

Abstract
Fibroblasts in the heart play a critical function in the secretion and modulation of extracellular matrix critical for optimal cellular architecture and mechanical stability required for its mechanical function. Fibroblasts are also intimately involved in both adaptive and nonadaptive responses to cardiac injury. Fibroblasts provide the elaboration of extracellular matrix and, as myofibroblasts, are responsible for cross-linking this matrix to form a mechanically stable scar after myocardial infarction. By contrast, during heart failure, fibroblasts secrete extracellular matrix, which manifests itself as excessive interstitial fibrosis that may mechanically limit cardiac function and distort cardiac architecture (adverse remodeling). This review examines the hypothesis that fibroblasts mediating scar formation and fibroblasts mediating interstitial fibrosis arise from different cellular precursors and in response to different autocoidal signaling cascades. We demonstrate that fibroblasts which generate scars arise from endogenous mesenchymal stem cells, whereas those mediating adverse remodeling are of myeloid origin and represent immunoinflammatory dysregulation.
INTRODUCTION
The deposition of extracellular matrix proteins and development of fibrous tissue play a critical role in the maintenance and pathophysiologic alteration of the architecture of the heart. In addition, fibrous tissue is critical to pathophysiologic adaptation in cardiac disease. Adequate generation of extracellular matrix proteins and collagen (primarily fibrillar) is a critical adaptation following myocardial infarction, which allows generation of an effective scar to ensure optimal mechanical stabilization of the myocardial wall [1,2]. Although pathologically significant abnormalities may occur, altering scar formation resulting in adverse mechanical consequences, this process is, in general, considered to be adaptive.
Fibrous tissue is also deposited in the heart on a reactive basis and is found in the interstitial space as well as in perivascular areas. This excessive collagen formation may result in muscle fiber entrapment, muscle atrophy, electrophysiologic abnormalities and, most commonly, abnormal cardiac function resulting from increased stiffness [3–6]. In general, this is considered to be an adverse or nonadaptive process and contributes to adverse remodeling of the pathological heart [3,7]. The former process is a normal response to myocardial infarction whereas the latter process, while sometimes idiopathic, usually accompanies ventricular overload and generalized inflammatory reactions.
It is important to point out, however, that the two processes frequently coexist under circumstances in which myocardial infarction occurs followed by heart failure and adverse remodeling [3]. Indeed, it is common to find inadequate scar formation (defective adaptive fibrosis) in the infarct zone coexisting with pathological interstitial fibrosis (excessive nonadaptive fibrosis) in the border zone near the infarct. The relationship between cardiac scar formation and nonadaptive fibrosis leading to cardiac dysfunction is not well resolved. This paper will present evidence that the two processes depend upon fibroblast populations arising from pathophysiologically distinct fibroblast precursors and mechanisms of induction. The potential significance of these findings with respect to potential therapeutic approaches will be discussed.
ORIGIN OF THE CARDIAC FIBROBLAST
One of the most plentiful cells in the adult myocardium is the structural fibroblast, which performs structural functions and also provides the source of continued renewal of matrix proteins. These cells are not ordinarily actively proliferating (although a slow rate of renewal must also be taking place) during normal cardiac function. It had long been presumed that these fibroblasts were the primary source of reactive myofibroblasts, which are seen to proliferate in response to tissue injury, express smooth muscle actin and secrete and process extracellular matrix to form a scar or nonadaptive fibrosis. However, attempts to culture structural fibroblasts from the heart demonstrate that they grow very slowly in cell culture and frequently do not survive many passages [8]. Recent evidence suggests that reactive fibroblasts in the myocardium arise from mesenchymal precursors of a variety of origins. Evidence for epithelial to mesenchymal transition has been more prominently demonstrated in other organs, but some evidence suggests their development in the heart under circumstances of chronic inflammation [9]. Similarly, endothelial to mesenchymal transition has been associated with cardiac fibrosis particularly that associated with heart valves [10]. Another source of fibroblasts is mesenchymal progenitor cells, which express markers of undifferentiated embryonic stem cells (e.g. Nanog), show an ability for self-renewal and differentiate into a variety of mesenchymal cells in culture [11,12]. While these mesenchymal stem cells are best characterized in the bone marrow where they occupy selective niches [11], endogenous mesenchymal stem cells are found in a variety of tissues including the heart where they have been described to play a major role in scar formation [11]. Another form of circulating fibroblast precursor, which is of hematopoietic origin (CD45+), is found in the circulating monocyte pool [13]. These cells have been implicated in fibrosis in several organs and are designated as “fibrocytes” because they are blood-borne and develop a combined cell surface phenotype and morphology when cultured [13,14]. These cells were associated with fibrosis in a variety of models of wound healing [15,16] as well as fibrosis in the lung [17,18] and experimental asthma [19]. They are attracted into tissues by chemokines [18,20].
In the remainder of this paper, we will focus on the origin and maturation of fibroblast precursors in the pathophysiology of the heart. We will focus on our work assigning specific pathophysiological functions to fibroblasts of distinct precursor origins and signaling characteristics: 1) myeloid-derived fibroblasts resulting from immune dysregulation and their role in interstitial fibrosis and 2) fibroblasts arising from endogenous mesenchymal stem cells in the heart to provide the myofibroblasts critical to the formation of a competent scar after myocardial infarction.
MYELOID ORIGIN OF FIBROBLASTS MEDIATING INTERSTITIAL FIBROSIS
In culture, bone marrow derived cells become spindle shaped when grown in the absence of serum, while serum markedly retarded the fibrocyte phenotype [21]. The factor in serum that prevented the fibrocyte phenotype was serum amyloid P (SAP) and this inhibition could be mimicked by aggregated IgG [22]. This suggests that fibrocytes represent a class of fibroblast precursors responsive to immune modulation via Fcγ receptors (FcγR) [21,22]. Further studies demonstrated that these cells matured as fibroblast-like cells more readily in the presence of Th2 lymphokines [23]. The latter finding correlates with the finding that Th2 lymphokines are frequently associated with reactive fibrosis [24].
