

Abstract
Helicobacter pylori is one of the most common infections in the world. Despite inciting inflammation, immunological clearance of the pathogen is often incomplete. CD4+CD25hiforkhead box protein 3 (FoxP3+) regulatory T cells (Tregs) are potent suppressors of different types of immune responses and have been implicated in limiting inflammatory responses to H. pylori. Investigating the influence of H. pylori on Treg function and proliferation, we found that H. pylori-stimulated dendritic cells (DCs) induced proliferation in Tregs and impaired their suppressive capability. This effect was mediated by interleukin (IL)-1β produced by H. pylori-stimulated DCs. These data correlated with in-vivo observations in which H. pylori+ gastric mucosa contained more Tregs in active cell division than uninfected stomachs. Inciting local proliferation of Tregs and inhibiting their suppressive function may represent a mechanism for the chronic gastritis and carcinogenesis attributable to H. pylori.
Keywords: Helicobacter pylori, human, interleukin-1, regulatory T cells, suppression
Introduction
Helicobacter pylori, a prevalent Gram-negative bacterium, is considered to be one of the most common infective organisms in the world. H. pylori predominantly colonizes the gastric antrum and establishes life-long chronic infection. While the majority of infections are asymptomatic, H. pylori infection has significant public health and economic implications as it is an important risk factor for gastritis, peptic ulcer disease, malignant transformation in the upper gastrointestinal (GI) tract and elevated cardiovascular risk [1–3]. As a result, antibiotic therapy to eradicate this bacterium is a key treatment of chronic gastritis and peptic ulceration occurring in the context of H. pylori [4].
H. pylori elicits an inflammatory response recruiting neutrophils, lymphocytes and dendritic cells (DCs) to the gastric mucosa [5]. The initial interaction between H. pylori and the innate host immune response is mediated through pattern recognition receptors, such as Toll-like receptors (TLR), expressed on gastric epithelial cells and through the H. pylori virulence factor cag pathogenicity island (cagPAI) [6,7]. The recruitment of DCs to the gastric lamina propria allows for antigen sampling by the extension of their dendrites through the epithelial cell layer [8,9]. We have shown previously that DC activation by H. pylori leads to the production of interleukin (IL)-10, IL-23 and limited amounts of IL-12 [10], and these H. pylori-treated DCs stimulate interferon (IFN)-γ production in naive T cells in vitro [10]. Biopsy material from H. pylori-infected individuals confirms both local infiltration of T helper type 1 (Th1) [11,12] as well as Th17 cells [13,14], suggesting that H. pylori has more than one effect on immunological cells.
CD4+CD25hiforkhead box protein 3 (FoxP3+) regulatory T cells (Treg) are naturally occurring T cells capable of suppressing CD4+CD25− effector T cell (Teff) proliferation and cytokine production [15]. These cells play a critical role in maintaining peripheral tolerance, with their absence resulting in severe multi-organ autoimmune diseases [16]. Tregs also moderate the immune response to pathogens by regulating the balance between immunity and inflammation – while Treg suppression needs to be overcome for effective anti-pathogen responses, excessive inflammation could result in disproportionate injury to healthy tissues [17]. Evidence has emerged to show a key role for Tregs in maintaining this balance, in some circumstances resulting in pathogen persistence in order to limit tissue injury [18,19]. For example, lesional sites in Leishmania major infection are characterized by the presence of both L. major and large numbers of Tregs that prevent the clearance of infection [18]. Similarly, Tregs limit the inflammatory response to H. hepaticus, thus limiting subsequent tissue damage [19]. In the case of H. pylori, infected individuals have H. pylori-specific circulating Tregs, impairing the memory response to H. pylori [20], and an elevated number of FoxP3+ cells in gastric biopsies [21]. This evidence suggests that H. pylori infection results in expansion of the Treg population and their recruitment to the site of infection in order to limit the inflammatory response.
