

Abstract
The present standard of care for B cell non-Hodgkin's lymphoma includes the anti-CD20 monoclonal antibody rituximab. Although combination treatments with chemotherapy and rituximab improved the duration of remissions and overall survival in indolent B cell lymphoma, the disease is essentially incurable. Thus, new therapeutic approaches are needed. One such approach is active immunization. Given that rituximab depletes both malignant and normal B cells, it is expected to impair humoral immune responses in vaccinated patients. Hence, optimal vaccination strategies for rituximab-treated patients require induction of effector T cells, which can be achieved by dendritic cell (DC) vaccines. We have demonstrated in a mouse model that chemotherapy combined with DC vaccines was therapeutically effective. However, efficacy was related to tumour size at the onset of treatment, decreasing in correlation with increasing tumour burdens. We therefore examined whether, in spite of its low efficacy in advanced disease, DC vaccination may synergize with anti-CD20 antibodies to enhance therapy. Lymphoma-bearing mice were treated with cyclophosphamide, anti-CD20 antibodies and an intratumoral DC vaccine. Results clearly demonstrated the enhanced therapeutic effect of this combination treatment. Thus, under conditions of disseminated disease, when either anti-CD20 antibody treatment or vaccination showed insufficient efficacy, their combination resulted in synergism that mediated long-term survival. We demonstrated further that the combination of antibody and vaccine induced T cell-mediated anti-tumour immune responses with long-term memory. Combination treatments including tumour cell-loaded DC vaccines may therefore provide a strategy for enhancing therapy in rituximab-treated patients.
Keywords: anti-CD20 antibodies, B cell depletion, B cell lymphoma, dendritic cell vaccines, synergism
Introduction
Indolent non-Hodgkin's lymphomas (NHL) remain incurable. The anti-CD20 monoclonal antibody (mAb) rituximab revolutionized the treatment of these malignancies and is currently utilized as a standard of care for NHL [1–3]. However, although rituximab significantly improves duration of remissions and overall survival, patients eventually relapse from residual resistant tumour cells. Thus, new therapeutic approaches are needed. One such approach is active immunization.
Active immunotherapy trials to date consist largely of vaccines that use the immunoglobulin (Ig) idiotypes (Id) as tumour-specific antigens [4]. The variable regions of surface Ig on a B cell form an antigen-binding site that is unique to each Ig and contain determinants termed Id. Because B cell lymphomas are clonal in nature, their Ig variable regions are distinct from those of their non-malignant counterparts. Therefore, Id of B cell lymphomas can serve as tumour-specific antigens for therapeutic vaccination. Although Phase I/II clinical trials of Id vaccines have shown encouraging clinical efficacy as well as clinical benefit, three large-scale Phase III trials were disappointing, as two of them failed to achieve their main endpoints of improved progression-free survival [5–7]. Detailed analysis of these trials indicated that a subset of patients with complete response following induction chemotherapy, in particular patients with an IgM vaccine isotype, may benefit from Id vaccine therapy. However, a more effective vaccine is needed for patients who do not achieve minimal residual disease.
The standard of care for follicular lymphoma changed during the course of clinical trials for Id vaccination and now includes the mAb rituximab. Because rituximab depletes both normal and malignant B cells, it is expected to impair humoral immune responses in vaccinated patients. Hence, it is crucial to determine its effect on anti-lymphoma T cell-mediated responses. Neelapu et al. reported that vaccination by Id protein plus granulocyte–macrophage colony-stimulating factor (GM-CSF) in patients with mantle cell lymphoma following rituximab-containing chemotherapy induced vigorous CD4 and CD8 anti-tumour type I responses in the absence of circulating B cells [8]. These results indicate that severe B cell depletion does not impair T cell priming, suggesting that vaccination may be used in combination with rituximab, provided that an effective T cell-inducing vaccine is utilized.
Dendritic cells (DCs) loaded with antigen are attractive vehicles for therapeutic cancer vaccines. Clinical studies of vaccination with autologous DCs loaded ex vivo with Id protein demonstrated significant immune and clinical responses [9,10]. More recently, an alternate approach of pulsing autologous DCs with apoptotic tumour cells showed impressive clinical responses in patients with relapsed indolent NHL, suggesting that immunization with tumour cell-loaded DCs represents a potentially effective strategy for the treatment of patients with relapsed and measurable disease [11]. In yet another approach that circumvents the requirement for ex-vivo antigen pulsing of DCs, naive DCs have been injected into the tumour after chemotherapy in murine lymphoma models. Antigens derived from dying tumour cells following chemotherapy are taken by the naive intratumorally injected DCs and cross-prime T cells against the lymphoma antigens, resulting in tumour regression [12,13]. This strategy has yet to be tested in patients.
We have demonstrated previously a synergistic effect of DC-based vaccination and anti-CD20 antibody treatment in the therapy of murine lymphoma [13]. Because mAbs against murine CD20 were not available at that time, we used anti-human CD20 mAbs and murine lymphoma cells engineered by retroviral transduction to express human CD20. In this experimental model, however, host B cells are not depleted because the anti-human CD20 mAbs do not react with murine CD20. As B cell-depleting anti-mouse CD20 mAbs are now available, we repeated this study with wild-type murine B cell lymphoma and anti-murine CD20 mAbs, which is a more clinically relevant setting. In this study we demonstrate an enhanced therapeutic effect of B cell-depleting anti-CD20 mAbs when combined with DC vaccination in advanced lymphoma.
Materials and methods
Mice
Female BALB/c and C3H/HeN mice (8 weeks of age) were purchased from Harlan Ltd (Jerusalem, Israel). All procedures were approved by the Institutional Animal Care and Use Committee.
Cell lines and antibodies
A20, a BALB/c-derived B cell lymphoma [14], was obtained from the American Type Culture Collection (Manassas, VA, USA). A C3H-derived B cell lymphoma, 38C-13, was generated in our laboratory [15]. L10, a BALB/c-derived B cell lymphoma [14], was provided by Dr R. Laskov (The Hebrew University, Jerusalem, Israel). The cells were maintained in RPMI-1640 medium supplemented with 10% heat-inactivated fetal calf serum, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin and 50 μM mercaptoethanol. The B cell-depleting mAb against murine CD20 (clone 5D2) was provided by Genentech Inc. (South San Francisco, CA, USA). The mAb against A20 Id (clone 1G6-B8) was provided by Dr R. Levy (Stanford University, Stanford, CA, USA).