Our studies of the relationship of interstitial fibrosis to immunologic and inflammatory factors began with observations arising from a model of daily ischemia (15 minutes) and reperfusion (I/RC) in closed chest mice. That model demonstrated the fibrotic cardiomyopathy associated with elevated macrophage infiltration and fibroblast numbers (day 5 peak) and interstitial fibrosis (day 7 peak) [25,26]. The model was designed to examine the pathological effects of generation of reactive oxygen species in a setting in which myocardial infarction did not occur. In myocardial infarction, ischemia and reperfusion is followed by a very brisk acute inflammatory reaction associated with elevation of multiple chemokines and cytokines thought to mediate the observed inflammatory infiltrate [27–30]. In the absence of infarction, to our surprise, there was no appreciable cytokine response and only one chemokine, monocyte chemoattractant protein-1 (MCP-1, CCL2) was found to be elevated [25]. MCP-1 became elevated shortly after the beginning of occlusion and reperfusion and peaked at 3 days while remaining significantly elevated for 10 days. Prior studies demonstrated that the predominant site of MCP-1 induction was in the venular endothelium in the area of ischemia [31]. Because we hypothesized that reactive oxygen had a major role in this induction, we demonstrated that the I/RC phenotype was totally abrogated in a model overexpressing extracellular SOD [25]. Similarly, genetic deletion of MCP-1 or administration of monoclonal antibodies to MCP-1 abrogated the I/RC response inducing the fibrosis, macrophage infiltration, and generation of fibroblasts [32]. We then isolated fibroblast populations from wild type and MCP-1−/− mice subjected to I/RC and found that the fibroblasts isolated from WT mouse hearts contained a large population of small spindle-shaped fibroblasts which, when cultured, displayed markedly increased proliferation when compared to fibroblasts from MCP-1−/− or sham controls [32]. By contrast, a direct application of MCP-1 to fibroblast cultures in any of the groups did not increase cardiac proliferation or further generation of spindle-shaped fibroblasts [32]. This led us to postulate the possibility that spindle-shaped fibroblasts had a critical role in the development of interstitial fibrosis in this model and originated from a population of blood borne precursors of bone marrow origin that had been attracted to the heart by MCP-1.
To pursue this hypothesis, we utilized bone marrow rescue techniques in which cells from the ROSA26 mouse (bearing the β-galactosidase/lacZ gene) were transplanted into irradiated WT animals. The presence of the β-galactosidase activity in cells found in tissues from the irradiated host (chimeric animals) indicated that the labeled cells were derived from the transplanted bone marrow. We subjected these animals to the I/RC protocol and examined the cells of the heart at 5 and 7 days, at which time fibroblast levels were highest [26]. In addition, hearts were taken and either examined histologically (7 days) or cells were isolated by proteolytic digestion (5 days) and cultured. Examination of the cultured cells demonstrated the presence of small spindle-shaped cells that were lacZ+ (and therefore of bone marrow origin) and also expressed CD34 (a marker of precursor cells undergoing asymmetric division), collagen type I and alpha smooth muscle actin. There were no lacZ+ fibroblasts in chimeric sham control animals. Spindle-shaped cells grew more rapidly than the structural fibroblasts from the same hearts [26]. Further examination of these marrow-derived cells demonstrated that they expressed CD45, confirming that they were of hematopoietic origin. Examination of the histologic sections of these animals demonstrated that CD45+ cells also expressed alpha smooth muscle actin. In vitro studies demonstrated that the cultured spindle-shaped fibroblasts expressed CD45 and collagen type I. Finally, cytometric analysis of primary dispersed cardiac cells after 5 days of I/RC demonstrated the unique appearance of CD45+CD34+ fibroblasts expressing collagen type I (Figure 1) as a marker of both cardiac fibroblasts and hematopoietic cells. CD45+ cells were not seen in sham operated animals [26]. These data suggested that the I/RC protocol induced prolonged MCP-1 expression mediating the uptake of mononuclear leukocytes and either directly or indirectly induced the formation of cells expressing both hematopoietic and stromal cell markers such as collagen. These cells are distinct from structural fibroblasts resident in the heart, and thus we termed them “myeloid-derived fibroblasts”.
Figure 1.
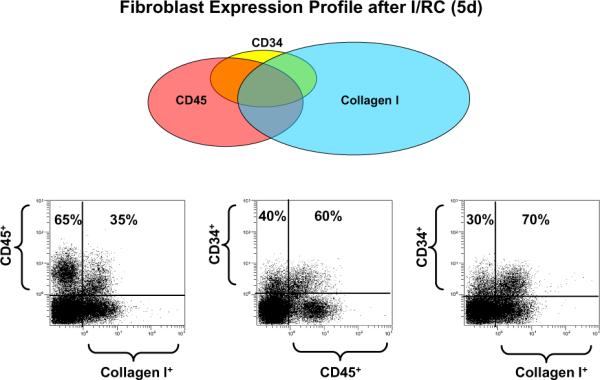
Distribution of various nonmyocyte cell types in the heart with I/RC. At the top is a Venn diagram of the phenotypes of cells with bone marrow-derived leukocyte markers (CD34 and CD45) and/or a fibroblast marker, collagen type I. The majority of cells with collagen type I and no leukocyte markers are structural fibroblasts. A substantial proportion of CD45+ leukocytes also make collagen type I, and these are bone marrow-derived fibroblasts. Histograms in the bottom row are representative of the types of flow cytometric analysis used to calculate the Venn.(reprinted with permission from Fibrocytes In Health and Disease, World Scientific Publishers)
The prolonged expression of MCP-1 in I/RC contrasts with the chemokine response to myocardial infarction which is rapidly suppressed [30,33]. Deletion of MCP-1 in I/RC completely obviates the fibrotic response whereas its deletion in acute myocardial infarction results in normal scar formation despite decreased macrophage infiltration. Thus, myeloid-derived fibroblasts appear to have a critical role in interstitial fibrosis in I/RC, while not having a major role in scar formation after myocardial infarction [26,8]. The origin of fibroblasts in infarct repair will be discussed in more detail further in the review.
MONOCYTE TO FIBROBLAST TRANSITION – AN IN VITRO MODEL TO FURTHER EXAMINE THE CELLULAR MECHANISMS IN I/RC
We had previously demonstrated that MCP-1 was induced in venular endothelial cells immediately upon reperfusion of a canine [27,28] or a murine [27] myocardial infarction model. While the I/RC model also resulted in MCP-1 induction, the absence of myocyte death or cardiac injury created a situation in which most of the mediators of the inflammatory cascade associated with infarction were absent. In the absence of cardiac injury and infarction, MCP-1 continued to be expressed for greater than a week, whereas chemokine production is ordinarily suppressed by the second day after a myocardial infarction [25,27]. One could thus consider I/RC and the resultant interstitial fibrosis as an example of inflammatory dysregulation resulting from the failure to suppress MCP-1.
This reasoning is predicated on the hypothesis that an MCP-1 sensitive mononuclear cell population is responsible for generating the CD45+ spindle-shaped fibroblasts found in the myocardium. We sought to model this inflammatory dysregulation in human cells utilizing a human mononuclear cell population and microvascular endothelial cells from human heart or human umbilical vein endothelial cells [35]. The model developed demonstrated the requirement for MCP-1-dependent transendothelial migration of mononuclear cells to generate monocyte to fibroblast transition; transition occurred only when monocytes transmigrated across intact endothelial monolayers [35]. The advantage of this model was two-fold: 1) it provided an additional way to study cellular and molecular mechanisms involved in generating spindle-shaped fibroblasts associated with interstitial fibrosis in the heart and 2) demonstrated that the phenomenon observed in mouse models also applied to human cells. We have utilized these advantages to extend our query into the mechanism of pathological cardiac interstitial fibrosis.