Pathogen-stimulated DCs have been implicated in the expansion of Tregs. Yamazaki et al. demonstrated that while splenic APCs are poor promoters of Treg proliferation, bone marrow-derived DCs are capable of inducing Tregs to proliferate to a degree comparable with Teff during the first 3 days of culture [22]. The underlying mechanisms are thought to be through both contact-dependent (e.g. CD86/80 co-stimulation [23]) and non-contact-dependent [cytokine production, in particular the inflammatory cytokines IL-1, IL-6 and tumour necrosis factor (TNF)-α] processes [24–28].
Based on reports of elevated Treg numbers in H. pylori-infected sites, we hypothesized that H. pylori instructs DCs to stimulate proliferation of Tregs locally. Furthermore, the presence of chronic inflammation despite the existence of elevated numbers of Tregs suggests that these Tregs have impaired ability to suppress local inflammation. We have investigated the direct and indirect effect of H. pylori on Treg proliferation and function in vitro as well as investigating Tregs in gastric tissue biopsies infected with H. pylori. We have found that H. pylori-stimulated DCs drive Treg proliferation, and impair their suppressive function through the production of IL-1β. This is corroborated by in-vivo data showing active division of Tregs in biopsy samples from infected individuals. Dissection of the long-term impact of Treg modulation and dysregulated immunpathology in the context of H. pylori may provide new insights into the mechanisms underlying the development of H. pylori-associated complications and/or potential targets for the local treatment of inflammation associated with H. pylori in the 15–20% of individuals unresponsive to eradication therapy.
Material and methods
Cells
Peripheral blood mononuclear cells (PBMCs) were separated from buffy coats provided by the National Blood Transfusion Centre (South Thames, London, UK). CD14+ and CD14− cells were then separated using CD14-Beads (Miltenyi Biotec, Woking, UK), according to the manufacturer's instructions. The CD14+ cells were then cultured in RPMI-1640 (Invitrogen, Paisley, UK) with 10% fetal calf serum (FCS; SeraQ, East Grinstead, UK), 50 IU/ml penicillin, 50 μg/ml streptomycin and 2 mM L-glutamine (PSG) (PAA Laboratories GmbH, Pasching, Austria). To develop DCs, IL-4 (10 ng/ml) (First Link, Birmingham, UK) and granulocyte–macrophage colony-stimulating factor (GM-CSF) (20 ng/ml) (kindly donated by Dr S. Brett, GlaxoSmithKline, Stevenage, UK) were added every 2 days before the cells were harvested at day 5. T cells were enriched from PBMCs derived from buffy coats by negative selection. CD4+ T cells were purified using a cocktail of antibodies against CD8, CD33, CD14, CD16, CD19, CD56 and γδ-T cell receptor (TCR). The CD4+ T cells were then divided into CD25+ and CD25− cells using anti-CD25 beads (Dynal Biotech, Oslo, Norway). For the CD25hi separation, CD4+ T cells were stained for CD4 and CD25 using anti-CD4-allophycocyanin (APC) (S3·5; Caltag, Buckingham, UK) and anti-CD25-phycoerythrin (PE) (3G10; Caltag). The CD4+CD25hi (top 2% for expression of CD25) were then separated from the CD4+CD25− T cell population by fluorescence-activated cell sorting (FACS) using a MoFlo high speed multi-laser cell sorter (Cytomation, Fort Collins, CO, USA) running Summit version 3·1 software (Cytomation).
T cell assay
Suppression assays were all carried out in complete medium (RPMI with PSG) containing 10% human serum (Biosera, Ringmer, UK) using 2 × 104 T cells with the following conditions: CD25− alone, CD25− : CD25+ at a 1:1 ratio and CD25+ alone. These cells were stimulated with CD3/CD28 expander beads (Dynal Biotech) in the presence of H. pylori. Alternatively, the T cells were stimulated with 2 × 103 allogeneic DCs treated previously with H. pylori, or medium alone for 8 h. Media were supplemented, or not, as described by IL-1 receptor antagonist (IL-1RA) (10 μg/ml, kindly donated by Dr Keith P. Ray, GlaxoSmithKline, Stevenage, UK), anti-IL-6 (10 μg/ml; R&D Systems, Abingdon, UK) or anti-TNFRII (0·2 μg per well; R&D Systems). All conditions were set up in triplicate with appropriate controls for cell density and incubated for 5 days before removal of supernatant for cytokine analysis and addition of 1 μCi/well [3H]-thymidine (GE Healthcare, Little Chalfont, UK). Twenty hours later the cells were harvested and the amount of incorporated thymidine was measured using a 1205 Betaplate liquid scintillation counter (LKB Wallac, Turku, Finland).