Generation of bone marrow-derived DCs
Primary DCs were obtained from mouse bone marrow progenitors, as described by Lutz et al. [16]. Briefly, bone marrow was flushed from the femurs and tibia of mice and resuspended in RPMI-1640 medium supplemented with 10% fetal calf serum, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 μM 2-mercaptoethanol and 20 ng/ml recombinant murine GM-CSF (ProSpec-Tany TechnoGene, Rehovot, Israel). Cells were plated in bacteriological Petri dishes (Falcon no. 1029; BD Biosciences, San Jose, CA, USA), and then fed with the same medium on days 3 and 6. On day 8, non-adherent cells were collected, centrifuged at room temperature, resuspended in medium containing 10 ng/ml GM-CSF, and plated for another 2 days in tissue culture plastic dishes (Falcon no. 3003; BD Biosciences). For antigen loading, cells were incubated for 18 h with tumour cells that were γ-irradiated with 100 Gy and cultured for 2 days after irradiation. Following 18 h incubation for antigen loading, DCs were harvested and washed.
Trimodal combination treatment of tumours
Animals were inoculated subcutaneously in the right flank with 7·5 × 106 A20 or 2 × 105 38C-13 tumour cells. In the A20 lymphoma model, treatment commenced when tumours reached a mean diameter of 11 mm (day 21). Cyclophosphamide (100 mg/kg body weight) was injected intraperitoneally twice on days 21 and 23. One day post-chemotherapy (day 24), mice were injected intraperitoneally with 200 μg of anti-CD20 mAbs. Two million DCs were given 2 and 5 days later (days 26 and 29), either by intratumoral injection of naive DCs or by subcutaneous injection (in the left flank) of tumour cell-loaded DCs. In the 38C-13 lymphoma model, treatment commenced when tumours reached a mean diameter of 8 mm (day 8), but other than that was similar to the combination treatment of A20.
Enzyme-linked immunospot (ELISPOT) assay for detection of interferon (IFN)-γ-secreting cells
ELISPOT assays were performed as we have described previously [17]. Briefly, nitrocellulose-based 96-well microtitre plates (Millipore Corporation, Bedford, MA, USA) were coated overnight at 4°C with 10 μg/ml of anti-mouse IFN-γ mAb R4-6A2 (BD Biosciences Pharmingen, San Diego, CA, USA). Following blocking, mixtures of spleen cells and irradiated tumour cells at ratios of 10:1 or 20:1 were added to plates and incubated for 24 h at 37°C. The cells were then removed, and biotinylated anti-mouse IFN-γ mAb XMG1·2 (BD Biosciences Pharmingen) was added at 4 μg/ml for 24 h at 4°C. Peroxidase-conjugated streptavidin (Jackson ImmunoResearch, West Grove, PA, USA) was then added for 2 h at room temperature followed by the 3-amino-9-ethylcarbazole substrate (Sigma-Aldrich, St Louis, MO, USA). Red spots representing single IFN-γ-secreting cells were counted using a dissecting microscope.
Flow cytometry
Tumour cells (1 × 106 cells per sample) were incubated for 45 min at 4°C with sera of immunized mice diluted 1:100 or with anti-Id antibodies (10 μg/ml). After washings, cells were incubated with fluorescein isothiocyanate-conjugated goat anti-mouse IgG1 (Jackson ImmunoResearch). Cells were then washed and analysed on a FACSort flow cytometer (BD Biosciences).
Statistical analysis
Differences between survival curves were assessed by the log-rank test. Values at P < 0·05 were considered statistically significant.
Results
The combination of anti-CD20 antibody treatment and DC vaccination results in a synergistic therapeutic effect
It has been demonstrated previously in lymphoma models that, although chemotherapy alone resulted in only transient tumour regression, cytoreduction by cyclophosphamide was required prior to anti-CD20 antibody treatment and DC vaccination in order to achieve a therapeutic effect [12,13]. Additionally, in the 38C-13 lymphoma model, we have demonstrated that intratumoral injection of naive DCs and subcutaneous injection of DCs loaded with irradiated tumour cells had comparable therapeutic effects, which were superior to vaccination with Id-loaded DCs [13]. In this study we compared intratumoral injection of naive DCs and subcutaneous injection of DCs loaded with irradiated tumour cells in the A20 lymphoma model. The latter was used in the present study because A20 cells, unlike 38C-13 cells, express surface CD20 ([18] and our unpublished observations). As shown in Fig. 1a, DCs loaded with tumour cells either ex vivo or in situ had a similar therapeutic effect against established tumours, resulting in long-term survival of a high percentage of treated animals (differences between the two vaccination regimens were not statistically significant). Because intratumoral injection of naive DCs does not require ex-vivo antigen loading, we used this approach during the present study.
Fig. 1.
