h after treatment of wild type and Gal-3−/− mice with APAP or PBS control (CTL). Left panel. Sections were stained with anti-PCNA antibody or IgG control. PCNA-positive hepatocyte nuclei were counted in six random fields per section (magnification 200x). Bars represent the mean ± SE (n=3-6 mice). bSignificantly different (p<0.05) from wild type mice. Right panel. Sections were stained with hematoxylin and eosin and hepatocytes containing mitotic figure enumerated in 10-12 random fields. Mitotic index was calculated as the number of positive cells/field.
Discussion
Classically and alternatively activated macrophage subpopulations have been shown to play distinct roles in the pathogenesis of APAP-induced hepatotoxicity. Thus, while classically activated macrophages release cytotoxic/proinflammatory mediators which contribute to injury, alternatively activated macrophages down regulate inflammation and promote tissue repair (reviewed in 8). However, mechanisms regulating phenotypic activation of macrophages in the liver have not been established. The present studies demonstrate that Gal-3 plays a role in promoting persistent activation of proinflammatory/cytotoxic macrophages in the liver following APAP intoxication. These findings are important as they suggest a novel mechanism for classical macrophage activation during the pathogenesis of APAP-induced hepatotoxicity.
Evidence suggests that classically and alternatively activated macrophages accumulating in the liver after APAP-induced injury arise from distinct precursors (4-7,24). The present studies provide additional support for this concept. We confirmed that APAP intoxication results in a time related increase in CD11b+ macrophages in the liver, which are distinct from resident Kupffer cells (6). Additionally, we demonstrated that these CD11b+ cells are heterogeneous with respect to their expression levels of the macrophage activation marker, Ly6C. Thus two subpopulations were identified: cells expressing high levels of Ly6C and cells expressing low levels of Ly6C. Moreover, these CD11b+ subpopulations are functionally distinct. Whereas CD11b+/Ly6Chi macrophages exhibited a classically activated proinflammatory phenotype, characterized by increased mRNA expression of TNF-α, iNOS, and CCR2, CD11b+/Ly6Clo macrophages expressed increased mRNA levels of the anti-inflammatory cytokine, IL-10, indicating that they are alternatively activated. Both CD11b+/Ly6Chi and CD11b+/Ly6Clo macrophage subpopulations were found to increase in number in the liver following APAP intoxication; the response of Ly6Chi macrophages was more robust than that of Ly6Clo macrophages. Accumulating Ly6Chi monocytes/macrophages have been reported to contribute to the development of tissue injury and inflammation in the liver induced by high-fat diet and carbon tetrachloride, as well as acute pancreatitis and myocardial infarction (25-28), and we speculate that they play a similar pathogenic role in APAP-induced hepatotoxicity.
APAP-induced increases in CD11b+/Ly6Clo macrophages in the liver were also associated with the appearance of Ym1-positive alternatively activated macrophages in the tissue. Hepatic expression of the anti-inflammatory lectin, Gal-1, which is thought to promote alternative macrophage activation (29-31), also increased after APAP administration, suggesting a mechanism mediating their appearance in the liver. In contrast, APAP had no major effect on expression of Fizz-1, another marker of alternative macrophage activation (23). These data support the concept of subpopulation heterogeneity in alternatively activated macrophages (32). Depletion of alternatively activated macrophages with clodronate liposomes is associated with a reduction in APAP-induced IL-10 expression in the liver and exaggerated hepatotoxicity (4). These data, along with reports that IL-10 plays a protective role in the liver following APAP intoxication (33,34), suggest that IL-10 producing CD11b+/Ly6Clo cells are key to tissue repair in this model. This is supported by our findings that in Gal-3−/− mice, increases in CD11b+/Ly6Clo cells were correlated with accelerated tissue repair, as measured by hepatocyte proliferation and reduced hepatotoxicity (18).
