

Abstract
Adaptive immunity requires signals from both the T cell antigen receptor and the costimulatory molecule CD28. These receptors activate multiple signaling pathways, including the cyclin-dependent kinase (CDK) cascade, and antigenic signals in the absence of costimulation results in a tolerant state that is enforced by the CDK inhibitory protein p27kip1. We find that CDK2, the major target of p27kip1, is highly active in T cells that infiltrate and reject cardiac allografts. To determine whether CDK2 is required for T cell alloimmunity, we utilized mice genetically deficient for CDK2. Blockade of CD28 costimulation alone was unable to inhibit the rejection of cardiac allografts by wild-type recipients, however, targeting this pathway in CDK2-deficient recipients led to long-term allograft survival. CDK2-deficient CD4+ T cells proliferated normally in response to stimulation in vitro and in vivo, however, genetic, shRNA, or small molecule-mediated antagonism of CDK2 resulted in decreased production of IL-2 and IFNγ. In addition, surviving grafts from CDK2-deficient recipients showed increased infiltration of Foxp3+ regulatory T cells (Treg), and Treg from CDK2-deficient mice exhibited increased suppressive activity in vitro and in an in vivo model of inflammatory bowel disease. These data suggest that that p27kip1 promotes peripheral tolerance through its ability to inhibit CDK2, which otherwise acts to promote conventional T cell differentiation and restrict Treg function.
Introduction
Naive T cells require signals from the costimulatory receptor CD28 for full activation, differentiation, and immune function. CD4+ T cells that experience TCR signaling in the absence of costimulation induce negative regulators of multiple TCR-coupled signaling pathways. For instance, diacylgycerol kinases, caspases, and E3 ubiquitin ligases target PLCg-1, Vav and PKC for proteolysis, resulting in defective activation of the MAPK, IKK and PI3K cascades upon re-encounter with antigen [reviewed in (1)]. This active inhibitory state is referred to as anergy, and a loss of any of these negative regulators leads to defects in the acquisition of peripheral tolerance. Anergic T cells also upregulate an inhibitor of the cyclin-dependent kinase cascade, p27kip1 (2, 3). T cells deficient for the gene encoding p27kip1 fail to become anergic when stimulated in the absence of costimulation (4), and mice with a loss of p27kip1 function are highly resistant to tolerance induced by costimuatory blockade in vivo (5, 6). Therefore, p27kip1 appears to be a key regulator of peripheral tolerance, but whether p27kip1 functions in this capacity as a CDK inhibitor, or influences tolerance as a result of other, CDK-independent functions, is unclear. p27kip1 also binds to RhoA and inhibits actin polymerization and focal adhesion (7), processes important for T cell activation and migration, and it can inhibit the activity of c-Jun, a crucial factor for transcription of the il2 gene, by binding to the c-Jun co-activator Jab1 (8). A deletion mutant of p27kip1 that lacks the CDK binding domain was shown to be defective in anergy induction (6), however, this mutant also lacks a serine residue that is critical for its interaction with Jab1 (9), for its subcelluar localization (10), and for protein stability (8). Therefore the mechanism by which p27kip1 contributes to tolerance, and the potential role of cyclin-dependent kinases in the balance between tolerance and immunity, have not been addressed.
In these current studies, we directly targeted CDK2, the major cyclin-dependent kinase regulated by p27kip1, to determine its role in T cell immunity vs. tolerance. Earlier biochemical work had indicated that CDK2 was critical for progression from the G1 phase to the S phase of the cell cycle, however, more recent studies in mice genetically deficient for individual or multiple CDK family members have shown that CDK1 is the major driver of cell cycle progression, while CDK2, 4 and 6 have supportive roles and specialized, cell cycle-independent functions [reviewed in (11)]. Using a combination of genetic and chemical approaches, we demonstrate that CDK2 is dispensable for CD4+ T cell proliferation, and instead contributes to optimal activation and differentiation. Loss of CDK2 function in vivo resulted in increased susceptibility to organ transplant tolerance induced by costimulatory blockade. Allograft survival in CDK2-deficient recipients was associated with reduced frequencies of donor-specific, IFNγ-producing T cells, and increased regulatory T cell function. Our results demonstrate that CDK2 controls the balance between immunity and tolerance by regulating both conventional and regulatory T cell function.
Materials and Methods
Mice
Female C57BL/6 (H-2b), BALB/c (H-2d), C57BL6×DBA F1 (H-2d/b), and Rag1-deficient mice on a B6 background were purchased from The Jackson laboratory and maintained in our specific pathogen free facility in accordance with ULAR- and AALAC-approved institutional guidelines on animal care and usage. CDK2-deficient mice on a 129S1/SvlmJxC57BL/6 background (12) were obtained from P. Kaldis and were back-crossed to B6 for 9–10 generations. All mice were used at 5–12 weeks of age.
Measurement of In Vitro T cell Activation and Cytokine Production
Single-cell suspensions of spleen and lymph node were depleted of CD8+ T cells using magnetic beads (Miltenyi Biotec, Inc.) and cultured for 24–72 hours with soluble, agonistic anti-CD3 mAb (2C11, 0.5 µg/mL, BioExpress) in the presence of either agonistic anti-CD28 (37–51, 0.5 µg/mL, BioExpress) or CTLA-4Ig (5 µg/mL, BioExpress). Following primary stimulation, CD4+ T cells were purified by positive selection on magnetic beads (Miltenyi Biotec, Inc.), rested overnight in medium, and restimulated on plate-bound anti-CD3/28 mAb for 4–24 hr. IL-2 and IFNγ in supernatants harvested during primary and secondary cultures was detected using quantitative ELISA kits (eBioscience, Inc.). In some experiments, the CDK2-selective inhibitors roscovitine (Calbiochem), alsterpaullone (Calbiochem), and NU6140 (4-[[6-(cyclohexylmethoxy)-1H-purin-2-yl]amino]-N,N-diethylbenzamide, Santa Cruz Biotech), and the CDK1-selective analog of indolylmethylene-2-indolinone (2H-indol-2-one, 3-[(2-chloro-1H-indol-3-yl)methylene]-1,3-dihydro, CAS No 220749-41-7, Santa Cruz Biotech) were added to cultures at the indicated concentrations.