As previously described, fibrocytes or myeloid-derived fibroblasts have been associated with localized fibrosis particularly in lung, skin and joints [15–19]. One of the striking features of the in vitro fibrocyte assay was its inhibition by aggregated IgG or serum amyloid P (SAP) [21,22], the principal hepatic acute phase reactant in the mouse. The similarity of the in vitro fibrocyte assay to our studies suggested that they would be related. We thus administered SAP to mice undergoing the I/RC protocol and demonstrated that SAP treatment completely abrogated I/RC fibrosis and cardiac dysfunction [26]. SAP treatment in mice also markedly reduced the presence of CD45+ fibroblasts in the heart and their proliferation in vitro. By contrast, SAP did not alter chemokine expression or the total number of macrophages seen in the heart after I/RC. We postulated that SAP may interact with a specific ligand on monocytic cells in a way that prevented monocyte to fibroblast transition. We also postulated that SAP might be functioning as a ligand to FcγR as part of its inhibitory action. Thus, we re-examined the I/RC model and the effects of SAP in mice lacking the FcγRγ chain (the common membrane protein signaling component for all murine activating FcγR). Removal of the FcγRγ chain completely eliminated the ability of SAP to protect against I/RC, so that the I/RC phenotype was unaffected [35].
These data suggested that there was a specific immune regulatory component to monocyte to fibroblast transition that could be affected by activating FcγR. Transendothelial migration of monocytes resulted in their maturation to macrophages, and ligands of activating FcγR such as IgG or SAP are generally associated with the M1 macrophage phenotype. In addition, recent evidence suggested that SAP also interfered with alternative macrophage activation (M2) by inhibiting many of the key M2 genes [36,37]. Combining these observations, we reasoned that alternative activation of macrophages to an M2 phenotype was critical for monocyte/macrophage to fibroblast transition. These data were further supported by experiments in other laboratories that demonstrated in a serum-free fibrocyte differentiation medium that IL-4 and IL-13 (Th2 lymphokines) also enhanced fibrocyte differentiation [23]; these lymphokines favor alternative activation to an M2 phenotype and suggest a role for T lymphocytes in the I/RC mechanism. Thus, we utilized our in vitro model of transendothelial migration of mononuclear cells to further study this phenomenon. Our studies demonstrated that production of IL-13 was necessary in the in vitro model of monocyte to fibroblast transition. The transition was completely inhibited by IL-13 blocking and stimulated by addition of higher levels of IL-13 [38]. By contrast, IL-12 (M1 cytokine) markedly inhibited monocyte to fibroblast transition [38]. In comparing cardiac tissue from normal mice and mice with increased interstitial fibrosis secondary to I/RC [38], there was a striking increase in the levels of IL-13 mRNA as well as IL-4 and IL-13 proteins associated with I/RC. Thus, our model now includes two cells in the mononuclear population; importantly, MCP-1 is a potent chemoattractant for both monocytes and some subsets of T lymphocytes.
IL-13 has been previously implicated in pathological fibrosis secondary to a direct effect on fibroblasts associated with the IL-13RA1-STAT 6-PDGF signaling pathway [39,40]. Data from our studies suggested that IL-13 is also important in the direct mediation of macrophage activation so that myeloid-derived fibroblasts may be dependent on (or identical to) one of the “alternative activation states” of macrophages (M2 phenotype). By contrast, inflammatory activation by an activating FcγR [41] or Th1/M1 stimulation [38] inhibits the myeloid-derived fibroblast induction. These findings are compatible with the inhibitory role of SAP discussed above. Figure 2 shows that I/RC is associated with marked increases in T lymphocytes (CD45+CD3+) and also M2 cells expressing CD45 and the M2a marker CD301. While the latter demonstrates alternative activation to M2 macrophages, in Figure 3 it is shown that they also become fibroblasts that are CD45+CD301+ and contain procollagen, demonstrating active collagen synthesis. In Figure 4, we show a postulated pathway for the cellular and immunologic basis for I/RC based on our current data.
Figure 2.
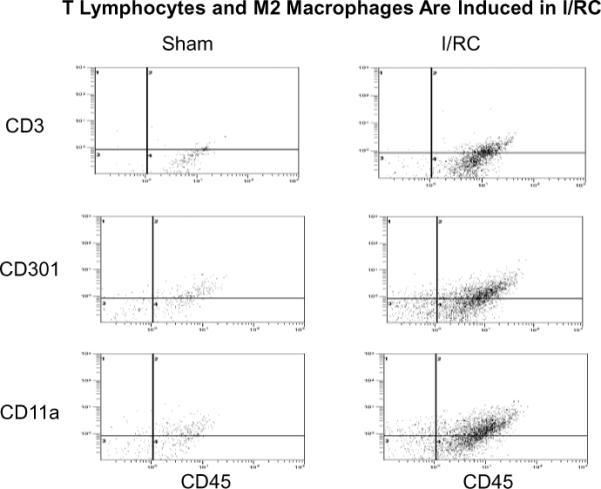
Representative cytometric diagrams for three color staining: Freshly dispersed, nonmyocyte cardiac cells from sham and I/RC-treated mouse hearts were gated for viable (calcein+) cells, and expression of the indicated marker proteins (y-axis;PE-labeled) and CD45 (x-axis, PE/Cy5 labeled) was measured (see also Figure 1). In sham treated hearts, few CD45+cells were detected. However after 5 days of I/RC, there was an increase in the number of CD3e+/CD45+ cells (T lymphocytes), CD301+/CD45+ cells (M2a macrophages), and CD11a+/CD45+cells (leukocytes) in the heart.
Figure 3.
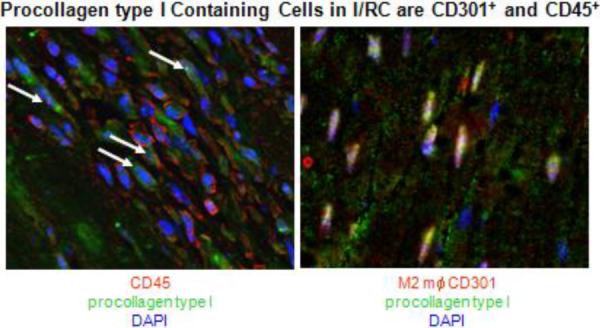
Immunofluorescence staining of mouse heart that had been subjected to I/RC for 5 days (procollagen type I and CD45) or 7 days (procollagen type I and CD301). CD45 is a marker for all hematopoietic cells and CD301 is a marker of M2a macrophages. The presence of procollagen type I indicates the active production of collagen within the cell. The white arrows point to double positive cells with nuclei in blue (DAPI).
Figure 4.