Following APAP intoxication we also observed a time related increase in Gal-3+ macrophages in the liver, which is in accord with our previous findings (18). The present studies show that these cells express high levels of Ly6C indicating a proinflammatory phenotype. Our observation that in the absence of Gal-3 the number of Ly6Chi macrophages appearing in the liver following APAP administration was significantly decreased, and that expression of the classical macrophage activation markers, iNOS, TNF-α and IL-12, and the proinflammatory chemokines and chemokine receptors, CCL2, CCL3, CCR1 and CCR2, was reduced, are in accord with this idea. The fact that CD11b+/Ly6Clo anti-inflammatory/wound repair macrophages increased in livers of APAP-treated Gal-3−/− mice, and that this was correlated with a more rapid appearance of Ym1-positive macrophages in the tissue, and a marked increase in Fizz-1 expression, suggest a shift in the balance of macrophage subpopulations leading to accelerated tissue repair. Analogous increases in alternatively activated macrophages and hepatoprotection have been described in Gal-3−/− mice treated with concanavalin A (17). Of note is our observation that in control mice Gal-3 is expressed at low levels by CD11b+/Ly6Clo cells, supporting previous reports that some liver resident macrophage subpopulations express this protein (18,35). Expression of Gal-3 on resident Kupffer cells is consistent with findings that these cells are constitutively activated due to continuous exposure to lipopolysaccharide in the portal circulation (36).
We also noted more rapid increases in peak expression of CX3CR1 and its ligand CX3CL1 in the livers of Gal-3−/− mice treated with APAP. CX3CR1 is highly expressed on anti-inflammatory/wound repair monocytes and macrophages (22), while CX3CL1 is produced primarily by hepatocytes and stellate cells following liver injury (37,38). CX3CL1 and CX3CR1 have been shown to play a protective role in toxin A-induced enteritis and carbon tetrachloride-induced liver inflammation and fibrosis, effects thought to be due to the recruitment of alternatively activated anti-inflammatory macrophages to sites of infection/injury (37,39). It remains to be determined if the CX3CL1/CX3CR1 signaling pathway is involved in hepatic recruitment of anti-inflammatory/wound repair macrophages following APAP intoxication.
A question arises as to the mechanisms underlying the appearance of proinflammatory macrophages in the liver following APAP intoxication. Our findings that peak expression of key proinflammatory mediators including TNF-α and IL-12, and chemokines like CCL2 and CCL4, precede maximal increases in Ly6Chi/Gal-3+ macrophages in the liver suggest that the proinflammatory hepatic microenvironment promotes the development of classically activated macrophages. However, at present we cannot exclude the possibility that macrophages are recruited to the liver in response to Gal-3 or other chemokines, or danger-associated molecular patterns, such as high mobility group box-1 (HMGB1), released from APAP-injured hepatocytes (40-42). It appears that once localized in the liver, Gal-3+ macrophages play a role in maintaining the proinflammatory/cytotoxic microenvironment. In this regard, Gal-3 has been shown to upregulate expression of TNF-α, iNOS, and IL-12 in primary microglia and human monocytes (11,13). Moreover, reduced susceptibility to injury induced by streptozotocin, concanavalin A, or antigen-induced arthritis in Gal-3−/− mice is associated with decreased expression of these inflammatory proteins (14,17,43).
In contrast to our findings, Gal-3 has previously been reported to induce alternative activation in cultured macrophages (44). Differences between these results and our findings may be due to analysis of bone marrow-derived macrophages activated in vitro with defined concentrations of cytokines versus freshly isolated liver macrophages from APAP-injured mice. This may be important as Gal-3 expression and its intracellular distribution have been shown to be altered when primary macrophages are cultured in vitro (45).
In summary, the present studies identify and characterize multiple macrophage subpopulations accumulating in the liver following APAP intoxication, and demonstrate a role for Gal-3 in promoting persistent classical macrophage activation which contributes to hepatotoxicity. A more detailed understanding of the mechanisms regulating the phenotype of activated macrophages during APAP-induced liver injury may lead to the development of novel approaches to mitigating toxicity caused by this widely utilized analgesic.
Abbreviations used in this paper
- APAP
acetaminophen
- Fizz-1
found in inflammatory zone-1
- GSH
glutathione
- iNOS
inducible nitric oxide synthase
- PCNA
proliferating cell nuclear antigen
Footnotes
This work was supported by NIH grants R01GM034310, R01ES004738, R01CA132624, U54AR055073 and P30ES005022.