Retroviral shRNA Transduction
An shRNA sequence (5‘CCGGAAGATGGACGGAGCTTG TTATCTCGAGATAACAAGCTCCGTCCATCTTTTTTTG3’) targeting the CDK2 transcript was cloned into the pMKO1 vector and used to transduce CD4+ T cells as described previously (13). A control, non-specific shRNA targeting human cyclin D3 (14) was also constructed. Knock down of CDK2 was confirmed by immunoblot analysis of transduced T cells using a CDK2-specific mAb (Cell Signaling Technologies, Inc.).
qPCR analysis of mRNA transcripts
CD4+CD25− (Tconv) and CD4+CD25+ (Treg) were purified from spleen and lymph node using magnetic beads (Miltenyi Biotec, Inc.). Total RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA), and reverse transcription was performed using the iScript Select cDNA synthesis kit (Biorad). Specific mRNA transcripts were quantified using Sybergreen on a StepOnePlus real-time thermal cycler (Applied Biosystems Inc., Foster City, USA). Expression levels were calculated by first normalizing to 18S rRNA for each sample, then normalized to unstimulated CD4+CD25− cells using the standard delta-delta CT method.
In Vivo Mixed Lymphocyte Response
Alloreactive T cell proliferation was assessed in an in vivo allogeneic mixed lymphocyte response, as described previously (15). Briefly, 2 × 106 CFSE-labeled donor splenocytes from CDK2-deficient or wild-type B6 littermates were adoptively transferred into semi-allogeneic B6×DBA F1 (H-2d/b) recipient mice. Recipients were treated with either CTLA4-Ig or control Ig fusion protein (i.p., 100 µg/mouse) on day 0 and 1. Recipient spleens were harvested on day 3, donor lymphocytes were identified by flow cytometry on the basis of their lack of H-2d, and CFSE dilution in the CD4+ subset was measured. Gating procedures for all parameters allowed less than 1% false positives in negative control samples.
Cardiac Transplantation
Studies were performed as described previously (5). Briefly, fully mismatched BALB/c (H-2d) hearts were transplanted heterotopically into wild-type or CDK2-deficient 129S1/SvlmJ×C57BL/6 (H-2b) mice. The donor ascending aorta and pulmonary artery were anastomosed end-to-side to recipient infrarenal aorta and inferior vena cava. CTLA-4Ig or control Ig fusion protein (BioExpress, 200 µg) was administered i.p. on days 0, 2, and 4 post-transplant. Graft survival was assessed by abdominal palpation, and rejection was defined as total cessation of cardiac contraction confirmed by histology. Statistical analysis of allograft survival was analyzed using Kaplan-Meier/log-rank methods.
Histology and Immunohistology
At time of harvest, portions of the grafts were fixed in formalin for paraffin sectioning or snap frozen for subsequent immunohistologic analysis. H&E-stained paraffin sections were evaluated using the revised International Society of Heart and Lung Transplantation standardized criteria for grading of cardiac allograft biopsies. Heart tissue was fixed, sectioned, and stained with eosin/hematoxilin or with Ab against CD3 (Epitomics), CD8 (Epitomics), CD4 (Epitomics), Foxp3 (eBiosciences), CDK2 (Santa Cruz Biotech), and cyclin A (Santa Cruz Biotech). Primary Ab were visualized using secondary HRP conjugates and Envision kits (Dako). Cells positive for each stain were counted within 10–15 high power fields (HPF) in sections from 4 allografts per group, and data were compared using an unpaired t-test.
ELISPOT Assay
ELISPOT assays were performed as previously described (5). Briefly, immunospot assays for IFNγ were performed by coating ELISPOT plates (BD Pharmingen) with anti-IFNγ mAb, followed by blocking, addition of splenocytes of transplant recipients (1×105, 2×105, or 4×105 cells/well) were placed in each well with donor splenocytes from BALB/c mouse as stimulator cells. After 24 h, cells were discarded and wells washed, followed by use of anti-IFNγ mAb, streptavidin-HRP and substrate reaction. Spots were counted automatically using ImmunoSpot Analyzer (Cellular Technology). p values were calculated by Student’s paired t test using Prism software (GraphPad).
In Vitro Treg Suppression
CD4+CD25+ (Treg) from CD90.2+ mice, CD4+CD25− (Tconv) from CD90.1+ mice, and CD90-negative APC were isolated from total splenocytes by using magnetic beads-conjugated mAbs (Miltenyi Biotec, Inc). APC (1 × 105) were irradiated (1000 rad) and plated onto 96-well plates along with effector cells (5 × 104/well) labeled with CFSE and 4μg/ml soluble anti-CD3 mAb. Treg cells were added to each well in varying ratios to effector cells and cultured for 72 h. After 3 days, suppression of responder cell proliferation was determined by flow cytometrically assessing the degree of inhibition of CFSE dilution.
Adoptive Transfer Colitis Model
To induce experimental colitis, conventional CD25−CD4+ T cells were purified from naive, wild-type B6–129 mice and adoptively transferred (1×106, i.p.) into Rag1-/-B6–129 recipients. Twenty days from the initial transfer, groups of five mice then received either PBS, CD25+CD4+ Treg purified from wild-type B6–129 donors (0.25×106, i.p.), or CD25+CD4+ Treg purified from CDK2-deficient B6–129 donors (0.25×106, i.p.). Recipients were weighed and observed for symptoms of diarrhea approximately every two days. At the end of the experiment, spleens and intestines were harvested for examination of gross pathology. Statistic differences between weight curves were determined by ANOVA (Prism).
Results
CDK2 is not required for CD4+ T cell cycle progression
As a primary approach to induce a loss of CDK2 function, we utilized mice deficient for CDK2 (12). Hematopoiesis, lymphoid development, and lymphocyte homeostasis is normal in CDK2-deficient mice (16), therefore these mice represent a good model in which to study the role of CDK2 in adaptive immune responses. We first tested whether the lack of CDK2 in CD4+ T cells resulted in defective cell cycle progression in response to mitogenic signaling. Wild-type and CDK2-deficient CD4+ T cells divided comparably in cultures of CD8-depleted spleen cells stimulated in vitro with agonistic anti-TCR antibody (Fig. 1 A and B). This is consistent with previous studies showing that fibroblasts from mice devoid of either CDK2 or cyclin E activity show a delay in S phase entry, but overall proliferate normally (12, 17). From these results we conclude that CDK2 is dispensable for essentially normal proliferation of activated CD4 cells.
Figure 1.