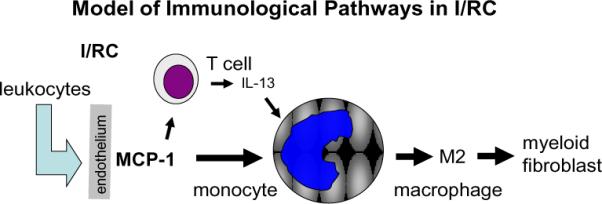
Graphical representation of monocyte-to-fibroblast maturation in the I/RC model of cardiac injury. MCP-1 expressed at the endothelium attracts leukocytes, including monocytes and some T lymphocytes. The T cell product, IL-13, induces maturation of the transmigrated monocytes into M2 macrophages that become fibroblasts.
These data do not preclude the possibility of endogenous fibroblasts also contributing to excess collagen deposition, potentially under the guidance of myeloid fibroblasts. We have observed increased collagen staining in resident fibroblasts as well as myeloid-derived fibroblasts [38, unpublished observations]; however, in the absence of myeloid fibroblasts, fibrosis is virtually eliminated in I/RC. It has also been found that conditioned media from myeloid-derived fibroblasts can promote the tissue remodeling capabilities of dermal fibroblasts; however, the myeloid-derived fibroblasts contributed little to the overall collagen production when cultured with dermal fibroblasts [42, 43]. Co-culture with monocyte-derived cell types from keloid patients can also stimulate fibroblast proliferation [44].
MONOCYTE TO FIBROBLAST TRANSITION MECHANISM IN OTHER MODELS OF CARDIAC INTERSTITIAL FIBROSIS
Angiotensin II
As part of our investigations regarding immune dysregulation in cardiac interstitial fibrosis, we have examined the role of myeloid-derived fibroblasts in other mouse models of cardiac interstitial fibrosis. We first chose a model of continuous infusion of angiotensin II (ang II) since ang II plays such a prominent role in virtually all causes of cardiac hypertrophy and failure. Ang II infusion resulted in rapid collagen deposition peaking at 2 weeks in the mouse. In the first week, we found highly significant elevations in cardiac myofibroblasts and monocytic cells, and immunohistochemical staining demonstrated MCP-1 expression in the small vessels of the heart similar to that found in I/RC. As in I/RC, the genetic deletion of MCP-1 resulted in a virtually complete abrogation of cardiac fibrosis and marked reduction in α-smooth muscle actin+ cells (myofibroblasts) along with a 50% reduction in macrophage infiltration [45]. We also found striking increases in collagen types I and III mRNA as well as a marked increase of MCP-1 mRNA.
Thus, ang II administration resulted in MCP-1 induction similar to that seen in I/RC and was accompanied by fibrotic cardiomyopathy and similar cellular components. Despite the fact that MCP-1−/− mice did not develop interstitial fibrosis in response to ang II infusion, they did develop similar degrees of hypertension and hypertrophy [45]. While these studies do not demonstrate MCP-1 as the sole factor controlling ang II-induced interstitial fibrosis, MCP-1 appears to be a necessary component of the fibrotic response.
Aging Mouse
We studied another pathologic model of interstitial fibrosis related to the aging mouse. Our studies demonstrated that aging mice develop increased interstitial fibrosis along with diastolic dysfunction [38]. We next demonstrated that the aging mouse heart contained an intrinsic renin angiotensin system (RAS) that increased with age. This was accompanied by increased expression of MCP-1 mRNA and an increase in venular MCP-1 protein. Subsequent studies demonstrated that interstitial fibrosis in the aging mouse was also associated with marked increases in CD45+ fibroblasts (Figure 5), Th2 lymphokines (Figure 6) and M2 fibroblasts (Figure 7) and marked increases in procollagen [38].
Figure 5.
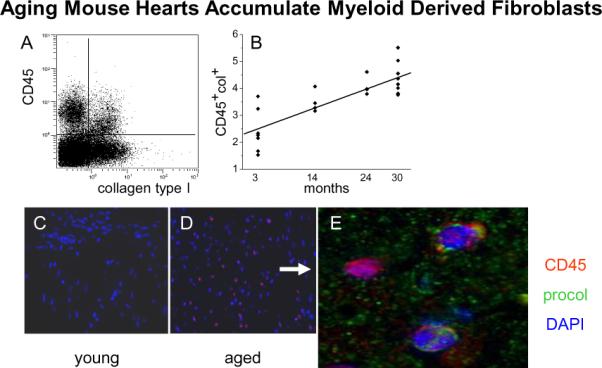
Expression of collagen type I in CD45+ cells. Nonmyocytes were isolated from hearts of different ages. (A) Flow cytometry of those cells demonstrated that a proportion of the nonmyocytes expressed both CD45 (hematopoietic marker) and collagen type I (fibroblast marker) as seen in the upper right quadrant (cells isolated from a 14 month old animal). (B) The proportion of the nonmyocytes that were double positives (CD45 and collagen type I) increased with age. Linear regression R2=0.66 and p=0.0001 (n=7, 4, 4, and 9 for 3, 14, 24, and 30 months). Immunofluorescence in paraffin sections of hearts with antibodies to CD45 (red) was negative in 3 month old hearts (C) and positive with a random distribution in 14 month old hearts (D). (E) Some CD45+ cells were also positive for procollagen type I (green). Cell nuclei are DAPI stained (blue).(reprinted with permission from JMCC 50:248-256, 2011)
Figure 6.
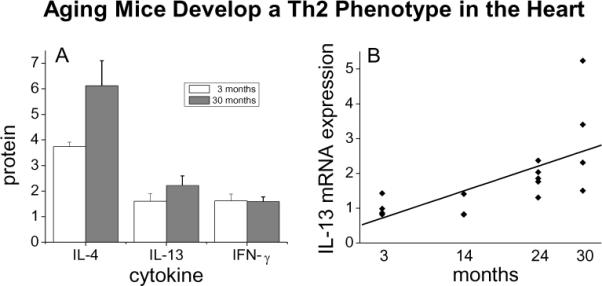
Measurement of cytokine mRNA and protein in hearts from mice of different ages. (A) Th2 (IL-4 and IL-13) and Th1 (IFN-γ) cytokines were measured by protein array in 3 month old versus 30 month old hearts (n=3). (B) mRNA expression of IL-13 by quantitative PCR, with a linear regression R2=0.43 and p=0.006; n=4, 3, 5, and 4 for 3, 14, 24, and 30 month old animals.(reprinted with permission from JMCC 50:248-256, 2011)
Figure 7.