References
- 1.Lee WM. Acute liver failure. Semin. Respir. Crit. Care Med. 2012;33:36–45. doi: 10.1055/s-0032-1301733. [DOI] [PubMed] [Google Scholar]
- 2.Nelson SD, Bruschi SA. Mechanisms of acetaminophen-induced liver disease. In: Kaplowitz N, DeLeve LD, editors. Drug-Induced Liver Disease. 2nd ed Informa Healthcare; New York, NY: 2007. pp. 353–388. [Google Scholar]
- 3.Laskin DL, Gardner CR, Price VF, Jollow DJ. Modulation of macrophage functioning abrogates the acute hepatotoxicity of acetaminophen. Hepatology. 1995;21:1045–1050. [PubMed] [Google Scholar]
- 4.Ju C, Reilly TP, Bourdi M, Radonovich MF, Brady JN, George JW, Pohl LR. Protective role of Kupffer cells in acetaminophen-induced hepatic injury in mice. Chem. Res. Toxicol. 2002;15:1504–1513. doi: 10.1021/tx0255976. [DOI] [PubMed] [Google Scholar]
- 5.Dambach DM, Watson LM, Gray KR, Durham SK, Laskin DL. Role of CCR2 in macrophage migration into the liver during acetaminophen-induced hepatotoxicity in the mouse. Hepatology. 2002;35:1093–1103. doi: 10.1053/jhep.2002.33162. [DOI] [PubMed] [Google Scholar]
- 6.Holt MP, Cheng L, Ju C. Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J. Leukoc. Biol. 2008;84:1410–1421. doi: 10.1189/jlb.0308173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gardner CR, Hankey P, Mishin V, Francis M, Yu S, Laskin JD, Laskin DL. Regulation of alternative macrophage activation in the liver following acetaminophen intoxication by stem cell-derived tyrosine kinase. Toxicol. Appl. Pharmacol. 2012;262:139–148. doi: 10.1016/j.taap.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laskin DL. Macrophages and inflammatory mediators in chemical toxicity: a battle of forces. Chem. Res. Toxicol. 2009;22:1376–1385. doi: 10.1021/tx900086v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laskin DL, Sunil VR, Gardner CR, Laskin JD. Macrophages and tissue injury: agents of defense or destruction? Annu. Rev. Pharmacol. Toxicol. 2011;51:267–288. doi: 10.1146/annurev.pharmtox.010909.105812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu FT, Hsu DK, Zuberi RI, Kuwabara I, Chi EY, Henderson WR., Jr. Expression and function of galectin-3, a beta-galactoside-binding lectin, in human monocytes and macrophages. Am. J. Pathol. 1995;147:1016–1028. [PMC free article] [PubMed] [Google Scholar]
- 11.Nishi Y, Sano H, Kawashima T, Okada T, Kuroda T, Kikkawa K, Kawashima S, Tanabe M, Goto T, Matsuzawa Y, Matsumura R, Tomioka H, Liu FT, Shirai K. Role of galectin-3 in human pulmonary fibrosis. Allergol. Int. 2007;56:57–65. doi: 10.2332/allergolint.O-06-449. [DOI] [PubMed] [Google Scholar]
- 12.Papaspyridonos M, McNeill E, de Bono JP, Smith A, Burnand KG, Channon KM, Greaves DR. Galectin-3 is an amplifier of inflammation in atherosclerotic plaque progression through macrophage activation and monocyte chemoattraction. Arterioscler. Thromb. Vasc. Biol. 2008;28:433–440. doi: 10.1161/ATVBAHA.107.159160. [DOI] [PubMed] [Google Scholar]
- 13.Jeon SB, Yoon HJ, Chang CY, Koh HS, Jeon SH, Park EJ. Galectin-3 exerts cytokine-like regulatory actions through the JAK-STAT pathway. J. Immunol. 2010;185:7037–7046. doi: 10.4049/jimmunol.1000154. [DOI] [PubMed] [Google Scholar]
- 14.Forsman H, Islander U, Andreasson E, Andersson A, Onnheim K, Karlstrom A, Savman K, Magnusson M, Brown KL, Karlsson A. Galectin 3 aggravates joint inflammation and destruction in antigen-induced arthritis. Arthritis Rheum. 2011;63:445–454. doi: 10.1002/art.30118. [DOI] [PubMed] [Google Scholar]
- 15.Fernandes Bertocchi AP, Campanhole G, Wang PH, Goncalves GM, Damiao MJ, Cenedeze MA, Beraldo FC, de Paula Antunes Teixeira V, Dos Reis MA, Mazzali M, Pacheco-Silva A, Camara NO. A Role for galectin-3 in renal tissue damage triggered by ischemia and reperfusion injury. Transpl. Int. 2008;21:999–1007. doi: 10.1111/j.1432-2277.2008.00705.x. [DOI] [PubMed] [Google Scholar]