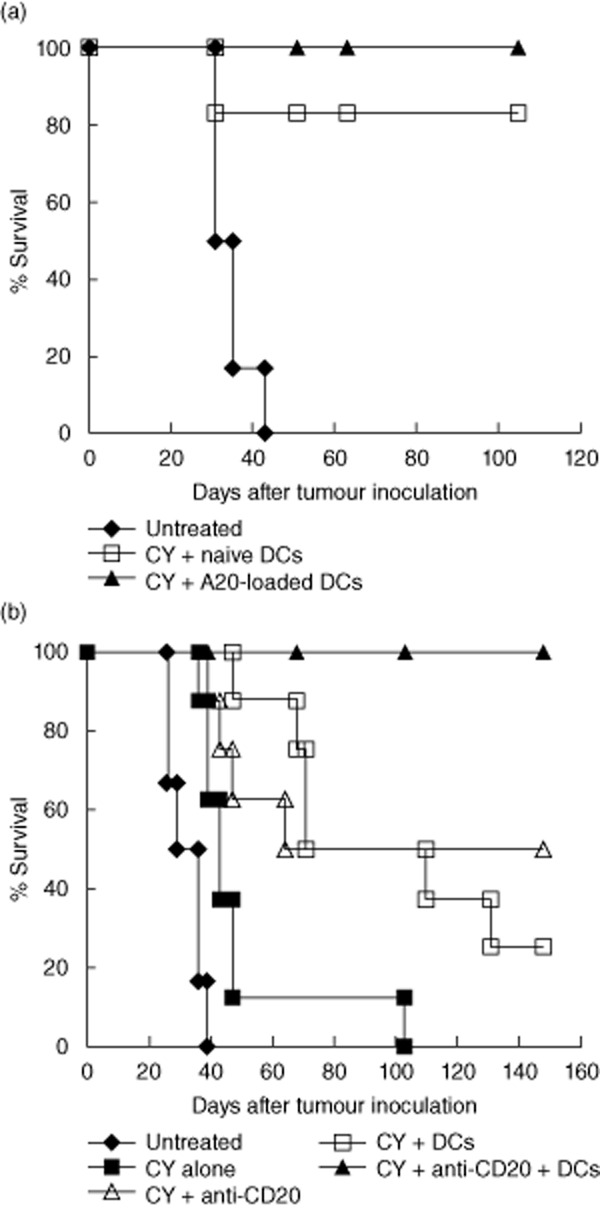
Dendritic cell (DC) vaccines can effectively eradicate only small tumours, but they synergize with CD20 monoclonal antibodies (mAbs) to enhance therapy in advanced disease. (a) Mice were inoculated subcutaneously in the right flank with 7·5 × 106 A20 tumour cells. Treatment commenced when tumours reached a mean diameter of 6 mm (day 14). Cyclophosphamide (CY) was injected intraperitoneally on days 14 and 15. Mice were then vaccinated on days 18 and 21 by subcutaneous injection in the left flank of 2 × 106 DCs loaded with irradiated A20 tumour cells, or by intratumoral injection of 2 × 106 naive DCs. Differences between the two vaccination regimens were not significant (P > 0·1). (b) Mice were inoculated subcutaneously in the right flank with 7·5 × 106 A20 tumour cells. Treatment commenced when tumours reached a mean diameter of 11 mm (day 21). Cyclophosphamide (CY) was injected intraperitoneally on days 21 and 23. On day 24, 200 μg of anti-CD20 mAbs were injected intraperitoneally. On days 26 and 29, 2 × 106 naive DCs were injected intratumorally. The combination of mAbs and DCs was superior to each of them alone (P < 0·005). Results are representative of three independent experiments.
Efficacy of DC vaccination was related to tumour size at the onset of treatment, decreasing in correlation with increasing tumour burdens. Thus, while 80–100% of mice with 6-mm tumours survived following vaccination (Fig. 1a), only 20–30% of mice with 11-mm tumours survived following the same treatment (Fig. 1b). This was also the case for anti-CD20 antibody treatment (data not shown). However, when DC vaccination was combined with anti-CD20 mAbs, the two agents synergized to yield an enhanced therapeutic effect in mice bearing large tumours. In a representative experiment depicted in Fig. 1b, mice bearing large A20 tumours (mean diameter of 11 mm on day 21 after tumour inoculation) were injected intraperitoneally twice with cyclophosphamide on days 21 and 23. One day post-chemotherapy (day 24), mice were injected intraperitoneally with anti-CD20 antibodies. Two and 5 days later (days 26 and 29), two injections of naive DCs were given into the tumour. This combination treatment induced a synergistic effect that led to 100% long-term survival (P < 0·005).
In-situ DC vaccination after chemotherapy and antibody treatment induces systemic long-term anti-tumour immunity
Untreated A20 tumour-bearing mice succumb to local and systemic tumour in lymph nodes, spleen and liver in 5–7 weeks. In fact, when treatment of tumour-bearing mice began (day 21), A20 tumour cells had already disseminated from local tumour to distant lymph nodes (Fig. 2a). Hence, the finding that treatment with chemotherapy, anti-CD20 mAbs and an in-situ DC vaccine resulted in long-term survival (> 5 months) indicated indirectly that local intratumoral DC injection induced a systemic anti-tumour response. To support this supposition, mice were inoculated with tumours either into the right flank or into both flanks. After systemic treatment with cyclophosphamide and anti-CD20 mAbs (as in Fig. 1b), the tumours in the right flank were injected with DCs, whereas the tumours in the left flank remained untreated. It was demonstrated that tumours in the right (injected) and left (non-injected) flanks regressed simultaneously. Moreover, DC injection into the right tumour of mice bearing tumours on both flanks induced long-term survival, which was similar to that observed in mice bearing tumours only in the right flank (Fig. 2b). These results indicated that intratumoral DC injection as part of the trimodal combination treatment induced an anti-tumour effect that was systemic, rather than local.
Fig. 2.
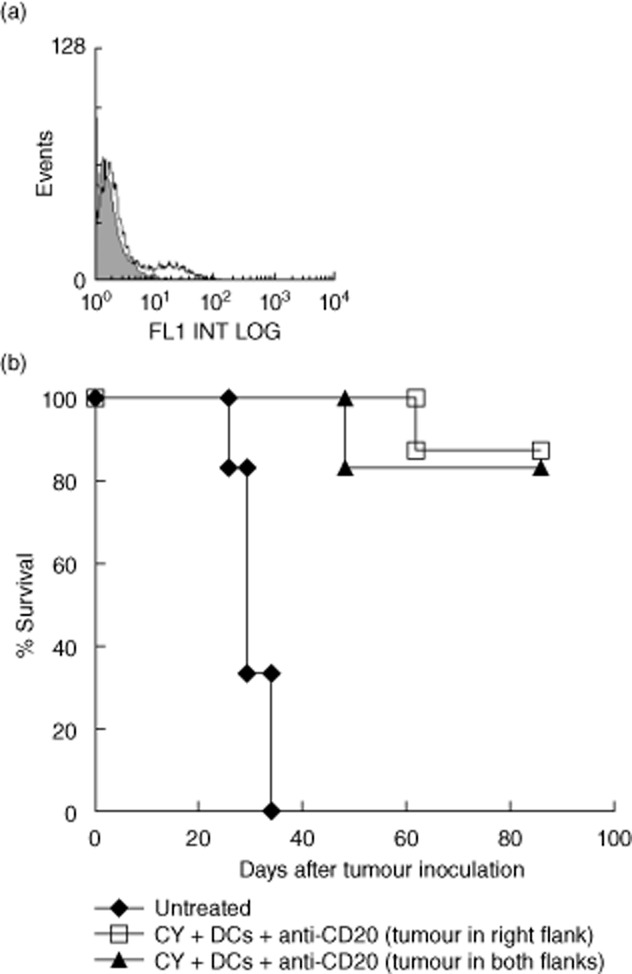
In-situ dendritic cell (DC) vaccination after chemotherapy and CD20 antibody treatment induces systemic anti-tumour immunity. (a) To show that subcutaneous A20 tumours of 11 mm are already disseminated in distant lymph nodes, fluorescence staining of lymph node cell suspensions with tumour-specific anti-Id monoclonal antibodies (mAbs) was performed. Shaded histogram depicts control antibody and open histogram depicts anti-Id antibody. (b) To show that unilateral intratumoral DC injection results in tumour regression on the contralateral side, mice were inoculated with 7·5 × 106 A20 tumour cells either into the right flank or into both flanks. After systemic treatment with cyclophosphamide and anti-CD20 mAbs (as in Fig. 1b), the tumours in the right flank where injected with 2 × 106 DCs, whereas the tumours in the left flank remained untreated. No difference between the two treatments could be demonstrated (P > 0·3). Results are representative of two independent experiments.