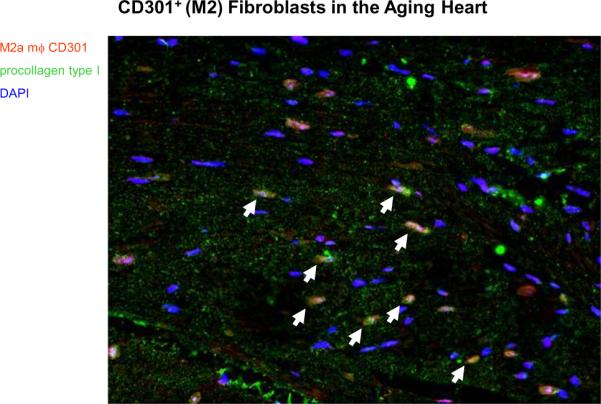
Immunofluorescence staining of a 14 month-old mouse heart with antibodies to CD301 (marker of M2a macrophages) and procollagen type I (marker for fibroblasts producing new collagen type I). The double positive cells are seen in mouse hearts from 14 to 30 months of age. The white arrows point to double positive cells with blue nuclei (DAPI).
This suggested that progressive fibrosis in the aging mouse was associated with ongoing collagen expression in myeloid fibroblasts. The experiments do not have the advantage (so far) of studies in aged MCP-1 knockout animals, so the same relationship between MCP-1 expression and fibrosis suggested in other models has yet to be confirmed. This model is also distinct from acute models in its prolonged induction of the molecular and cellular components of the myeloid-based fibrotic reaction.
ADAPTIVE FIBROTIC RESPONSES - CARDIAC SCAR FORMATION
Myeloid-derived fibroblasts
The common role of myeloid fibroblast generation arising from immune dysregulation and the production of MCP-1 suggested that this might be a common mechanism for adverse or nonadaptive fibrosis. Recent work with our collaborators has suggested a similar mechanism occurring in chronic renal fibrosis with the exception that it is associated with dysregulation of a different chemokine pair [46]. We then applied the same methods to question the potential role of myeloid fibroblasts in cardiac scar formation. Myocardial infarction and reperfusion resulted in localized increase of MCP-1 similar to that seen in I/RC; in myocardial infarction, generation of MCP-1 is more intense but very short lived, disappearing within a day or two, suppressed by activation of latent TGF-B [27]. Examination of infarcted hearts confirmed the presence of myeloid fibroblasts in the area of infarction, suggesting that they may play a role in scar formation. However, we found that myocardial infarction in MCP-1−/− mice allowed formation of a normal scar with no difference in infarct size. Deletion of MCP-1 resulted in a modest delay in phagocytosis reflecting the reduction in monocytes infiltrating the myocardium; however, the scar formation was ultimately unimpaired. Experiments did demonstrate, however, that interstitial fibrosis remote from the scar, which formed after large myocardial infarctions as a result of adverse remodeling, was markedly reduced in MCP-1−/− mice [34].
These data suggest that a different precursor cell population than described above has a role in scar formation. In addition, we have found that myocardial infarction in aged [47,48] or obese [49] mice resulted in scars with low cellularity and poor cross-linking, but the heart had increased tendencies to adverse remodeling and interstitial fibrosis. We adopted the hypothesis that adverse remodeling was pathophysiologically linked with myeloid fibroblast formation, but that scar formation was produced by fibroblasts originating from a different source.
Endogenous mesenchymal stem cells
While looking for the precursor cell population responsible for scar formation after infarction, we have discovered that, in the mouse, there was a rapid appearance of fibroblasts into the infarct wound following occlusion/reperfusion and initial wound debridement. Our work demonstrated that these fibroblasts arose from CD44+CD45neg (mesenchymal) precursors that appeared rapidly and proliferated (day 3 peak) in the wound. Fibroblasts derived from these precursor cells are not structural fibroblasts (which are CD44neg), but arose from small precursor cells that expressed primitive markers (TERT, Nanog, CD34). The small precursors also had evidence of collagen type I in their Golgi apparatus and expressed discoidin domain receptor 2 (DDR2), a marker that is specific for fibroblasts in cardiac tissue [50]. Both in vivo and in vitro, we have demonstrated that these small mesenchymal cells became fibroblasts and proliferated very actively [8]. While fibroblast number reached its peak at 3 days after infarct, transition into myofibroblasts occurred towards the end of the first week and the cells participated in collagen cross-linking, followed by cell-free collagen bundles. In vitro, we isolated these CD44+ precursor cells from infarcted hearts and also, in small numbers, in control tissue and demonstrated that they show extended self renewal capability and maintain expression of the primitive marker Nanog. When placed into differentiation media, they were capable of forming osteocytes, chondrocytes, adipocytes and fibroblasts, suggesting that they were endogenous mesenchymal stem cells. Fibroblasts formed from these precursors in vitro differed from structural fibroblasts in that they were CD44+, proliferated robustly, and were able to be serially cultured for many passages beyond their Hayflick number [8]. Thus, scar formation appeared to be mediated by endogenous mesenchymal stem cells that were induced to proliferate in the area of the scar and then differentiated into fibroblasts and myofibroblasts responsible for scar formation and cross-linking (Figure 8).
Figure 8.
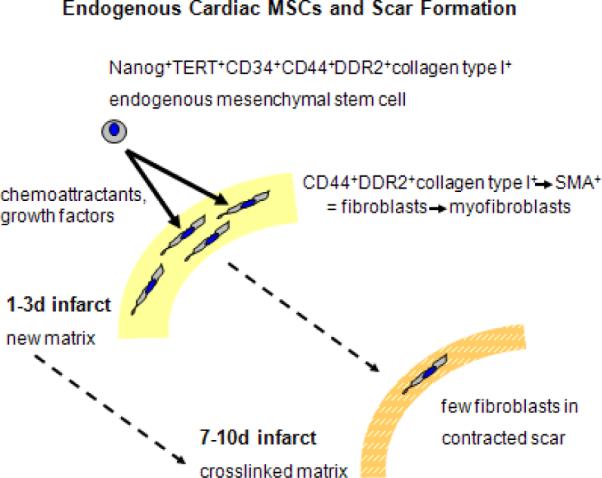
Schematic model of the participation of a resident mesenchymal stem cell in scar formation after a myocardial infarction In the uninjured heart, CD44+ cells with primitive markers (Nanog, TERT, and CD34) also express fibroblast markers such as collagen type I and DDR2. These cells proliferate and mature into fibroblasts and then myofibroblasts (adding the expression of α-smooth muscle actin,SMA). After they deposit matrix and cross-link it to form the scar, their numbers decrease.(modified from with permission from Fibrocytes In Health and Disease, World Scientific Publishers)
Pathophysiology of endogenous cardiac mesenchymal stem cells in aged mice
As briefly described above, we have observed that the aging mouse developed progressively increased interstitial fibrosis and diastolic dysfunction associated with myeloid-derived fibroblast formation [38]. However, previous work in our laboratory has demonstrated that the aging mouse, beginning as early as 12 months, demonstrated poor scar formation and greater infarct expansion after myocardial infarction [47,48]. We postulated that this might result from defective proliferation of endogenous mesenchymal stem cells and/or production of defective fibroblasts in response to myocardial infarction. Examination of cardiac mesenchymal stem cells revealed that there was a marked reduction in the pluripotency marker, Nanog, in cells from the aging mouse that were associated with defective lineage choice [51].