Proliferation of CD4+ T cells from wild-type vs. CDK2-deficient mice. Purified CD4+CD25− T cells from wild-type (dark gray) or CDK2-deficient (light gray) mice were labeled with CFSE and stimulated in vitro with soluble anti-CD3 (0.5 µg/mL) in the presence of Thy1.2-depleted spleen cells for 72 hours, and cell division was assessed by flow cytometry (A). The total number of cell divisions achieved by each population was calculated as described previously (52) (B). The data depicted are representative of 4 independent experiments.
CDK2 promotes cytokine production by CD4+ T cells
The inhibitory partner of CDK2, p27kip1, acts as a sensor of mitogenic signals from CD28 and IL-2R, and CD4+ T cells genetically deficient for p27kip1 differentiate even in the absence of costimulation (1). Therefore, we predicted that CD4+ T cells deficient for CDK2 would show an opposite phenotype, i.e., these cells should have problems integrating signals from the TCR and CD28 for cytokine production. Despite normal activation, proliferation, and survival of CDK2-deficient CD4+ T cells exhibited decreased production of IL-2 and IFNγ in response to primary stimulation, both in the presence and absence of CD28 costimulation (Fig. 2 A and B). This does not reflect a developmental defect, because acute inhibition of CDK2 activity in wild-type CD4+ T cells also repressed the production of IL-2 and IFNγ. Treatment of wild-type T cells with three different small molecule inhibitors of CDK2 (roscovitine, NU6140 & alsterpaullone), but not with a CDK1 inhibitor (indolylmethylene-2-indolinone), led to a dose-dependent decrease in IL-2 production (Fig. 2 C). Similarly, knock-down of CDK2 in wild-type CD4+ T cells using short hairpin RNAs (shRNA) also led to reduced expression of IL-2 and IFNγ at both the protein (Fig. 2 D) and mRNA level (not shown).
Figure 2.

Cytokine production by wild-type vs. CDK2-deficient CD4+ T cells. CD8-depleted spleen and lymph node cells from wild-type (dark gray) or CDK2-deficient (light gray) mice were stimulated with soluble anti-CD3 plus either anti-CD28 or CTLA-4Ig, and supernatants were tested for IL-2 (A) and IFNγ (B) by ELISA. Wild-type cells were stimulated with anti-CD3 in the presence of three CDK2-selective inhibitory drugs (roscovitine, NU6140 & alsterpaullone) or the CDK1-selective drug indolylmethylene-2-indolinone (I2I), and IL-2 secretion was measured by ELISA (C). Purified wild-type CD4+ T cells were transduced with retroviruses encoding either a control shRNA that targets human cyclin D3, or shRNA targeting two distinct regions of the CDK2 transcript, and CDK2 protein levels were measured by immunoblotting (D, top left panel), and IL-2 and IFNγ in the supernatants were quantified by ELISA (D, lower panels). Wild-type or CDK2-deficient cells were stimulated as in A and B, and IL-2 (E) and IFNγ (G) in primary supernatants were measured by ELISA. Cultures were rested, CD4+ T cells were purified and restimulated on plate-bound anti-CD3 and anti-CD28, and IL-2 (F) and IFNγ (H) in secondary supernatants were measured ELISA. Error bars in A–F depict the standard error of the mean. The data depicted are representative of 3–4 independent experiments.
T cells that have been primed through the TCR and CD28 (Fig. 2 F and H, dark grey circles) are able to secrete IL-2 and IFNγ more rapidly and robustly than naive cells (Fig. 2 E and G, dark grey circles) when stimulated. However, primed CDK2-deficient cells exhibited a significant defect in the production of IL-2 (Fig. 2 F, light grey circles), comparable to the anergic response observed in wild-type cells primed in the absence of CD28 costimulation (Fig. 2 F, dark grey squares). CDK2-deficient CD4 cells also showed a marked delay in IFNγ secretion (Fig. 2 H, light grey circles), and blockade of costimulation during priming completely abrogated the differentiation of these cells into IFNγ producers (Fig. 2 H, light grey squares). The mechanism by which CDK2 is coupled to cytokine gene expression is currently unclear. CDK2-deficient CD4+ T cells showed no defects in the expression of factors involved in the transactivation of the il2 promoter such as NFAT1, NFkB p65, Fos, or c-Jun at the mRNA level, nor did these cells show elevated expression of anergy-associated genes such as Egr-2, Egr-3, Itch, Grail, Cbl-b or Ikaros (our unpublished observations). Additional experiments will be needed to establish how CDK2 contributes to the control of cytokine gene expression.
CDK2 promotes acute alloimmune T cell responses and contributes to costimulatory blockade-resistant allograft rejection in vivo
Our previous studies showed that the CDK2 inhibitory protein p27kip1 is required for transplantation tolerance induced by combined costimulatory blockade (5). To address whether CDK2 itself has a role in the alloimmune response, we first used a parent-into-F1 mixed lymphocyte reaction model to measure allospecific T cell activation and expansion in vivo (15). A high frequency of alloreactive donor CD4+ T cells from wild-type B6 mice showed robust expansion when adoptively transferred into semi-allogeneic B6xDBA2 recipients (Fig. 3 A and B), and blockade of CD28 costimulation resulted in a marked decrease in alloreactive T cell division (Fig. 3 A) and expansion (Fig. 3 B). Donor alloreactive CD4+ T cells lacking CDK2 divided as well as wild-type donor cells in recipients treated with control Ig (Fig. 3 A), further emphasizing that CDK2 activity is not required for efficient T cell cycle progression. However, CDK2-deficient alloreactive T cells failed to accumulate in the spleen (Fig. 3 B), suggesting that CDK2 may contribute to the survival or trafficking of proliferating T cells in vivo. While alloreactive T cells lacking p27kip1 enjoy a significant proliferative advantage over wild-type cells under conditions of costimulatory blockade (4), CDK2-deficient T cells instead exhibited a near complete failure to divide and expand in CTLA-4Ig-treated recipients (Fig. 3 A and B).
Figure 3.

In vivo mixed lymphocyte responses by wild-type vs. CDK2-deficient CD4+ T cells. Spleen and lymph node cells from wild-type or CDK2-deficient mice (H-2b) were labeled with CFSE, adoptively transferred into B6×DBA F1 recipients (H-2bxd), and the recipients were treated with CTLA-4Ig or control Ig. After 3 days, spleens were harvested and cell division by H-2d-negative donor CD4+ T cells was assessed by flow cytometry (A). The average number of divided CD4+ T cells (+/− SEM, p < 0.01) from three recipients per group is depicted in B and is representative of two independent experiments.