Long-term surviving mice (5 months post-trimodal combination treatment) were rechallenged with the same dose of tumour cells. As shown in Fig. 3, surviving mice were able to resist the rechallenge (P < 0·005), indicating that combined treatment induced immune-mediated tumour rejection with long-term memory.
Fig. 3.
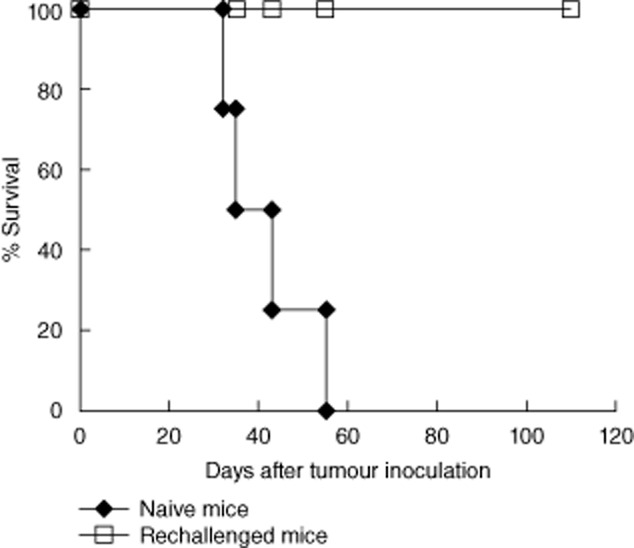
Long-term surviving mice resist tumour rechallenge. Five months post-combination therapy, surviving mice were rechallenged subcutaneously with 7·5 × 106 A20 tumour cells. Naive mice were similarly challenged. P < 0·005. Results are representative of three independent experiments.
The combination treatment induces tumour-specific T cells but no antibody production
To determine whether tumour eradication induced by the combination treatment was antibody-dependent, sera of treated mice were analysed by immunofluorescence for tumour binding. Figure 4a demonstrates that sera of mice with complete response to anti-CD20 mAbs and DC vaccines (as in Fig. 1b) did not contain antibodies that bind to tumour cells, indicating that tumour rejection did not involve active anti-tumour antibody production.
Fig. 4.
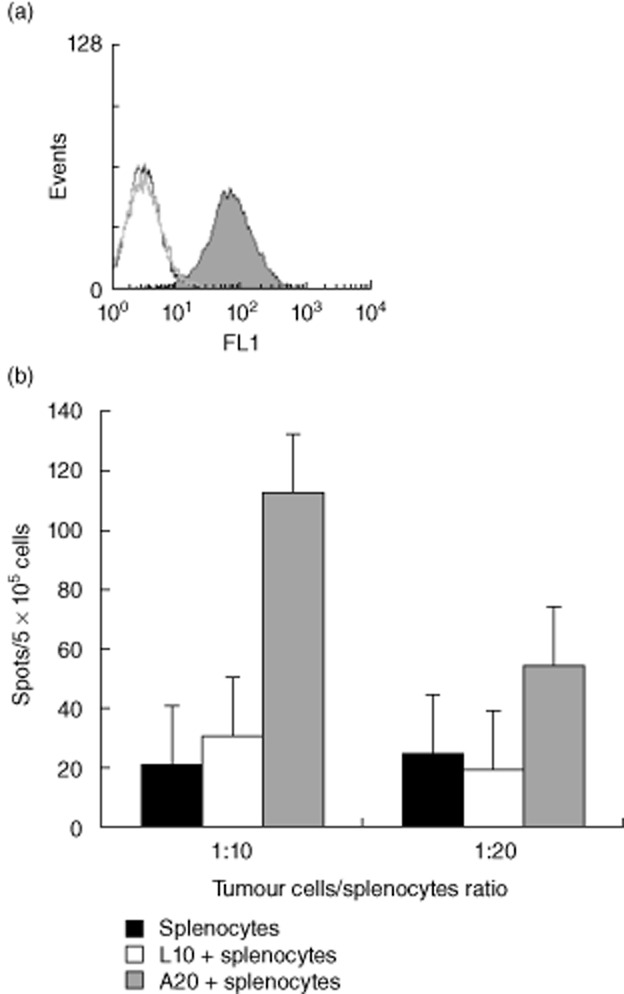
The combination treatment induces tumour-specific T cells but no antibody production against A20 lymphoma. (a) Sera of mice responding to the combination treatment were diluted 1:100 and analysed by immunofluorescence for tumour binding. A representative mouse is presented. Open black and grey histograms depict sera before tumour inoculation and after treatment, respectively. Shaded histogram depicts a positive control of anti-A20 Id. (b) spleen cells of surviving mice were incubated with irradiated A20 or control L10 tumour cells at a 10:1 or 20:1 ratio. The number of interferon (IFN)-γ-secreting cells was determined by enzyme-linked immunospot assay. Responses to A20 tumour were enhanced compared to controls (P < 0·05). Results are representative of three independent experiments.