Mesenchymal stem cells exhibited a favored adipocytic commitment over myofibroblast differentiation. In addition, fibroblasts derived from these aging mesenchymal stem cells had reduced expression of TGF-β receptors I (TβRI) and II (TβRII), diminished Smad3 phosphorylation and had a poor ability to contract collagen pad and decreased directed motility. Overexpression of a constitutively active TβRI partially ameliorated their defective motility but did not improve their contractility. This suggested that reduced TGF-β responsiveness resulted in these defective fibroblasts and provided a potential explanation for defective scar in the aging mouse. Further in vitro studies have demonstrated that the aging fibroblasts can be partially “rescued” in vitro through stimulation of non-canonical TGF-β pathways through Tak1 (TGF-β activated kinase 1) and p38 activated protein kinase via stimulation of AMP kinase by AICAR, an AMPK activator [51]. TGF-β signaling via this pathway partially compensates for the paucity of TGF-β receptors and improves function in vitro (Figure 9).
Figure 9.
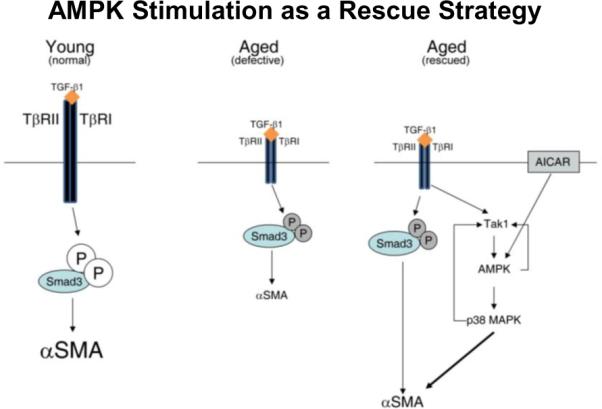
Rescue of the aged fibroblast to myofibroblast differentiation by amplification of TGF-β signaling via the Tak1/AMPK/p38MAPK pathway. Stimulation of cardiac fibroblasts derived from young animals (Young) with TGF-β1 resulted in phosphorylation of Smad3 and increased expression of β-SMA(left panel). Cardiac fibroblasts derived from 30-month-old animals (Aged) demonstrated reduced expression of both TβRI and TβRII, which resulted in decreased signal transduction and diminished α-SMA expression (middle panel). AICAR/TGF-β1 synergistically activated Tak1, AMPK, and p38MAPK, resulting in up-regulation of α-SMA expression (right panel). Large and small font sizes indicate, respectively, up-regulation or reduction of expression. (reprinted with permission from Am J Pathol 179:179-1806, 2011)
Thus, myocardial scar appears to result from a pathophysiologically distinct biological process involving endogenous mesenchymal stem cells, which is highly dependent on TGF-β signaling. Improving the efficiency of TGF-β signaling in endogenous mesenchymal stem cells may lead to potential interventions for improving scar quality in myocardial repair.
THE ROLE OF DEVELOPMENTAL PRECURSORS IN DICTATING CARDIAC FIBROBLAST FUNCTION - AN HYPOTHESIS
We have presented our studies suggesting that myeloid fibroblasts play a major role in nonadaptive fibrosis and adverse remodeling in the heart. By contrast, fibroblasts mediating cardiac scar formation mature from endogenous mesenchymal stem cells present within the heart that rapidly proliferate after cardiac necrosis. Developmental pathways from precursors to mature fibroblasts in each case are quite complex and distinctive. The myeloid fibroblast is controlled by chemokines and cytokines known to alter and mediate chronic inflammation in which fibrosis is often a consequence and may become the most serious outcome of chronic inflammatory states. Endogenous mesenchymal stem cells in the normal mouse heart differentiate into fibroblasts at the site of injury; significant literature exists suggesting that the fibroblast is the preferred end stage of mesenchymal stem cell differentiation [12]. Fully understanding the regulation of each of these processes will increase the potential for intervention. In the context of the special volume on “Myocardial Cellular Cross-Talk in Heart Failure”, we propose that this contribution be viewed as strong evidence that it will be possible to intercede in nonadaptive fibrosis in a manner that does not impair cardiac scar formation. Conversely, it is possible that a better understanding of the potential pathological issues that might impair the role of the mesenchymal stem cell in cardiac scar formation (as seen in our studies on aging and obesity cited above) might lead to specific therapies for patients at risk. Our in vitro studies have demonstrated that the aging fibroblast can be partially rescued in vitro through stimulation of non-canonical TGF-β pathways through Tak1 and p38 activated protein kinase [51]. TGF-β signaling via this pathway partially compensates for the paucity of TGF-β receptors and improves function in vitro. Thus, myocardial scar appears to result from a pathophysiologically distinct biological process that is highly dependent on TGF-β signaling. Improving the efficiency of TGF-β signaling in endogenous mesenchymal stem cells presents potential approaches for improving scar quality in myocardial repair.
ACKNOWLEGEMENTS
This work is supported by National Institutes of Health RO1 HL-089792 (MLE), The American Heart Association 10SDG4280031 (SBH), the Hankamer Foundation and The Medallion Foundation. The authors wish to thank Ms. Sharon Malinowski for her editorial assistance with the manuscript.
Footnotes
ETHICAL CONSIDERATIONS The Guide for the Care and Use of Animals was followed when performing the experiments described in this paper. The experiments performed comply with the current laws of the country in which they were performed.