Cytokine production
CD40L-transfected L cells (CD40Ltx) [29] were grown in RPMI with 10% FCS and PSG. Once growth was confluent, cells were harvested by incubating them with Versene [ethylenediamine tetraacetic acid (EDTA)] (Cambrex, Verviers, Belgium) for 15 min. They were then removed from the flask, washed in phosphate-buffered saline (PBS) and resuspended in RPMI with 10% FCS and PSG. DCs (6 × 104/well) were incubated in 500 μl RPMI-1640 with 10% FCS and PSG, alone or in the presence of CD40L transfectants (1·5 × 105/cells). These DCs were then incubated with H. pylori [106 colony-forming units (cfu)/ml] or medium alone and supernatants collected 24 h later. Supernatants were analysed using a proinflammatory I 4-plex for IFN-γ, IL-1β, TNF-α and IL-6 (Meso Scale Discovery, Gaithersburg, MD, USA) in accordance with the manufacturer's protocol using a SECTOR™ Imager 2400 (Meso Scale Discovery).
Biopsy material and sectioning
Human gastric biopsy specimens were obtained from the gastric antra of subjects referred to the gastroenterology clinic for endoscopy. CLOtest® (Kimberly-Clark, West Malling, UK) was used to determine H. pylori status. Biopsies were snap-frozen in octreotide (OCT) (Lab-Tek Products, Miles Laboratories, Naperville, IL, USA), sectioned at 6–8 μm on a cryostat and fixed in 4% paraformaldehyde solution. Immunohistochemical analysis was performed in these sections double-stained with the primary antibodies mouse anti-human FoxP3 (259D/C7; BD Pharmingen, Oxford, UK) and rabbit polyclonal against the marker Ki-67 (MM1; Leica Microsystems, Germany). Secondary antibodies were goat anti-mouse IgG antibody conjugated with AlexaFluor 555 and goat anti-rabbit IgG conjugated with AlexaFluor 488 (both from Invitrogen, Paisley, UK). Prolong Gold AntiFade Reagent with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen) staining was used to counterstain nuclei. Serial images were obtained with a fluorescence microscope.
Statistical analysis
Statistical analyses were carried out on Microsoft Excel for Windows 2003 (Microsoft Corporation, Redmond, WA, USA). Percentage suppression was calculated as the reduction in proliferation in the presence of Tregs expressed as a percentage of Teff proliferation in the absence of Tregs. Parametric and non-parametric data were calculated as the mean ± standard deviation (s.d.) and median with associated interquartile range, respectively. For comparison of parametric data, paired and unpaired t-tests were used (for paired and unpaired data sets). Comparisons of central tendency for non-parametric data sets were made by using the Wilcoxon's signed-rank test and Mann–Whitney U-tests for paired and unpaired data, respectively. One-sided tests were used for comparison of small sample sizes (n < 5). A P-value of < 0·05 was considered significant in call cases.
Results
H. pylori expands Tregs in the gastric mucosa
Elevated Treg numbers have been observed in response to H. pylori infection, both at the site of infection and circulating in the periphery [20,21]. To determine whether the elevated number of Tregs was due to active proliferation at the site of infection, we stained gastric biopsy specimens from patients with and without confirmed H. pylori infection for FoxP3 and the proliferation marker Ki67 (four sections from each patient and four patients). As expected from previous publications, H. pylori-positive biopsy specimens had greater numbers of FoxP3+ cells than H. pylori-negative specimens (Fig. 1a). In the presence of H. pylori, a greater percentage of Tregs stained positively for Ki67 (10·2 ± 1·5% versus 2·4 ± 2·0% of FoxP3+ cells, P < 0·05; Fig. 1a,b), suggesting that Tregs proliferate in vivo in the presence of H. pylori.