Because tumour immunity did not involve antibodies, by using IFN-γ-ELISPOT assays we determined if the combination treatment induced an anti-tumour T cell response. Spleen cells of surviving mice were incubated with irradiated tumour cells. As shown in Fig. 4b, T lymphocytes of surviving mice reacted specifically to A20 tumour by IFN-γ secretion (P < 0·05).
The combination treatment has no effect on CD20-negative lymphoma
Previous studies demonstrated an enhanced ability of T cells to reject tumours and enhanced vaccine-induced anti-tumour immune responses in the absence of B cells [19–22]. Because the combination of anti-CD20 mAbs and DC vaccine synergized to enhance therapy, it was possible that the augmented therapeutic outcome resulted in part from CD20 mAb-mediated depletion of host B cells. To determine if this was the case, we used the B cell lymphoma 38C-13 that does not express surface CD20. Neither the mouse anti-mouse CD20 mAb 5D2 (used along this study) nor the rat anti-mouse CD20 mAb AISB12 (eBioscience, San Diego, CA, USA) reacted with 38C-13 cells (data not shown). In the model of 38C-13 tumour-bearing mice, anti-CD20 antibodies do not react with tumour cells but they do deplete host B cells. Results depicted in Fig. 5 demonstrate not only that anti-CD20 mAbs alone had no therapeutic effect, but also that their combination with vaccination was ineffective. A control group of mice injected with the same dose of anti-CD20 antibodies ascertained that B cell depletion in blood, spleen and lymph nodes took place (data not shown). Hence, no augmentation of vaccination by B cell depletion could be demonstrated.
Fig. 5.
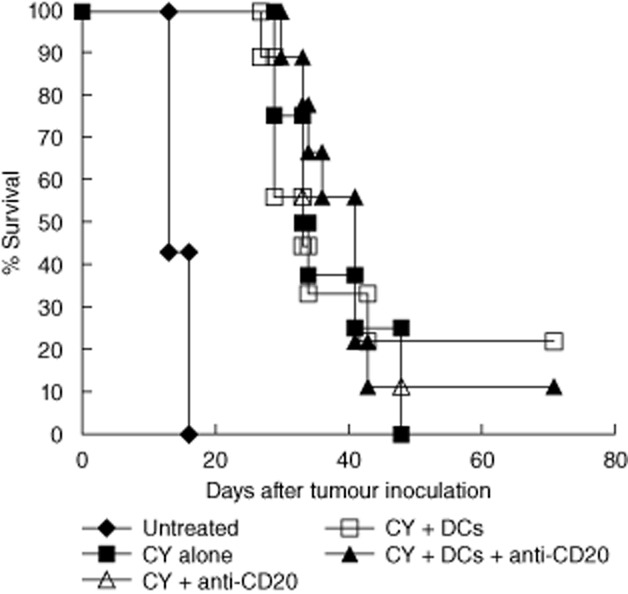
The combination treatment is ineffective for CD20-negative lymphoma. C3H Mice were inoculated subcutaneously in the right flank with 2 × 105 38C-13 tumour cells. Treatment commenced when tumours reached mean diameter of 8 mm (day 8). Cyclophosphamide (CY) was injected intraperitoneally on days 8 and 9. On day 10, 200 μg of anti-CD20 monoclonal antibodies (mAbs) were injected intraperitoneally. On days 12 and 15, 2 × 106 naive dendritic cells (DCs) were injected intratumorally. Results are representative of three independent experiments.
Discussion
The main objective of this study was to evaluate the therapeutic potential of a triple combination treatment consisting of chemotherapy, anti-CD20 mAbs and DC vaccination, when compared to chemotherapy plus anti-CD20 mAbs or chemotherapy plus DC vaccination in a lymphoma model. We demonstrated enhanced therapeutic effects of B cell-depleting anti-CD20 mAbs upon combination with DC vaccination in advanced lymphoma.
Unlike the majority of preclinical and clinical studies, we did not use the Id protein as antigen for DC loading. We have demonstrated previously that DCs loaded with tumour cells ex vivo or in situ were more potent against pre-existing tumours than Id-loaded DCs [13]. The greater efficacy of DCs loaded with tumour cells compared to DCs loaded with tumour-specific Id protein may be explained by recognition of one or more non-Id antigens by the immune system. It has been reported that Id was not the major target of tumour immunity in mice vaccinated by DC–lymphoma or DC–myeloma cell vaccines [12,23,24]. In addition to greater efficacy, DC–lymphoma cell vaccines have the advantage of circumventing the expensive and time-consuming preparation of Id protein by hybridoma or recombinant DNA technologies. Furthermore, intratumoral injection of naive DCs, as applied in the present study, does not require ex-vivo antigen loading, thus offering an additional shortcut to the production of DC vaccines. Vaccination with whole tumour cells may, however, carry the risk of inducing autoimmunity. It has been reported that immunization by DCs loaded with lymphoma cell-derived peptides together with CD40 ligand and interleukin (IL)-2-expressing fibroblasts resulted in a systemic autoimmune disorder that resembled graft-versus-host disease [25]. In contrast, we and others found no evidence for autoimmunity when using whole lymphoma cell vaccines in previous studies [13,23]. Similarly, in the present study we did not observe any signs of illness (behavioural changes, weight loss, hair loss, wasting). All survivor animals appeared healthy up to 8 months after treatment. With regard to the feasibility of intratumoral injection in patients, clinical trials demonstrated the feasibility of in-situ vaccination in lymphoma [26]. Even deep lymphoma sites can be injected by techniques of interventional radiology. Disseminated disease does not preclude in-situ vaccination, because injection of DCs into a single tumour site induces systemic immune responses resulting in systemic tumour regression (Fig. 2 and references [12,13]).