Next, we transplanted wild-type (WT) or CDK2-deficient mice with fully mismatched BALB/c cardiac allografts. In the absence of treatment, the kinetics of allograft survival in both groups were equivalent, with both WT and CDK2-deficient mice rejecting their grafts within 7 days post-transplant (Fig. 4 A, black symbols). Blockade of the CD28 costimulatory pathway in WT recipients using CTLA-4Ig provided a significant benefit, but all grafts eventually rejected with a mean survival of ~30 days (Fig. 4 A, blue symbols). The frequency of donor-specific, IFNγ-producing CD4+ T cells was similar in the spleens of untreated recipients (Fig. 4 B), however, CDK2-deficient recipients treated with CTLA-4Ig exhibited 5-fold fewer donor-specific IFNγ-producing cells than WT recipients (Fig. 4 C). Histologic analysis of cardiac allografts harvested at 30 days post-transplant from WT recipients treated with CTLA-4Ig showed diffuse mononuclear cell infiltration with arteritis and areas of focal myocyte necrosis (Fig. 4 D, left panel), and infiltrates consisted of a mixture of CD4+ and CD8+ T cells, plus small numbers of Foxp3+ mononuclear cells (Fig. 4 E, left panels). Mononuclear cells infiltrating the grafts in WT recipients treated with CTLA-4Ig showed strong expression of CDK2 and the CDK2-dependent protein cyclin A (Fig. 4 F, left panels), indicating that the CDK pathway is highly active in immune cells during allograft rejection. In contrast, costimulatory blockade in CDK2-deficient recipients resulted in long-term allograft acceptance, with all recipients retaining their allografts for greater than 100 days (p<0.001) (Fig. 4 A, red symbols). At 30 days post-transplant, these grafts exhibited normal cardiac histology with well-preserved vasculature and an absence of myocyte necrosis (Fig. 4 D, right panel). Infiltrates of CD4+ and CD8+ T cell infiltrates were observed at 30 days post-transplant, but tended to occur in perivascular areas rather than more diffusely as was seen in WT recipients, and were accompanied by a two-fold increase in the numbers of infiltrating Foxp3+ mononuclear cells (Fig. 4 E, right panels, 5.8 ± 4.9 Foxp3+ cells/HPF in WT recipients vs. 12.7 ± 7.1 Foxp3+ cells/HPF grafts in CDK2-deficient mice - mean ± SD, p<0.005). The increase in Treg infiltration in CDK2-deficient recipients was not due to an increase in the size of the regulatory T cell pool, as CDK2-deficient mice showed normal frequencies of CD4+CD25+Foxp3+ cells in the thymus, lymph nodes and spleen (data not shown). In addition, mononuclear cells infiltrating the allografts in CDK2-deficient recipients not only lacked expression of CDK2, but were almost completely devoid of cyclin A expression (Fig. 4 F, right panels). These results demonstrate that the CDK2 pathway is active during an alloimmune response, where it has a role in driving the differentiation and effector function of alloreactive CD4+ T cells. As predicted, this is opposite of the role of the inhibitor p27kip1 (5), suggesting that p27kip1 is required for allograft tolerance largely because of its ability to restrain CDK2 activity.
Figure 4.
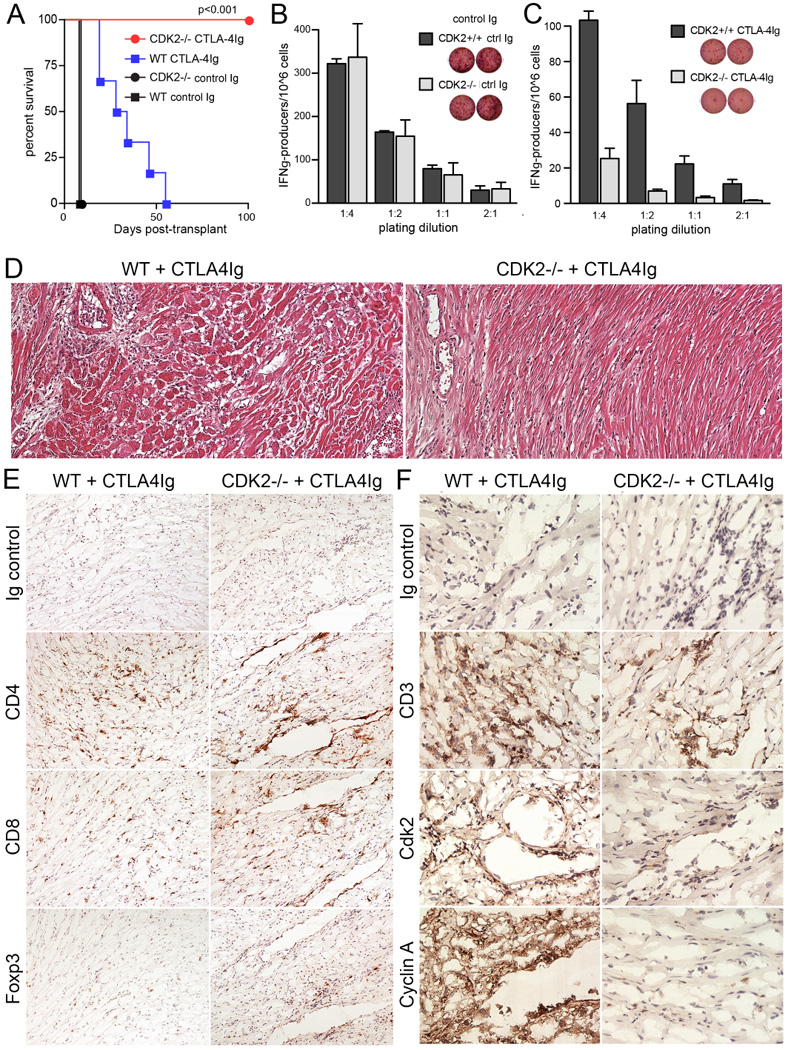
Cardiac allograft response in wild-type vs. CDK2-deficient mice. Wild-type (squares) or CDK2-deficient (circles) mice were transplanted with BALB/c cardiac grafts, treated with control Ig (n=3, black lines) or CTLA-4Ig (n=6–7, colored lines), and graft survival was observed over a 100 day period (A). In a separate experiment, groups of three wild-type or CDK2-deficient mice were transplanted and treated as in A, and sacrificed at day 32 post-transplant. p values were calculated by ANOVA. CD4+ T cells purified from the spleens of control Ig-treated (B) and CTLA-4Ig-treated (C) recipients were stimulated with BALB/c splenocytes in vitro, and the frequency of donor-specific cells was determined by ELISPOT. Allograft tissue harvested from CTLA-4Ig-treated recipients was sectioned and stained with H&E (D), antibodies against CD4, CD8, and Foxp3 (E), or antibodies against CD3, CDK2 and cyclin A (F).