- 16.Doverhag C, Hedtjarn M, Poirier F, Mallard C, Hagberg H, Karlsson A, Savman K. Galectin-3 contributes to neonatal hypoxic-ischemic brain injury. Neurobiol. Dis. 2010;38:36–46. doi: 10.1016/j.nbd.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 17.Volarevic V, Milovanovic M, Ljujic B, Pejnovic N, Arsenijevic N, Nilsson U, Leffler H, Lukic ML. Galectin-3 deficiency prevents concanavalin A-induced hepatitis in mice. Hepatology. 2012;55:1954–1964. doi: 10.1002/hep.25542. [DOI] [PubMed] [Google Scholar]
- 18.Dragomir AC, Sun R, Mishin V, Hall LB, Laskin JD, Laskin DL. Role of galectin-3 in acetaminophen-induced hepatotoxicity and inflammatory mediator production. Toxicol. Sci. 2012;127:609–619. doi: 10.1093/toxsci/kfs117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen LC, Gordon RE, Laskin JD, Laskin DL. Role of TLR-4 in liver macrophage and endothelial cell responsiveness during acute endotoxemia. Exp. Mol. Pathol. 2007;83:311–326. doi: 10.1016/j.yexmp.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lloyd CM, Phillips AR, Cooper GJ, Dunbar PR. Three-colour fluorescence immunohistochemistry reveals the diversity of cells staining for macrophage markers in murine spleen and liver. J. Immunol. Methods. 2008;334:70–81. doi: 10.1016/j.jim.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 21.Dziennis S, Van Etten RA, Pahl HL, Morris DL, Rothstein TL, Blosch CM, Perlmutter RM, Tenen DG. The CD11b promoter directs high-level expression of reporter genes in macrophages in transgenic mice. Blood. 1995;85:319–329. [PubMed] [Google Scholar]
- 22.Robbins CS, Swirski FK. The multiple roles of monocyte subsets in steady state and inflammation. Cell. Mol. Life Sci. 2010;67:2685–2693. doi: 10.1007/s00018-010-0375-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raes G, De Baetselier P, Noel W, Beschin A, Brombacher F, Hassanzadeh Gh G. Differential expression of FIZZ1 and Ym1 in alternatively versus classically activated macrophages. J. Leukoc. Biol. 2002;71:597–602. [PubMed] [Google Scholar]
- 24.Si Y, Tsou CL, Croft K, Charo IF. CCR2 mediates hematopoietic stem and progenitor cell trafficking to sites of inflammation in mice. J. Clin. Invest. 2010;120:1192–1203. doi: 10.1172/JCI40310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deng ZB, Liu Y, Liu C, Xiang X, Wang J, Cheng Z, Shah SV, Zhang S, Zhang L, Zhuang X, Michalek S, Grizzle WE, Zhang HG. Immature myeloid cells induced by a high-fat diet contribute to liver inflammation. Hepatology. 2009;50:1412–1420. doi: 10.1002/hep.23148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karlmark KR, Weiskirchen R, Zimmermann HW, Gassler N, Ginhoux F, Weber C, Merad M, Luedde T, Trautwein C, Tacke F. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology. 2009;50:261–274. doi: 10.1002/hep.22950. [DOI] [PubMed] [Google Scholar]
- 27.Perides G, Weiss ER, Michael ES, Laukkarinen JM, Duffield JS, Steer ML. TNF-alpha-dependent regulation of acute pancreatitis severity by Ly-6C(hi) monocytes in mice. J. Biol. Chem. 2011;286:13327–13335. doi: 10.1074/jbc.M111.218388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Correa SG, Sotomayor CE, Aoki MP, Maldonado CA, Rabinovich GA. Opposite effects of galectin-1 on alternative metabolic pathways of L-arginine in resident, inflammatory, and activated macrophages. Glycobiol. 2003;13:119–128. doi: 10.1093/glycob/cwg010. [DOI] [PubMed] [Google Scholar]
- 30.Cooper D, Iqbal AJ, Gittens BR, Cervone C, Perretti M. The effect of galectins on leukocyte trafficking in inflammation: sweet or sour? Ann. N. Y. Acad. Sci. 2012;1253:181–192. doi: 10.1111/j.1749-6632.2011.06291.x. [DOI] [PubMed] [Google Scholar]