Cytoreduction by chemotherapy was a prerequisite in any combination treatment that we tested. In addition to cytoreduction, chemotherapy may enhance immune reactivity. Several anti-neoplastic drugs, including cyclophosphamide, can exert both immunosuppressive and immunomodulating effects depending on the dosage and the temporal relationship between drug administration and antigen challenge [27,28]. It has been shown that a single administration of cyclophosphamide depleted CD4+CD25+ regulatory T cells and delayed tumour growth [29]. Moreover, cyclophosphamide has been shown to augment the anti-tumour effect of DC vaccines by reducing the level of regulatory T cells [30]. The latter not only inhibit effector T cell activity, but also down-regulate antigen uptake and presentation by DCs, thus decreasing the efficacy of DC vaccination [31]. Low-dose cyclophosphamide may also enhance immunity by modulating the balance of DC subsets in lymphoid organs, as it has a selective cytotoxic effect on CD8+ lymphoid tissue-resident DCs that have tolerogenic properties, thus negatively regulating immune responses [32].
The mechanisms underlying synergism between anti-CD20 mAbs and DC vaccines are not yet clear. We suggest that cross-presentation of tumour antigens may be enhanced by the antibodies due to targeting of IgG-complexed antigens into DCs via Fc receptors. It has been reported that targeting of antibody-coated apoptotic tumour cells to DCs via Fc receptors promotes uptake and cross-presentation of tumour antigens to T cells [23,33]. In-vitro studies indicated that rituximab promotes uptake of apoptotic lymphoma cells by DCs, thus generating tumour-specific cytotoxic T lymphocytes [34]. In vivo, it has been demonstrated in a mouse model that anti-CD20 mAbs exerted therapeutic effects through the induction of an adaptive cellular immune response [35], and clinical data also suggested a ‘vaccinal’ effect of anti-CD20 antibodies [36]. Hence, promotion of antigen cross-presentation and induction of a protective T cell response may account, at least in part, for the superior anti-tumour outcome of anti-CD20 antibody treatment combined with in-situ DC injection.
Murine B cell lymphomas, including A20, as well as various human B cell lymphomas, produce IL-10, which has been implicated in growth and immune evasion of these tumours [37–40]. Among its broad anti-inflammatory properties, IL-10 inhibits the differentiation, maturation, migration and antigen-presenting function of DCs [41–44]. Induction of anti-tumour immune responses by DC vaccines may therefore be impaired by tumour-derived IL-10. It has been demonstrated that rituximab inhibits secretion of IL-10 by lymphoma cells [40]. Hence, synergism between anti-CD20 mAbs and DC vaccines may also result from reduced IL-10-mediated suppression of the injected DCs.
Previous studies have demonstrated the enhanced ability of T cells to reject tumours in the absence of B cells, suggesting that B cell depletion may enhance vaccine-induced anti-tumour immune responses [19–22]. It was therefore possible that the augmented therapeutic outcome demonstrated in the present study resulted in part from CD20 mAb-mediated depletion of host B cells. This was tested by using the CD20-negative 38C-13 B cell lymphoma. The 38C-13 is more aggressive and harder to cure than the A20 lymphoma, which is reflected by the inefficacy of anti-CD20 treatment or DC vaccination alone. In a previous study using anti-human CD20 mAbs and 38C-13 tumour cells expressing human CD20, a model in which tumour cells but not host B cells are affected by the mAbs, we demonstrated that under conditions of large primary tumours that had already spread to lymph nodes, when anti-CD20 antibody treatment and DC vaccination alone had no effect, synergism between these two modalities upon their combination resulted in significant long-term survival [13]. Conversely, when CD20-negative wild-type 38C-13 tumour cells and anti-mouse CD20 mAbs were used in the present study, no enhanced therapeutic effect could be demonstrated. Thus, enhancement of DC vaccines by anti-CD20 mAbs requires tumour perturbation by the antibodies, while depletion of host B cells per se is insufficient to evoke tumour rejection.
In contrast to previous studies in non-haematopoietic murine tumour models that demonstrated augmented ability of T cells to reject tumours in the absence of B cells [19–22], a recent study demonstrated that B cell depletion by CD20 mAbs prior to melanoma inoculation caused an increase in tumour volume and lung metastasis associated with impaired anti-tumour T cell-mediated immunity [45]. We observed neither a negative nor a positive effect of B cell depletion on 38C-13 tumour growth, which may be due to the different type of tumour (lymphoma versus non-haematopoietic tumours) and therapeutic protocol (injection of CD20 mAbs subsequent to chemotherapy in advanced disease). While the ability of B cells to regulate anti-tumour immune responses negatively has been attributed to provision of IL-10 by regulatory B10 cells [46], other B cell subpopulations can, conversely, contribute positively to tumour immunity [45]. The balance between the negative and the positive regulatory activity of B cells most probably affects the development of tumour immunity. This balance may vary at different stages of tumour progression or in response to different tumours.
In conclusion, in B cell lymphoma, anti-CD20 antibody treatment and DC vaccination synergize to improve the therapeutic outcome compared to the treatment with each one of them alone. Combination treatments including tumour cell-loaded DC vaccines may therefore provide a strategy for enhancing therapy in rituximab-treated patients.
Disclosure
None of the authors has conflicts of interest to declare.
References
- 1.Czuczman MS, Weaver R, Alkuzweny B, Berlfein J, Grillo-Lopez AJ. Prolonged clinical and molecular remission in patients with low-grade or follicular non-Hodgkin's lymphoma treated with rituximab plus CHOP chemotherapy: 9-year follow-up. J Clin Oncol. 2004;22:4711–4716. doi: 10.1200/JCO.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 2.Fisher RI, LeBlanc M, Press OW, Maloney DG, Unger JM, Miller TP. New treatment options have changed the survival of patients with follicular lymphoma. J Clin Oncol. 2005;23:8447–8452. doi: 10.1200/JCO.2005.03.1674. [DOI] [PubMed] [Google Scholar]
- 3.Hiddemann W, Kneba M, Dreyling M, et al. Frontline therapy with rituximab added to the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) significantly improves the outcome for patients with advanced follicular lymphoma compared with therapy with CHOP alone: results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood. 2005;106:3725–3732. doi: 10.1182/blood-2005-01-0016. [DOI] [PubMed] [Google Scholar]
- 4.Hollander N. Current vaccination strategies for the treatment of B-cell lymphoma and multiple myeloma. Crit Rev Immunol. 2009;29:399–418. doi: 10.1615/critrevimmunol.v29.i5.30. [DOI] [PubMed] [Google Scholar]
- 5.Levy R, Robertson MJ, Ganjoo K, Leonard J, Vose J, Denney D. Results of a phase 3 trial evaluating safety and efficacy of specific immunotherapy, recombinant idiotype (Id) conjugated to KLH (Id-KLH) with GM-CSF, compared to non-specific immunotherapy, KLH with GM-CSF, in patients with follicular non-Hodgkin's lymphoma. 2008. The 99th Annual Meeting of the American Association for Cancer Research, San Diego, CA. Abstract LB-204.