CDK2 opposes regulatory T cell function
To address whether the regulatory T cells in CDK2-deficient mice exhibit a gain of suppressive function, we isolated CD4+CD25+ cells from naive, wild-type or CDK2-deficient mice and measured their ability to inhibit naive CD4+ T cell proliferation in a standard, in vitro suppression assay. At all ratios tested, CDK2-deficient Treg exhibited an enhanced capacity to suppress the proliferation of wild-type CD4+ T cells, representing a ~3-fold increase in efficiency (Fig. 5 A, solid red line). In addition, conventional CD4+ T cells from CDK2-deficient mice were slightly more susceptible to suppression by wild-type Treg compared to wild-type Tconv (Fig. 5 A, dotted blue line). The combination of CDK2-deficient Treg against CDK2-deficient Tconv, which simulates the circumstance in intact CDK2-deficient mice, showed the strongest level of suppression (Fig. 5 A, dotted red lines). These data demonstrate that CDK2 acts in a cell intrinsic manner to restrict Treg suppressive capacity in vitro. The mechanistic basis for this is not clear, however, regulatory T cells lacking CDK2 exhibited a moderate increase in the expression of several genes involved in Treg function, including CD25, STAT5, GITR, PD-1, SMAD3, and TGFβ (Fig. 5 B).
Figure 5.

Regulatory T cell function in wild-type vs. CDK2-deficient mice. CD4+CD25+ Treg purified from naive wild-type or CDK2-deficient mice were cultured at different ratios with CD4+CD25− wild-type or CDK2-deficient Tconv in a standard anti-CD3-based in vitro suppression system. Plotted is the percent suppression of Tconv cell division as measured by CFSE (A). Gene expression by resting or 4-hour stimulated wild-type and CDK2-deficient Treg and Tconv was assessed by qRT-PCR (B). Naive CD4+CD25− T cells were purified from wild-type mice and adoptively transferred into Rag1-deficient recipients (C). At day 20 post-transfer, Rag1-deficient recipients (5 per group) received purified Treg from wild-type mice (green symbols), Treg from CDK2-deficient mice (blue symbols), or PBS as a control (red symbols). Recipients were subsequently weighed and monitored for symptoms of colitis every two days. The difference in weight kinetics between each treatment group was statistically significant (p < 0.05, ANOVA).
To determine whether CDK2 controls Treg function in vivo, we tested the capacity of wildtype vs. CDK2-deficient regulatory T cells to suppress colitis mediated by naive CD4+ T cells in a murine model of inflammatory bowel disease (18). Colitis was first induced in immunodeficient (Rag1-/-) mice by adoptive transfer of wild-type naive CD4+ T cells. Recipients began to exhibit symptoms of disease around 18 days post-transfer, and at day 20 animals were randomized and received PBS as a control, or a therapeutic dose of CD4+CD25+ Treg from either wild-type or CDK2-deficient mice. Mock-treated animals continued to lose weight and exhibit severe diarrhea and wasting over the next 40 days (Fig. 5 C, red line), while recipients of wild-type Treg began to recover within 10 days and showed markedly reduced symptoms of disease by day 50 (Fig. 5 C, green line). Mice that received CDK2-deficient Treg began to recover rapidly within 5 days (Fig. 5 C, blue line), no longer suffered from wasting or diarrhea, and by day 30 their weight was indistinguishable from control animals receiving no naive CD4+ T cells (Fig. 5 C, black line). These mice showed significantly reduced gross and histologic pathology in the colon (e.g., reduced thickening and hemorrhaging of the intestinal wall) compared to recipients of wildtype Treg (data not shown). Together, these data show that CDK2 acts in a cell-intrinsic manner to control the capacity of regulatory T cells to inhibit conventional T cell proliferation and immunopathogenesis.
Discussion
TCR signaling in the absence of costimulation results in active suppression of several kinase cascades. For example, diacylgycerol kinases deplete the secondary messenger diacyglycerol, and E3 ubiquitin ligases such as Itch and Cbl-b target PLCγ-1 and PKC for proteolysis, together resulting in defective activation of downstream MAPK pathways (1). In addition, the phosphatase PTEN opposes the activity of PI3K (19), and p27kip1 binds to and inhibits the activity of the CDK2-cyclin E holoenzyme (3). Like the other negative regulators listed above, p27kip1 is required for the induction of anergy (4), but its exact role in this process has been unclear. While p27kip1 is clearly a potent CDK inhibitor, it also has CDK-independent functions in the cell. For instance, p27kip1 has been shown to bind RhoA and inhibit actin polymerization and focal adhesion (7), and it can inhibit the activity of c-Jun by binding to its co-activator, Jab1 (8). Our results demonstrate that CDK2 activity is required for normal cytokine production by CD4+ T cells in vitro, and for full alloimmune T cell differentiation and effector function in vivo. This indicates that dysregulated CDK2 activity in p27kip1-deficient mice (20–22) directly contributes to the enhanced T cell responses in these animals (4, 23, 24), and to their resistance to tolerance induction (5, 25).