Fig. 1.
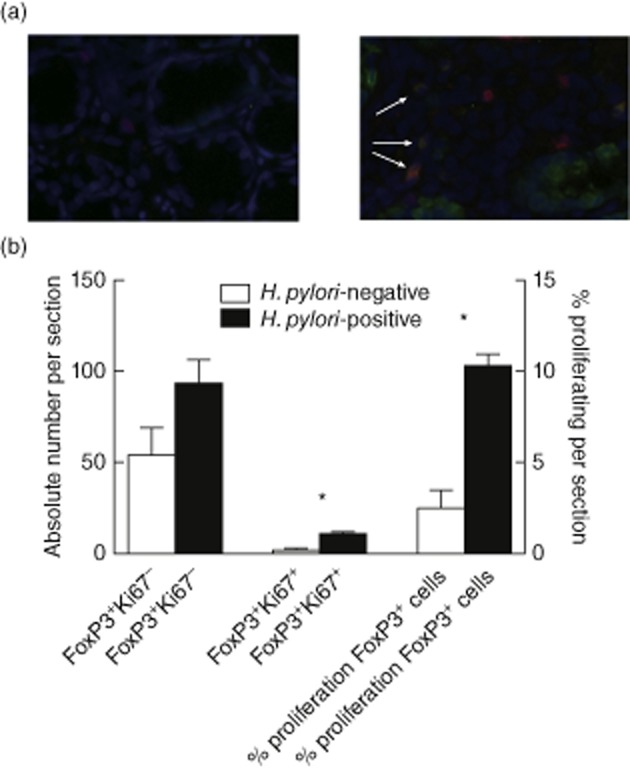
Regulatory T cells (Tregs) in Helicobacter pylori-infected gastric mucosa proliferate in vivo. Gastric antrum biopsy samples from patients with (a, right panel) and without (a, left panel) H. pylori infection were stained for forkhead box protein 3 (FoxP3) (green), Ki67 (red) and 4′,6-diamidino-2-phenylindole (DAPI) (blue). FoxP3+Ki67− (non-proliferating Tregs, green cells) and FoxP3+Ki67+ (proliferating Tregs, brown cells, arrowed) were counted and the percentage of proliferating Tregs per section calculated (b). (a) Representative examples; (b) pooled data from four sections. *P < 0·05.
H. pylori-stimulated DCs induce expansion of human Tregs
DCs play a critical role in presenting pathogens to the adaptive immune response. Murine models have indicated that pathogen-stimulated DCs can alter Treg function [22,26] and their presence in the gastric mucosa indicates that they have the opportunity to influence Treg function [13]. To determine whether H. pylori-stimulated DCs (HpDCs) can influence Treg proliferation and can, at least in part, explain the expansion of Tregs seen at biopsy sites of H. pylori-infected individuals [10,13], DCs were generated from peripheral blood monocytes using GM-CSF and IL-4, and incubated with H. pylori [106−4 cfu/ml corresponding to multiplicity of infection (MOI) of 0·75] for 8 h before being washed and placed in co-culture with allogeneic Tregs for 5 days (Fig. 2), as described previously by us [10]. Allogeneic Tregs were used, as published previously [10], to ensure that the frequency of responding Tregs was not dependent on previous H. pylori exposure and relied purely on the high frequency of alloreactive Tregs [30]. HpDC-induced Treg proliferation was assessed by [3H]-thymidine incorporation; an example is shown in Fig. 2a. This was confirmed through cumulative experiments with HpDCs (106 cfu/ml), in which the differences between Treg proliferation in the presence and absence of H. pylori were found to be statistically significant (Fig. 2b). Tregs were enriched using magnetic beads and, although the purity reached 90%, to ensure further that proliferation measured was not due to non-Treg (e.g. CD4+CD25int T cells) ‘contamination’ of Treg preparations, Tregs were purified to >98% purity by FACS sorting (to ensure that only the CD25hi cells were selected) and cultured with DCs as before. HpDCs expanded allogeneic CD25hi cells, confirming that the proliferation observed was not due to impurities (Fig. 2c). We also ruled out the possibility that H. pylori acts directly on Tregs by stimulating Tregs with anti-CD3/CD28 microbeads in the presence of H. pylori and observing no change in Treg proliferation under these conditions (data not shown), concluding that enhanced Treg proliferation was DC-dependent.