- 6.Freedman A, Neelapu SS, Nichols C, et al. Placebo-controlled phase III trial of patient-specific immunotherapy with mitumprotimut-T and granulocyte–macrophage colony-stimulating factor after rituximab in patients with follicular lymphoma. J Clin Oncol. 2009;27:3036–3043. doi: 10.1200/JCO.2008.19.8903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schuster SJ, Neelapu SS, Gause BL, et al. Vaccination with patient-specific tumor-derived antigen in first remission improves disease-free survival in follicular lymphoma. J Clin Oncol. 2011;29:2787–2794. doi: 10.1200/JCO.2010.33.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neelapu SS, Kwak LW, Kobrin CB, et al. Vaccine induced tumor-specific immunity despite severe B-cell depletion in mantle cell lymphoma. Nat Med. 2005;11:986–991. doi: 10.1038/nm1290. [DOI] [PubMed] [Google Scholar]
- 9.Hsu FJ, Benike C, Fagnoni F, et al. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat Med. 1996;2:52–58. doi: 10.1038/nm0196-52. [DOI] [PubMed] [Google Scholar]
- 10.Timmerman JM, Czerwinski DK, Davis TA, et al. Idiotype-pulsed dendritic cell vaccination for B-cell lymphoma: clinical and immune responses in 35 patients. Blood. 2002;99:1517–1526. doi: 10.1182/blood.v99.5.1517. [DOI] [PubMed] [Google Scholar]
- 11.Di Nicola M, Zappasodi R, Carlo-Stella C, et al. Vaccination with autologous tumor-loaded dendritic cells induces clinical and immunologic responses in indolent B-cell lymphoma patients with relapsed and measurable disease: a pilot study. Blood. 2009;113:18–27. doi: 10.1182/blood-2008-06-165654. [DOI] [PubMed] [Google Scholar]
- 12.Song W, Levy R. Therapeutic vaccination against murine lymphoma by intratumoral injection of naïve dendritic cells. Cancer Res. 2005;65:5958–5964. doi: 10.1158/0008-5472.CAN-05-0406. [DOI] [PubMed] [Google Scholar]
- 13.Gadri Z, Kukulansky T, Bar-Or E, Haimovich J, Hollander N. Synergistic effect of dendritic cell vaccination and anti-CD20 antibody treatment in the therapy of murine lymphoma. J Immunother. 2009;32:333–340. doi: 10.1097/CJI.0b013e31819b7c17. [DOI] [PubMed] [Google Scholar]
- 14.Kim KJ, Kanellopoulos-Langevin C, Merwin RM, Sachs DH, Asofsky R. Establishment and characterization of BALB/c lymphoma lines with B cell properties. J Immunol. 1979;122:549–554. [PubMed] [Google Scholar]
- 15.Bergman Y, Haimovich J. Characterization of a carcinogen-induced murine B cell line of C3H/eB origin. Eur J Immunol. 1977;7:413–417. doi: 10.1002/eji.1830070702. [DOI] [PubMed] [Google Scholar]
- 16.Lutz MB, Kukutsch N, Ogilvie ALJ, et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 17.Heyfets A, Haimovich J, Hollander N. Determination of idiotype-specific T cells in idiotype-vaccinated mice. Immunol Lett. 2002;80:207–213. doi: 10.1016/s0165-2478(01)00321-2. [DOI] [PubMed] [Google Scholar]
- 18.Uchida J, Lee Y, Hasegawa M, et al. Mouse CD20 expression and function. Int Immunol. 2004;16:119–129. doi: 10.1093/intimm/dxh009. [DOI] [PubMed] [Google Scholar]
- 19.Qin Z, Richter G, Schüler T, Ibe S, Cao X, Blankenstein T. B cells inhibit induction of T-cell dependent tumor immunity. Nat Med. 1998;4:627–630. doi: 10.1038/nm0598-627. [DOI] [PubMed] [Google Scholar]
- 20.Shah S, Divekar AA, Hilchey SP, et al. Increased rejection of primary tumors in mice lacking B cells: inhibition of anti-tumor CTL and TH1 cytokine responses by B cells. Int J Cancer. 2005;117:574–586. doi: 10.1002/ijc.21177. [DOI] [PubMed] [Google Scholar]
- 21.Perricone MA, Smith KA, Claussen KA, et al. Enhanced efficacy of melanoma vaccines in the absence of B lymphocytes. J Immunother. 2004;27:273–281. doi: 10.1097/00002371-200407000-00003. [DOI] [PubMed] [Google Scholar]
- 22.Kim S, Fridlender ZG, Dunn R, et al. B-cell depletion using anti-CD20 antibody augments antitumor immune responses and immunotherapy in nonhematopoetic murine tumor models. J Immunother. 2008;31:446–457. doi: 10.1097/CJI.0b013e31816d1d6a. [DOI] [PubMed] [Google Scholar]
- 23.Franki SN, Steward KK, Betting DJ, Kafi K, Yamada RE, Timmerman JM. Dendritic cells loaded with apoptotic antibody-coated tumor cells provide protective immunity against B-cell lymphoma in vivo. Blood. 2008;111:1504–1511. doi: 10.1182/blood-2007-03-080507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cohen S, Haimovich J, Hollander N. Dendritic cell-based therapeutic vaccination against myeloma: vaccine formulation determines efficacy against light-chain myeloma. J Immunol. 2009;182:1667–1673. doi: 10.4049/jimmunol.182.3.1667. [DOI] [PubMed] [Google Scholar]
- 25.Roskrow MA, Dilloo D, Suzuki N, Zhong W, Rooney CM, Brenner MK. Autoimmune disease induced by dendritic cell immunization against leukemia. Leuk Res. 1999;23:549–557. doi: 10.1016/s0145-2126(99)00045-4. [DOI] [PubMed] [Google Scholar]