Unicellular eukaryotes such as S. pombe encode a single, ancestral cyclin-dependent kinase, Cdc2a, which is sufficient for mitotic cell division in these organisms [reviewed in (26)]. Multicellular eukaryotes encode an orthologue of Cdc2a, termed CDK1, but also express multiple additional CDK (e.g., CDK2, 4, 5, 6). These interphase CDK are thought to play successive roles in promoting cell cycle progression, however, recent studies on mice lacking individual interphase CDK have indicated that these factors are not required for cellular proliferation. Indeed, embryonic development and organogenesis proceeds through E13.5 in mice with compound genetic deficiency for CDK2, 3, 4, 5 and 6, and MEF from these animals can proliferate and undergo transformation in vitro. These results show that CDK1 is sufficient for essential cell cycle progression, while interphase CDK appear to have evolved primarily to promote the differentiation of specialized tissues (27). For example, mice lacking CDK2 show normal growth and development, but cannot undergo meiosis and therefore exhibit defects in ovarian development and spermatogenesis (12, 28). Similarly, our results indicate that CDK2 is not required for the proliferation or the survival of CD4+ T cells, but is necessary for normal T cell differentiation. Based upon the phenotype of CD4+ T cells lacking the CDK-inhibitory protein p27kip1, which behave as though they have received an anergy avoidance signal even when they have not (4, 25), CDK2 has been proposed to act as a cell-intrinsic anergy avoidance sensor (1, 25, 29). We now show direct genetic evidence that, in the absence of CDK2 activity, CD4+ T cells enter an hypo-responsive state even when stimulated in the presence of signals that normally allow T cells to escape anergy. Still, whether the reduced capacity of T cells to rapidly produce IL-2 and IFNγ when primed in the absence of CDK2 is truly reflective of anergy is not clear. Anti-CD3/28-activated CDK2-deficient cells do not exhibit an anergic transcript profile, and addition of exogenous IL-2 does not rescue the defect in cytokine production (our unpublished observations). Therefore, it is possible that the defect may instead reflect a failure in differentiation and memory - i.e., the capacity of cells to epigenetically or transcriptionally poise cytokine loci for rapid expression upon restimulation. More studies will be required to distinguish between these two possibilities.
It is clear from multiple studies that complete blockade of CD28 costimulation, either by the saturating doses of CTLA-4Ig used in this study or by genetic deletion of CD28, is not sufficient to block allograft rejection (e.g., see Fig. 4A). Thus, in wild-type mice, CDK2 must be able to cooperate with other receptors to promote graft rejection. These may include alternative costimulatory receptors such as CD40L or ICOS, or growth factor receptors for IL-2 or other γc-coupled cytokines. In fact, T helper differentiation is highly influenced by IL-2 receptor-coupled signaling. IL-2 inhibits Th17 and Tfh differentiation (30–32), while it promotes anergy avoidance (1), drives Th1 and Th2 development (33–36), and cooperates with TGFβ to induce Foxp3 in iTreg (37, 38). IL-2 is thought to mediate these effects largely through its capacity to activate STAT5, which directly promotes the expression of IL-4, IL-12R, Tbet, IFNγ, and Foxp3, and can antagonize the binding of STAT3 and the expression of Bcl-6. Our studies now demonstrate that receptors for other factors distinct from CD28 must also signal through CDK2 to drive Th1 differentiation and control Treg function.
How might CDK2 be acting to regulate conventional T cell differentiation and regulatory T cell function? In addition to proteins involved in DNA replication and mitosis, CDK2 is known to phosphorylate DNA binding proteins such as c-Jun (39), Runx1 (40, 41), Sp1 (42, 43), and Smad3 (44), all of which can regulate transcription from the il2 promoter. CDK2 has also been shown to regulate transcriptional and chromatin regulatory co-factors such as CBP/p300 (45), EZH2 (46), and histone H1 (47, 48). Studies in CDK1-only mice have led to the hypothesis that interphase CDK must act on crucial, cell type-specific substrates (27). While cyclin-dependent kinases have been shown to regulate the lymphocyte-specific Rag proteins during lymphocyte development (49), mature T lymphocyte-specific substrates have not been reported. In the case of Treg, the most obvious lineage-specific substrate would be Foxp3. Interestingly, the murine Foxp3 protein contains four putative CDK substrate motifs (S/TP followed by one or more basic residues), and we have preliminary evidence that CDK2-cyclin E can phosphorylate Foxp3 in vitro (our unpublished observations). Additional studies will be required to establish the biochemical mechanism(s) by which CDK2 influences T cell differentiation and Treg function.
These findings are therapeutically relevant to transplantation and autoimmune disease, as many kinase inhibitors have been successfully implemented in the clinic, and roscovitine (seliciclib) is currently in clinical trials for the treatment of cancer. While the short half-life of roscovitine has hampered in vivo studies in preclinical models (50), this CDK2 inhibitory drug was recently shown to block alloimmune pathology in a murine model of graft-vs.-host disease (51). Cyclin-dependent kinases may therefore serve as novel immunomodulatory targets in the treatment of immunopathologic disease.
Acknowledgements
The authors thank P. Morawski, C. Hunter, and H. Hu for helpful discussion.
Footnotes
This work was supported by NIH grant AI054643 to ADW. ADW and WWH are members of the Fred & Suzanne Biesecker Pediatric Liver Center.