Fig. 2.
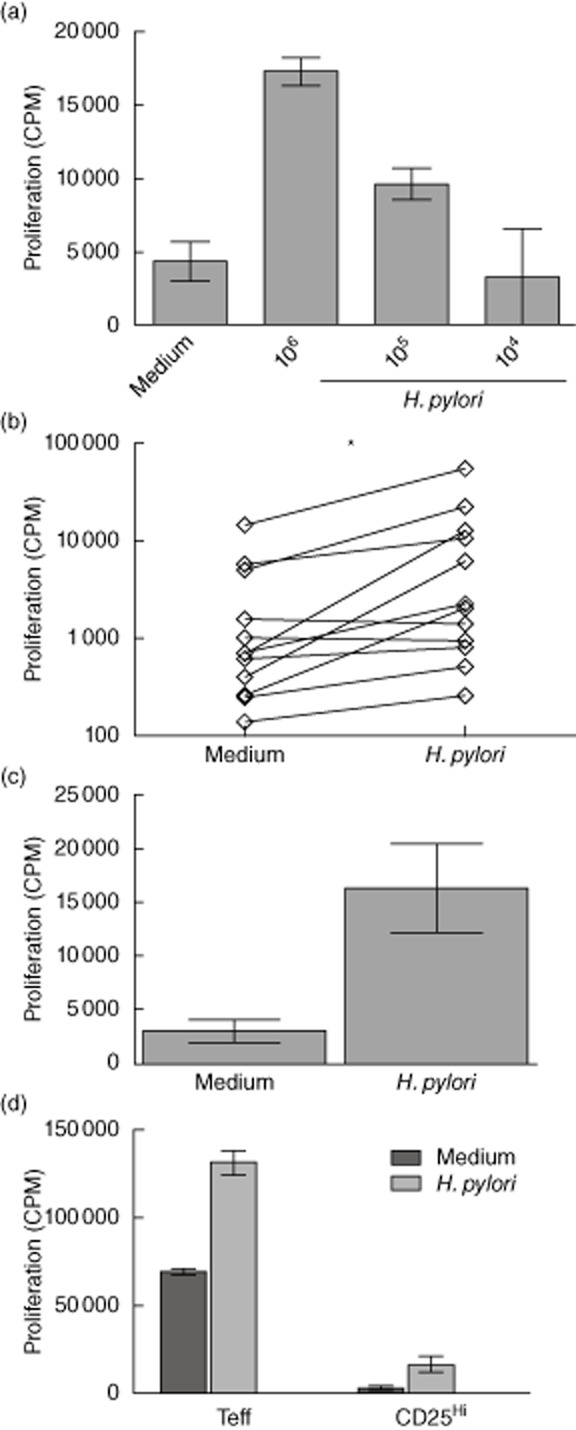
Helicobacter pylori-stimulated dendritic cells (DCs) induce regulatory T cell (Treg) proliferation: (a) H. pylori (104–6 colony-forming units/ml)-activated allogeneic DCs were co-cultured with Tregs for 5 days and their proliferation determined by [3H]-thymidine incorporation. The plot shows a representative example from 12 independent experiments. (b) Cumulative day 5 proliferation from 12 independent experiments in which immature DCs (ImmDCs) or H. pylori-stimulated DCs (HpDCs) were used to stimulate allogeneic Tregs. Note that (b) is plotted on a log10 scale. (c) Proliferation of fluorescence activated cell sorted (FACS) CD25hi cells were determined in response to HpDCs. Plot shows a representative example from two independent experiments. *P < 0·05.