- 31.Liu FT, Rabinovich GA. Galectins: regulators of acute and chronic inflammation. Ann. N. Y. Acad. Sci. 2010;1183:158–182. doi: 10.1111/j.1749-6632.2009.05131.x. [DOI] [PubMed] [Google Scholar]
- 32.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bourdi M, Masubuchi Y, Reilly TP, Amouzadeh HR, Martin JL, George JW, Shah AG, Pohl LR. Protection against acetaminophen-induced liver injury and lethality by interleukin 10: role of inducible nitric oxide synthase. Hepatology. 2002;35:289–298. doi: 10.1053/jhep.2002.30956. [DOI] [PubMed] [Google Scholar]
- 34.Bourdi M, Eiras DP, Holt MP, Webster MR, Reilly TP, Welch KD, Pohl LR. Role of IL-6 in an IL-10 and IL-4 double knockout mouse model uniquely susceptible to acetaminophen-induced liver injury. Chem. Res. Toxicol. 2007;20:208–216. doi: 10.1021/tx060228l. [DOI] [PubMed] [Google Scholar]
- 35.Nibbering PH, Leijh PC, van Furth R. Quantitative immunocytochemical characterization of mononuclear phagocytes. II. Monocytes and tissue macrophages. Immunology. 1987;62:171–176. [PMC free article] [PubMed] [Google Scholar]
- 36.Ahmad N, Chen LC, Gordon MA, Laskin JD, Laskin DL. Regulation of cyclooxygenase-2 by nitric oxide in activated hepatic macrophages during acute endotoxemia. J. Leukoc. Biol. 2002;71:1005–1011. [PubMed] [Google Scholar]
- 37.Aoyama T, Inokuchi S, Brenner DA, Seki E. CX3CL1-CX3CR1 interaction prevents carbon tetrachloride-induced liver inflammation and fibrosis in mice. Hepatology. 2010;52:1390–1400. doi: 10.1002/hep.23795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karlmark KR, Zimmermann HW, Roderburg C, Gassler N, Wasmuth HE, Luedde T, Trautwein C, Tacke F. The fractalkine receptor CX(3)CR1 protects against liver fibrosis by controlling differentiation and survival of infiltrating hepatic monocytes. Hepatology. 2010;52:1769–1782. doi: 10.1002/hep.23894. [DOI] [PubMed] [Google Scholar]
- 39.Inui M, Ishida Y, Kimura A, Kuninaka Y, Mukaida N, Kondo T. Protective roles of CX3CR1-mediated signals in toxin A-induced enteritis through the induction of heme oxygenase-1 expression. J. Immunol. 2011;186:423–431. doi: 10.4049/jimmunol.1000043. [DOI] [PubMed] [Google Scholar]
- 40.Laskin DL, Pilaro AM, Ji S. Potential role of activated macrophages in acetaminophen hepatotoxicity. II. Mechanism of macrophage accumulation and activation. Toxicol. Appl. Pharmacol. 1986;86:216–226. doi: 10.1016/0041-008x(86)90052-9. [DOI] [PubMed] [Google Scholar]
- 41.Sano H, Hsu DK, Yu L, Apgar JR, Kuwabara I, Yamanaka T, Hirashima M, Liu FT. Human galectin-3 is a novel chemoattractant for monocytes and macrophages. J. Immunol. 2000;165:2156–2164. doi: 10.4049/jimmunol.165.4.2156. [DOI] [PubMed] [Google Scholar]
- 42.Dragomir AC, Laskin JD, Laskin DL. Macrophage activation by factors released from acetaminophen-injured hepatocytes: potential role of HMGB1. Toxicol. Appl. Pharmacol. 2011;253:170–177. doi: 10.1016/j.taap.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mensah-Brown EP, Al Rabesi Z, Shahin A, Al Shamsi M, Arsenijevic N, Hsu DK, Liu FT, Lukic ML. Targeted disruption of the galectin-3 gene results in decreased susceptibility to multiple low dose streptozotocin-induced diabetes in mice. Clin. Immunol. 2009;130:83–88. doi: 10.1016/j.clim.2008.08.024. [DOI] [PubMed] [Google Scholar]
- 44.MacKinnon AC, Farnworth SL, Hodkinson PS, Henderson NC, Atkinson KM, Leffler H, Nilsson UJ, Haslett C, Forbes SJ, Sethi T. Regulation of alternative macrophage activation by galectin-3. J. Immunol. 2008;180:2650–2658. doi: 10.4049/jimmunol.180.4.2650. [DOI] [PubMed] [Google Scholar]
- 45.Dumic J, Lauc G, Hadzija M, Flogel M. Transfer to in vitro conditions influences expression and intracellular distribution of galectin-3 in murine peritoneal macrophages. Z. Naturforsch. C. 2000;55:261–266. doi: 10.1515/znc-2000-3-419. [DOI] [PubMed] [Google Scholar]