References
- 1.Wells AD. New insights into the molecular basis of T cell anergy: anergy factors, avoidance sensors, and epigenetic imprinting. J Immunol. 2009;182:7331–7341. doi: 10.4049/jimmunol.0803917. [DOI] [PubMed] [Google Scholar]
- 2.Polyak K, Lee M-H, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, Massagué J. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78:59–66. doi: 10.1016/0092-8674(94)90572-x. [DOI] [PubMed] [Google Scholar]
- 3.Boussiotis V, Freeman G, Taylor P, Berezovskaya A, Grass I, Blazar B, Nadler L. p27kip1 functions as an anergy factor inhibiting interleukin 2 transcription and clonal expansion of alloreactive human and mouse helper T lymphocytes. Nat Med. 2000;6:290–297. doi: 10.1038/73144. [DOI] [PubMed] [Google Scholar]
- 4.Rowell EA, Walsh MC, Wells AD. Opposing roles for the cyclin-dependent kinase inhibitor p27kip1 in the control of CD4+ T cell proliferation and effector function. J Immunol. 2005;174:3359–3368. doi: 10.4049/jimmunol.174.6.3359. [DOI] [PubMed] [Google Scholar]
- 5.Rowell EA, Wang L, Hancock WW, Wells AD. The cyclin-dependent kinase inhibitor p27kip1 is required for transplantation tolerance induced by costimulatory blockade. J Immunol. 2006;177:5169–5176. doi: 10.4049/jimmunol.177.8.5169. [DOI] [PubMed] [Google Scholar]
- 6.Li L, Iwamoto Y, Berezovskaya A, Boussiotis VA. A pathway regulated by cell cycle inhibitor p27Kip1 and checkpoint inhibitor Smad3 is involved in the induction of T cell tolerance. Nat Immunol. 2006;7:1157–1165. doi: 10.1038/ni1398. [DOI] [PubMed] [Google Scholar]
- 7.Besson A, Gurian-West M, Schmidt A, Hall A, Roberts J. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004;18:862–876. doi: 10.1101/gad.1185504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tomoda K, Kubota Y, Kato J. Degradation of the cyclin-dependent-kinase inhibitor p27Kip1 is instigated by Jab1. Nature. 1999;398:160–165. doi: 10.1038/18230. [DOI] [PubMed] [Google Scholar]
- 9.Chopra S, Fernandez De Mattos S, Lam EW-F, Mann DJ. Jab1 co-activation of c-Jun is abrogated by the serine 10-phosphorylated form of p27Kip1. J Biol Chem. 2002;277:32413–32416. doi: 10.1074/jbc.C200311200. [DOI] [PubMed] [Google Scholar]
- 10.Ishida N, Hara T, Kamura T, Yoshida M, Nakayama K, Nakayama KI. Phosphorylation of p27Kip1 on serine 10 is required for its binding to CRM1 and nuclear export. J Biol Chem. 2002;277:14355–14358. doi: 10.1074/jbc.C100762200. [DOI] [PubMed] [Google Scholar]
- 11.Satyanarayana A, Kaldis P. Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene. 2009;28:2925–2939. doi: 10.1038/onc.2009.170. [DOI] [PubMed] [Google Scholar]
- 12.Berthet C, Aleem E, Coppola V, Tessarollo L, Kaldis P. Cdk2 knockout mice are viable. Curr Biol. 2003;13:1775–1785. doi: 10.1016/j.cub.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 13.Chen C, Rowell EA, Thomas RM, Hancock WW, Wells AD. Transcriptional regulation by Foxp3 is associated with direct promoter occupancy and modulation of histone acetylation. J Biol Chem. 2006;281:36828–36834. doi: 10.1074/jbc.M608848200. [DOI] [PubMed] [Google Scholar]
- 14.Sicinska E, Aifantis I, Le Cam L, Swat W, Borowski C, Yu Q, Ferrando AA, Levin SD, Geng Y, von Boehmer H, Sicinski P. Requirement for cyclin D3 in lymphocyte development and T cell leukemias. Cancer Cell. 2003;4:451–461. doi: 10.1016/s1535-6108(03)00301-5. [DOI] [PubMed] [Google Scholar]
- 15.Suchin E, Langmuir P, Palmer E, Sayegh M, Wells A, Turka L. Quantifying the frequency of alloreactive T cells in vivo: new answers to an old question. J Immunol. 2001;166:973–981. doi: 10.4049/jimmunol.166.2.973. [DOI] [PubMed] [Google Scholar]
- 16.Berthet C, Rodriguez-Galan MC, Hodge DL, Gooya J, Pascal V, Young HA, Keller J, Bosselut R, Kaldis P. Hematopoiesis and thymic apoptosis are not affected by the loss of Cdk2. Mol Cell Biol. 2007;27:5079–5089. doi: 10.1128/MCB.00029-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geng Y, Yu Q, Sicinska E, Das M, Schneider JE, Bhattacharya S, Rideout WM, Bronson RT, Gardner H, Sicinski P. Cyclin E ablation in the mouse. Cell. 2003;114:431–443. doi: 10.1016/s0092-8674(03)00645-7. [DOI] [PubMed] [Google Scholar]
- 18.Powrie F, Correa-Oliveira R, Mauze S, Coffman RL. Regulatory interactions between CD45RBhigh and CD45RBlow CD4+ T cells are important for the balance between protective and pathogenic cell-mediated immunity. J Exp Med. 1994;179:589–600. doi: 10.1084/jem.179.2.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buckler J, Walsh P, Porrett P, Choi Y, Turka L. Cutting edge: T cell requirement for CD28 costimulation is due to negative regulation of TCR signals by PTEN. J Immunol. 2006;177:4262–4266. doi: 10.4049/jimmunol.177.7.4262. [DOI] [PubMed] [Google Scholar]
- 20.Fero M, Rivkin M, Tasch M, Porter P, Carow C, Firpo E, Polyak K, Tsai L, Broudy V, Perlmutter R, Kaushansky K, Roberts J. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell. 1996;85:733–744. doi: 10.1016/s0092-8674(00)81239-8. [DOI] [PubMed] [Google Scholar]
- 21.Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh D. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996;85:707–720. doi: 10.1016/s0092-8674(00)81237-4. [DOI] [PubMed] [Google Scholar]
- 22.Kiyokawa H, Kineman R, Manova-Todorova K, Soares V, Hoffman E, Ono M, Khanam D, Hayday A, Frohman L, Koff A. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1) Cell. 1996;85:721–732. doi: 10.1016/s0092-8674(00)81238-6. [DOI] [PubMed] [Google Scholar]
- 23.Wolfraim L, Letterio J. Cutting edge: p27Kip1 deficiency reduces the requirement for CD28-mediated costimulation in naive CD8+ but not CD4+ T lymphocytes. J Immunol. 2005;174:2481–2484. doi: 10.4049/jimmunol.174.5.2481. [DOI] [PubMed] [Google Scholar]
- 24.Singh A, Jatzek A, Plisch EH, Srinivasan R, Svaren J, Suresh M. Regulation of memory CD8 T-cell differentiation by cyclin-dependent kinase inhibitor p27Kip1. Mol Cell Biol. 2010;30:5145–5159. doi: 10.1128/MCB.01045-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li L, Iwamoto Y, Berezovskaya A, Boussiotis V. A pathway regulated by cell cycle inhibitor p27(Kip1) and checkpoint inhibitor Smad3 is involved in the induction of T cell tolerance. Nat Immunol. 2006;7:1157–1165. doi: 10.1038/ni1398. [DOI] [PubMed] [Google Scholar]