Tregs stimulated with HpDC have similar rates of proliferation to Teff in the first 3 days of stimulation
The efficiency of Treg suppression of Teffs is dependent on their relative ratio within the same environment. Thus, proliferation of Tregs induced by HpDCs has the potential to favour Treg suppression by altering this ratio. To gauge the relative ratio of Tregs to Teffs, we therefore compared the kinetics of Treg proliferation against that of Teffs, starting with the same number of cells. Tregs and Teffs were stimulated by HpDCs for 1, 2, 3, 4, 5 and 8 days and their proliferation determined by [3H]-thymidine incorporation. We found that Treg proliferation was enhanced by HpDCs as early as day 2, and was comparable to Teff proliferation. However, after day 4, Teff proliferation continued to increase whereas the proliferation of Tregs plateaued and then declined (Fig. 3). This suggests that while Teff have a greater capacity for expansion, Treg expansion in response to HpDCs is short-lived, this follows similar observations in mouse models [22] that showed a short-lived burst of expansion in Tregs in response to activated DCs, and that the efficiency of Treg-mediated suppression might be expected to decline after day 3 due to significant changes in relative numbers altering the Treg : Teff ratio.
Fig. 3.
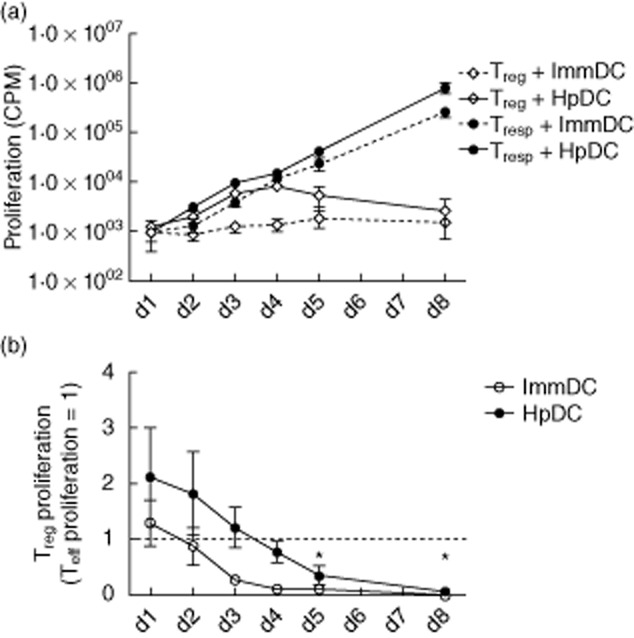
Regulatory T cell (Treg) proliferation induced by Helicobacter pylori-stimulated DCs (HpDCs) is similar to the effector T cell (Teff) population during the first 3 days: (a) Teffs and Tregs were stimulated with HpDCs and immature DCs (ImmDCs) and proliferation determined at days 1, 2, 3, 4, 5 and 8. The plot is represented on a log10 scale. (b) Treg proliferation was normalized at each time-point to corresponding Teff proliferation (where proliferation of the latter = 1). The dotted line shows the point of equivalence, where proliferation of Teffs and Tregs is identical. (a,b) Cumulative mean data from three experiments. Error bars show standard deviation.
H. pylori-treated DCs produce IL-1β, IL-6 and TNF-α and enhance Treg proliferation via IL-1β
We have demonstrated previously that H. pylori induces DCs to produce IL-23 but only small amounts of IL-12 [10,13]. Because inflammatory cytokines, in particular IL-1, IL-6 and TNF-α, have been implicated in the modulation of Treg function [24–28], we sought to determine whether Treg proliferation induced by DCs treated with H. pylori could be caused by production of inflammatory cytokines. To investigate the cytokines produced by DCs in response to H. pylori, DCs were treated for 24 h with H. pylori (106 cfu/ml) and supernatant concentrations of IL-1β, IL-6 and TNF-α determined. H. pylori stimulated IL-1β, IL-6 and TNF-α release by DCs (Fig. 4). As it has been demonstrated previously that ligation of CD40 on DCs further enhanced cytokine release mediated by TLR engagement [31], DCs were cultured with H. pylori in the presence or absence of murine L cells transfected with human CD40L (CD40Ltx cells) [29]. The cytokine production was amplified by the presence of CD40Ltx cells (Fig. 4). Altogether, IL-6 and TNF-α were produced in higher quantities than IL-1β in response to H. pylori, with an interquartile range of 14–20, 1800–8800 and 130–1400 pg/ml for IL-1β, IL-6 and TNF-α, respectively, in the absence of CD40L and 120–250, 12 000–42 000, 8900–19 000 pg/ml for IL-1β, IL-6 and TNF-α, respectively, with CD40Ltx (Fig. 4).