REFERENCE LIST
- 1.Weber KT. Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol. 1989;13:1637–1652. doi: 10.1016/0735-1097(89)90360-4. [DOI] [PubMed] [Google Scholar]
- 2.Sun Y, Kiani MF, Postlethwaite AE, Weber KT. Infarct scar as living tissue. Basic Res Cardiol. 2002;97:343–347. doi: 10.1007/s00395-002-0365-8. [DOI] [PubMed] [Google Scholar]
- 3.Weber KT, Brilla CG, Janicki JS. Myocardial fibrosis: functional significance and regulatory factors. Cardiovasc Res. 1993;27:341–348. doi: 10.1093/cvr/27.3.341. [DOI] [PubMed] [Google Scholar]
- 4.Jalil JE, Janicki JS, Pick R, Abrahams C, Weber KT. Fibrosis-induced reduction of endomyocardium in the rat after isoproterenol treatment. Circ Res. 1989;65:258–264. doi: 10.1161/01.res.65.2.258. [DOI] [PubMed] [Google Scholar]
- 5.Thiedemann KU, Holubarsch C, Medugorac I, Jacob R. Connective tissue content and myocardial stiffness in pressure overload hypertrophy. A combined study of morphologic, morphometric, biochemical, and mechanical parameters. Basic Res Cardiol. 1983;78:140–155. doi: 10.1007/BF01906668. [DOI] [PubMed] [Google Scholar]
- 6.Bing OH, Matsushita S, Fanburg BL, Levine HJ. Mechanical properties of rat cardiac muscle during experimental hypertrophy. Circ Res. 1971;28:234–245. doi: 10.1161/01.res.28.2.234. [DOI] [PubMed] [Google Scholar]
- 7.Weber KT, Pick R, Jalil JE, Janicki JS, Carroll EP. Patterns of myocardial fibrosis. J Mol Cell Cardiol. 1989;21(Suppl 5):121–131. doi: 10.1016/0022-2828(89)90778-5. [DOI] [PubMed] [Google Scholar]
- 8.Carlson S, Trial J, Soeller C, Entman ML. Cardiac Mesenchymal Stem Cells Contribute to Scar Formation After Myocardial Infarction. Cardiovasc Res. 2011;91:99–107. doi: 10.1093/cvr/cvr061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 11.Esposito MT, Di Noto R, Mirabelli P, Gorrese M, Parisi S, Montanaro D, Del Vecchio L, Pastore L. Culture conditions allow selection of different mesenchymal progenitors from adult mouse bone marrow. Tissue Eng Part A. 2009;15:2525–2536. doi: 10.1089/ten.tea.2008.0509. [DOI] [PubMed] [Google Scholar]
- 12.Sarugaser R, Hanoun L, Keating A, Stanford WL, Davies JE. Human mesenchymal stem cells self-renew and differentiate according to a deterministic hierarchy. PLoS One. 2009;4:e6498. doi: 10.1371/journal.pone.0006498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med. 1994;1:71–81. [PMC free article] [PubMed] [Google Scholar]
- 14.Pilling D, Fan T, Huang D, Kaul B, Gomer RH. Identification of markers that distinguish monocyte-derived fibrocytes from monocytes, macrophages, and fibroblasts. PLoS One. 2009;4:e7475. doi: 10.1371/journal.pone.0007475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abe R, Donnelly SC, Peng T, Bucala R, Metz CN. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol. 2001;166:7556–7562. doi: 10.4049/jimmunol.166.12.7556. [DOI] [PubMed] [Google Scholar]
- 16.Yang L, Scott PG, Giuffre J, Shankowsky HA, Ghahary A, Tredget EE. Peripheral blood fibrocytes from burn patients: identification and quantification of fibrocytes in adherent cells cultured from peripheral blood mononuclear cells. Lab Invest. 2002;82:1183–1192. doi: 10.1097/01.lab.0000027841.50269.61. [DOI] [PubMed] [Google Scholar]
- 17.Hashimoto N, Jin H, Liu T, Chensue SW, Phan SH. Bone marrow-derived progenitor cells in pulmonary fibrosis. J Clin Invest. 2004;113:243–252. doi: 10.1172/JCI18847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Phillips RJ, Burdick MD, Hong K, Lutz MA, Murray LA, Xue YY, Belperio JA, Keane MP, Strieter RM. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. 2004;114:438–446. doi: 10.1172/JCI20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmidt M, Sun G, Stacey MA, Mori L, Mattoli S. Identification of circulating fibrocytes as precursors of bronchial myofibroblasts in asthma. J Immunol. 2003;171:380–389. doi: 10.4049/jimmunol.171.1.380. [DOI] [PubMed] [Google Scholar]
- 20.Moore BB, Kolodsick JE, Thannickal VJ, Cooke K, Moore TA, Hogaboam C, Wilke CA, Toews GB. CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am J Pathol. 2005;166:675–684. doi: 10.1016/S0002-9440(10)62289-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pilling D, Buckley CD, Salmon M, Gomer RH. Inhibition of fibrocyte differentiation by serum amyloid P. J Immunol. 2003;171:5537–5546. doi: 10.4049/jimmunol.171.10.5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pilling D, Tucker NM, Gomer RH. Aggregated IgG inhibits the differentiation of human fibrocytes. J Leukoc Biol. 2006;79:1242–1251. doi: 10.1189/jlb.0805456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shao DD, Suresh R, Vakil V, Gomer RH, Pilling D. Pivotal Advance: Th-1 cytokines inhibit, and Th-2 cytokines promote fibrocyte differentiation. J Leukoc Biol. 2008;83:1323–1333. doi: 10.1189/jlb.1107782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dewald O, Frangogiannis NG, Zoerlein M, Duerr GD, Klemm C, Knuefermann P, Taffet G, Michael LH, Crapo JD, Welz A, Entman ML. Development of murine ischemic cardiomyopathy is associated with a transient inflammatory reaction and depends on reactive oxygen species. Proceedings of the National Academy of Sciences, USA. 2003;100:2700–2705. doi: 10.1073/pnas.0438035100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haudek SB, Xia Y, Huebener P, Lee JM, Carlson S, Crawford JR, Pilling D, Gomer RH, Trial J, Frangogiannis NG, Entman ML. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc Natl Acad Sci U S A. 2006;103:18284–18289. doi: 10.1073/pnas.0608799103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dewald O, Ren G, Duerr GD, Zoerlein M, Klemm C, Gersch C, Tincey S, Michael LH, Entman ML, Frangogiannis NG. Of mice and dogs: species-specific differences in the inflammatory response following myocardial infarction. Am J Pathol. 2004;164:665–677. doi: 10.1016/S0002-9440(10)63154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar AG, Ballantyne CM, Michael LH, Kukielka GL, Youker KA, Lindsey ML, Hawkins HK, Birdsall HH, Mackay CR, LaRosa GJ, Rossen RD, Smith CW, Entman ML. Induction of monocyte chemoattractant protein-1 in the small veins of the ischemic and reperfused canine myocardium. Circulation. 1997;95:693–700. doi: 10.1161/01.cir.95.3.693. [DOI] [PubMed] [Google Scholar]