- 26.Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci. 2005;30:630–641. doi: 10.1016/j.tibs.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 27.Santamaria D, Barrière C, Cerqueira A, Hunt S, Tardy C, Newton K, Cáceres JF, Dubus P, Malumbres M, Barbacid M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature. 2007;448:811–815. doi: 10.1038/nature06046. [DOI] [PubMed] [Google Scholar]
- 28.Ortega S, Prieto I, Odajima J, Martín A, Dubus P, Sotillo R, Barbero JL, Malumbres M, Barbacid M. Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat Genet. 2003;35:25–31. doi: 10.1038/ng1232. [DOI] [PubMed] [Google Scholar]
- 29.Wells AD. Cyclin-dependent kinases: molecular switches controlling anergy and potential therapeutic targets for tolerance. Semin Immunol. 2007;19:173–179. doi: 10.1016/j.smim.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, Shevach EM, O'Shea JJ. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 31.Yang X-P, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, Hirahara K, Sun H-W, Wei L, Vahedi G, Kanno Y, O'Shea JJ, Laurence A. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. 2011;12:247–254. doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med. 2012 doi: 10.1084/jem.20111174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi M, Lin TH, Appell KC, Berg LJ. Janus-kinase-3-dependent signals induce chromatin remodeling at the Ifng locus during T helper 1 cell differentiation. Immunity. 2008;28:763–773. doi: 10.1016/j.immuni.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pepper M, Pagán AJ, Igyártó BZ, Taylor JJ, Jenkins MK. Opposing signals from the bcl6 transcription factor and the interleukin-2 receptor generate T helper 1 central and effector memory cells. Immunity. 2011;35:583–595. doi: 10.1016/j.immuni.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liao W, Lin J-X, Wang L, Li P, Leonard WJ. Modulation of cytokine receptors by IL-2 broadly regulates differentiation into helper T cell lineages. Nat Immunol. 2011;12:551–559. doi: 10.1038/ni.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cote-Sierra J, Foucras G, Guo L, Chiodetti L, Young HA, Hu-Li J, Zhu J, Paul WE. Interleukin 2 plays a central role in Th2 differentiation. Proc Natl Acad Sci USA. 2004;101:3880–3885. doi: 10.1073/pnas.0400339101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng SG, Wang J, Wang P, Gray JD, Horwitz DA. IL-2 is essential for TGF-beta to convert naive CD4+CD25− cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J Immunol. 2007;178:2018–2027. doi: 10.4049/jimmunol.178.4.2018. [DOI] [PubMed] [Google Scholar]
- 38.Davidson TS, DiPaolo RJ, Andersson J, Shevach EM. Cutting Edge: IL-2 is essential for TGF-beta-mediated induction of Foxp3+ T regulatory cells. J Immunol. 2007;178:4022–4026. doi: 10.4049/jimmunol.178.7.4022. [DOI] [PubMed] [Google Scholar]
- 39.Vanden Bush TJ, Bishop GA. CDK-Mediated Regulation of Cell Functions via c-Jun Phosphorylation and AP-1 Activation. PLoS ONE. 2011;6:e19468. doi: 10.1371/journal.pone.0019468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Biggs JR, Peterson LF, Zhang Y, Kraft AS, Zhang D-E. AML1/RUNX1 phosphorylation by cyclin-dependent kinases regulates the degradation of AML1/RUNX1 by the anaphase-promoting complex. Mol Cell Biol. 2006;26:7420–7429. doi: 10.1128/MCB.00597-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo H, Friedman AD. Phosphorylation of RUNX1 by cyclin-dependent kinase reduces direct interaction with HDAC1 and HDAC3. J Biol Chem. 2011;286:208–215. doi: 10.1074/jbc.M110.149013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haidweger E, Novy M, Rotheneder H. Modulation of Sp1 activity by a cyclin A/CDK complex. Journal of Molecular Biology. 2001;306:201–212. doi: 10.1006/jmbi.2000.4406. [DOI] [PubMed] [Google Scholar]
- 43.Banchio C, Schang LM, Vance DE. Phosphorylation of Sp1 by cyclin-dependent kinase 2 modulates the role of Sp1 in CTP:phosphocholine cytidylyltransferase alpha regulation during the S phase of the cell cycle. J Biol Chem. 2004;279:40220–40226. doi: 10.1074/jbc.M406468200. [DOI] [PubMed] [Google Scholar]
- 44.Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nat Cell Biol. 2004;430:226–231. doi: 10.1038/nature02650. [DOI] [PubMed] [Google Scholar]
- 45.Perkins ND, Felzien LK, Betts JC, Leung K, Beach DH, Nabel GJ. Regulation of NF-kappaB by cyclin-dependent kinases associated with the p300 coactivator. Science. 1997;275:523–527. doi: 10.1126/science.275.5299.523. [DOI] [PubMed] [Google Scholar]
- 46.Chen S, Bohrer LR, Rai AN, Pan Y, Gan L, Zhou X, Bagchi A, Simon JA, Huang H. Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2. Nat Cell Biol. 2010;12:1108–1114. doi: 10.1038/ncb2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hale TK, Contreras A, Morrison AJ, Herrera RE. Phosphorylation of the linker histone H1 by CDK regulates its binding to HP1alpha. Mol Cell. 2006;22:693–699. doi: 10.1016/j.molcel.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 48.Contreras A, Hale TK, Stenoien DL, Rosen JM, Mancini MA, Herrera RE. The dynamic mobility of histone H1 is regulated by cyclin/CDK phosphorylation. Mol Cell Biol. 2003;23:8626–8636. doi: 10.1128/MCB.23.23.8626-8636.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Z, Dordai DI, Lee J, Desiderio S. A conserved degradation signal regulates RAG-2 accumulation during cell division and links V(D)J recombination to the cell cycle. Immunity. 1996;5:575–589. doi: 10.1016/s1074-7613(00)80272-1. [DOI] [PubMed] [Google Scholar]
- 50.Nutley B, Raynaud F, Wilson S, Fischer P, Hayes A, Goddard P, McClue S, Jarman M, Lane D, Workman P. Metabolism and pharmacokinetics of the cyclin-dependent kinase inhibitor R-roscovitine in the mouse. Mol Cancer Ther. 2005;4:125–139. [PubMed] [Google Scholar]
- 51.Li L, Wang H, Kim JS, Pihan G, Boussiotis V. The cyclin dependent kinase inhibitor (R)-roscovitine prevents alloreactive T cell clonal expansion and protects against acute GvHD. Cell Cycle. 2009;8:1794–1802. doi: 10.4161/cc.8.11.8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wells A, Gudmundsdottir H, Turka L. Following the fate of individual T cells throughout activation and clonal expansion. Signals from T cell receptor and CD28 differentially regulate the induction and duration of a proliferative response. J Clin Invest. 1997;100:3173–3183. doi: 10.1172/JCI119873. [DOI] [PMC free article] [PubMed] [Google Scholar]