- 26.Brody JD, Ai WZ, Czerwinski DK, et al. In situ vaccination with TLR9 agonist induces systemic lymphoma regression: a phase I/II study. J Clin Oncol. 2010;28:4324–4332. doi: 10.1200/JCO.2010.28.9793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Terando A, Mule JJ. On combining antineoplastic drugs with tumor vaccines. Cancer Immunol Immunother. 2003;52:680–685. doi: 10.1007/s00262-003-0426-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8:59–73. doi: 10.1038/nri2216. [DOI] [PubMed] [Google Scholar]
- 29.Ghiringhelli F, Larmonier N, Schmitt E, et al. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol. 2004;34:336–344. doi: 10.1002/eji.200324181. [DOI] [PubMed] [Google Scholar]
- 30.Liu JY, Wu Y, Zhang XS, et al. Single administration of low dose cyclophosphamide augments the antitumor effect of dendritic cell vaccine. Cancer Immunol Immunother. 2007;56:1597–1604. doi: 10.1007/s00262-007-0305-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Navarrete AM, Delignat S, Teillaud JL, Kaveri SV, Lacroix-Desmazes S, Bayry J. CD4+CD25+ regulatory T cell-mediated changes in the expression of endocytic receptors and endocytosis process of human dendritic cells. Vaccine. 2011;9:2649–2652. doi: 10.1016/j.vaccine.2011.01.095. [DOI] [PubMed] [Google Scholar]
- 32.Nakahara T, Uchi H, Lesokhin AM, et al. Cyclophosphamide enhances immunity by modulating the balance of dendritic cell subsets in lymphoid organs. Blood. 2010;115:4384–4392. doi: 10.1182/blood-2009-11-251231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dhodapkar KM, Krasovsky J, Williamson B, Dhodapkar MV. Antitumor monoclonal antibodies enhance cross-presentation of cellular antigens and the generation of myeloma-specific killer T cells by dendritic cells. J Exp Med. 2002;195:125–133. doi: 10.1084/jem.20011097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Selenco N, Majdic O, Draxler S, et al. CD20 antibody (C2B8)-induced apoptosis of lymphoma cells promotes phagocytosis by dendritic cells and cross-priming of CD8+ cytotoxic T cells. Leukemia. 2001;15:1619–1626. doi: 10.1038/sj.leu.2402226. [DOI] [PubMed] [Google Scholar]
- 35.Abes R, Gelize E, Fridman WH, Teillaud JL. Long-lasting anti-tumor protection by anti-CD20 antibody through cellular immune response. Blood. 2010;116:926–934. doi: 10.1182/blood-2009-10-248609. [DOI] [PubMed] [Google Scholar]
- 36.Cartron G, Watier H, Golay J, Solal-Celigny P. From the bench to the bedside: ways to improve rituximab efficacy. Blood. 2004;104:2635–2642. doi: 10.1182/blood-2004-03-1110. [DOI] [PubMed] [Google Scholar]
- 37.O'Garra A, Stapleton G, Dhar V, et al. Production of cytokines by mouse B cells: B lymphomas and normal B cells produce interleukin 10. Int Immunol. 1990;2:821–832. doi: 10.1093/intimm/2.9.821. [DOI] [PubMed] [Google Scholar]
- 38.Bost KL, Bieligk SC, Jaffe BM. Lymphokine mRNA expression by transplantable murine B lymphocytic malignancies. Tumor-derived IL-10 as a possible mechanism for modulating the anti-tumor response. J Immunol. 1995;154:718–729. [PubMed] [Google Scholar]
- 39.Elpek KG, Lacelle C, Singh NP, Yolcu ES, Shirwan H. CD4+CD25+ T regulatory cells dominate multiple immune evasion mechanisms in early but not late phases of tumor development in a B cell lymphoma model. J Immunol. 2007;178:6840–6848. doi: 10.4049/jimmunol.178.11.6840. [DOI] [PubMed] [Google Scholar]
- 40.Alas S, Emmanouilides C, Bonavida B. Inhibition of interleukin 10 by rituximab results in down-regulation of bcl-2 and sensitization of B-cell non-Hodgkin's lymphoma to apoptosis. Clin Cancer Res. 2001;7:709–723. [PubMed] [Google Scholar]
- 41.Enk AH, Angeloni VL, Udey MC, Katz SI. Inhibition of Langerhans cell antigen-presenting function by IL-10. A role for IL-10 in induction of tolerance. J Immunol. 1993;151:2390–2398. [PubMed] [Google Scholar]
- 42.De Smedt T, Van Mechelen M, De Becker G, Urbain J, Leo O, Moser M. Effect of interleukin-10 on dendritic cell maturation and function. Eur J Immunol. 1997;27:1229–1235. doi: 10.1002/eji.1830270526. [DOI] [PubMed] [Google Scholar]
- 43.Demangel C, Bertolino P, Britton W. Autocrine IL-10 impairs dendritic cell (DC)-derived immune responses to mycobacterial infection by suppressing DC trafficking to draining lymph nodes and local IL-12 production. Eur J Immunol. 2002;32:994–1002. doi: 10.1002/1521-4141(200204)32:4<994::AID-IMMU994>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 44.Steinbrink K, Graulich E, Kubsch S, Knop J, Enk AH. CD4+ and CD8+ anergic T cells induced by interleukin-10-treated human dendritic cells display antigen-specific suppressor activity. Blood. 2002;99:2468–2476. doi: 10.1182/blood.v99.7.2468. [DOI] [PubMed] [Google Scholar]
- 45.DiLillo DJ, Yanaba K, Tedder TF. B cells are required for optimal CD4+ and CD8+ T cell tumor immunity: therapeutic B cell depletion enhances B16 melanoma growth in mice. J Immunol. 2010;184:4006–4016. doi: 10.4049/jimmunol.0903009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DiLillo DJ, Matsushita T, Tedder TF. B10 cells and regulatory B cells balance immune responses during inflammation, autoimmunity, and cancer. Ann NY Acad Sci. 2010;1183:38–57. doi: 10.1111/j.1749-6632.2009.05137.x. [DOI] [PubMed] [Google Scholar]