- 29.Kukielka GL, Smith CW, LaRosa GJ, Manning AM, Mendoza LH, Hughes BJ, Youker KA, Hawkins HK, Michael LH, Rot A, Entman ML. Interleukin-8 gene induction in the myocardium after ischemia and reperfusion in vivo. Journal of Clinical Investigation. 1995;95:89–103. doi: 10.1172/JCI117680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frangogiannis NG, Mendoza LH, Lewallen M, Michael LH, Smith CW, Entman ML. Induction and suppression of interferon-inducible protein (IP)-10 in reperfused myocardial infarcts may regulate angiogenesis. FASEB Journal. 2001;15:1428–1430. doi: 10.1096/fj.00-0745fje. [DOI] [PubMed] [Google Scholar]
- 31.Lakshminarayanan V, Lewallen M, Frangogiannis NG, Evans AJ, Wedin KE, Michael LH, Entman ML. Reactive oxygen intermediates induce monocyte chemotactic protein-1 in vascular endothelium after brief ischemia. American Journal of Pathology. 2001;159:1301–1311. doi: 10.1016/S0002-9440(10)62517-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frangogiannis NG, Dewald O, Xia Y, Ren G, Haudek S, Leucker T, Kraemer D, Taffet G, Rollins BJ, Entman ML. Critical role of monocyte chemoattractant protein-1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation. 2007;115:584–592. doi: 10.1161/CIRCULATIONAHA.106.646091. [DOI] [PubMed] [Google Scholar]
- 33.Frangogiannis NG, Ren G, Dewald O, Zymek P, Koerting A, Winkelmann K, Michael LH, Lawler J, Entman ML. Critical role of endogenous thrombospondin (TSP)-1 in preventing expansion of healing myocardial infarcts. Circulation. 2005;111:2935–2942. doi: 10.1161/CIRCULATIONAHA.104.510354. [DOI] [PubMed] [Google Scholar]
- 34.Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Michael LH, Rollins BJ, Entman ML, Frangogiannis NG. CCL2/monocyte chemoattractant protein (MCP)-1 regulates inflammatory responses critical to healing myocardial infarcts. Circulation Research. 2005;96:881–889. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- 35.Haudek SB, Trial J, Xia Y, Gupta D, Pilling D, Entman ML. Fc receptor engagement mediates differentiation of cardiac fibroblast precursor cells. Proc Natl Acad Sci U S A. 2008;105:10179–10184. doi: 10.1073/pnas.0804910105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moreira AP, Cavassani KA, Hullinger R, Rosada RS, Fong DJ, Murray L, Hesson DP, Hogaboam CM. Serum amyloid P attenuates M2 macrophage activation and protects against fungal spore-induced allergic airway disease. J Allergy Clin Immunol. 2010;126:712–721. doi: 10.1016/j.jaci.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 37.Murray LA, Chen Q, Kramer MS, Hesson DP, Argentieri RL, Peng X, Gulati M, Homer RJ, Russell T, van RN, Elias JA, Hogaboam CM, Herzog EL. TGF-beta driven lung fibrosis is macrophage dependent and blocked by Serum amyloid P. Int J Biochem Cell Biol. 2011;43:154–162. doi: 10.1016/j.biocel.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 38.Cieslik KA, Taffet GE, Carlson S, Hermosillo J, Trial J, Entman ML. Immune-inflammatory Dysregulation Modulates the Incidence of Progressive Fibrosis and Diastolic Stiffness in the Aging Heart. J Mol Cell Cardiol. 2011;50:248–256. doi: 10.1016/j.yjmcc.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Friedrich K, Brändlein S, Ehrhardt I, Krause S. Interleukin-4- and Interleukin-13 receptors trigger distinct JAK/STAT activation patterns inmouse lymphocytes. Signal Transduction. 2003;1–2:26–32. [Google Scholar]
- 40.Ingram JL, Rice AB, Geisenhoffer K, Madtes DK, Bonner JC. IL-13 and IL-1beta promote lung fibroblast growth through coordinated up-regulation of PDGF-AA and PDGF-Ralpha. Faseb J. 2004;18:1132–1134. doi: 10.1096/fj.03-1492fje. [DOI] [PubMed] [Google Scholar]
- 41.Haudek SB, Gupta D, Dewald O, Schwartz RJ, Wei L, Trial J, Entman ML. Rho Kinase-1 mediates cardiac fibrosis by regulating fibroblast precursor cell differentiation. Cardiovascular Research. 2009;83:511–518. doi: 10.1093/cvr/cvp135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang JF, Jiao H, Stewart TL, Shankowsky HA, Scott PG, Tredget EE. Fibrocytes from burn patients regulate the activities of fibroblasts. Wound Repair Regen. 2007;15:113–121. doi: 10.1111/j.1524-475X.2006.00192.x. [DOI] [PubMed] [Google Scholar]
- 43.Medina A, Ghahary A. Fibrocytes can be reprogrammed to promote tissue remodeling capacity of dermal fibroblasts. Mol Cell Biochem. 2010 doi: 10.1007/s11010-010-0524-4. [DOI] [PubMed] [Google Scholar]
- 44.Liao WT, Yu HS, Arbiser JL, Hong CH, Govindarajan B, Chai CY, Shan WJ, Lin YF, Chen GS, Lee CH. Enhanced MCP-1 release by keloid CD14+ cells augments fibroblast proliferation: role of MCP-1 and Akt pathway in keloids. Exp Dermatol. 2010;19:e142–e150. doi: 10.1111/j.1600-0625.2009.01021.x. [DOI] [PubMed] [Google Scholar]
- 45.Haudek SB, Cheng J, Du J, Wang Y, Hermosillo-Rodriguez J, Trial J, Taffet GE, Entman ML. Monocytic fibroblast precursors mediate fibrosis in angiotensin-II-induced cardiac hypertrophy. J Mol Cell Cardiol. 2010;49:499–507. doi: 10.1016/j.yjmcc.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen G, Lin SC, Chen J, He L, Dong F, Xu J, Han S, Du J, Entman ML, Wang Y. CXCL16 recruits bone marrow-derived fibroblast precursors in renal fibrosis. J Am Soc Nephrol. 2011;22:1876–1886. doi: 10.1681/ASN.2010080881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gould KE, Taffet GE, Michael LH, Christie RM, Konkol DL, Pocius JS, Zachariah JP, Chaupin DF, Daniel SL, Sandusky GE, Jr., Hartley CJ, Entman ML. Heart failure and greater infarct expansion in middle-aged mice: a relevant model for postinfarction failure. Am J Physiol Heart Circ Physiol. 2002;282:H615–H621. doi: 10.1152/ajpheart.00206.2001. [DOI] [PubMed] [Google Scholar]
- 48.Bujak M, Kweon HJ, Chatila K, Li N, Taffet G, Frangogiannis NG. Aging-related defects are associated with adverse cardiac remodeling in a mouse model of reperfused myocardial infarction. J Am Coll Cardiol. 2008;51:1384–1392. doi: 10.1016/j.jacc.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thakker GD, Frangogiannis NG, Bujak M, Zymek P, Gaubatz JW, Reddy AK, Taffet G, Michael LH, Entman ML, Ballantyne CM. Effects of diet-induced obesity on inflammation and remodeling after myocardial infarction. Am J Physiol Heart Circ Physiol. 2006;291:H2504–H2514. doi: 10.1152/ajpheart.00322.2006. [DOI] [PubMed] [Google Scholar]
- 50.Goldsmith EC, Hoffman A, Morales MO, Potts JD, Price RL, McFadden A, Rice M, Borg TK. Organization of fibroblasts in the heart. Dev Dyn. 2004;230:787–794. doi: 10.1002/dvdy.20095. [DOI] [PubMed] [Google Scholar]
- 51.Cieslik KA, Trial J, Entman ML. Defective Mesenchymal Stem Cell Differentiation in Aging Murine Heart: Rescue by Tak1/AMPK/p38 MAPK Pathway. Am J Pathol. 2011;179:1792–1806. doi: 10.1016/j.ajpath.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]