Fig. 4.
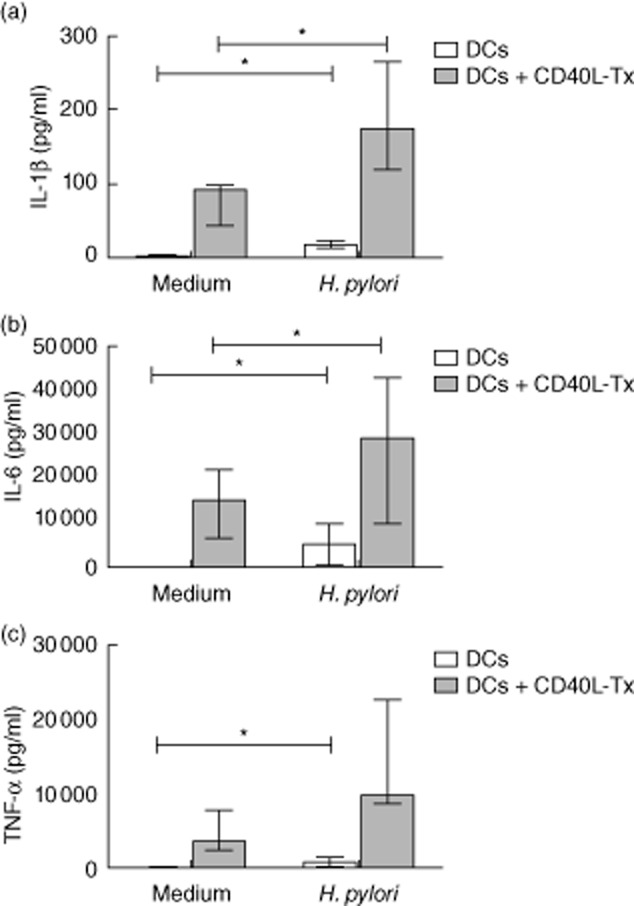
Helicobacter pylori-treated dendritic cells (DCs) produce interleukin (IL)-1β, IL-6 and tumour necrosis factor (TNF)-α. DCs were cultured alone or stimulated with H. pylori (106 colony-forming units/ml) for 24 h in the presence or absence of CD40L transfected L (CD40LTx) cells. Supernatants were taken and concentrations of IL-1β, IL-6 and TNF-α (a,b,c, respectively) determined. Graphs show median and interquartile ranges of cumulative data from seven independent experiments. *P < 0·05.
Having found that HpDCs produce IL-1β, IL-6 and TNF-α, we investigated whether these cytokines influenced Treg proliferation. Tregs were stimulated initially by allogeneic immature DCs (ImmDCs) in the presence of each of these cytokines at 1 ng/ml and 10 ng/ml. We found that only IL-1β was capable of inducing significant Treg proliferation (Fig. 5a). In addition, IL-1β was capable of mediating its affect in the absence of DCs and could amplify anti-CD3/CD28-mediated Treg proliferation at concentrations as low as 100 pg/ml, lower than the amount of IL-1β produced naturally by H. pylori-treated DCs (Fig. 5b). We confirmed the role of IL-1β in HpDC-induced Treg proliferation by stimulating Tregs with HpDCs in the presence of a neutralizing IL-1RA. The addition of IL-1RA inhibited Treg proliferation, while anti-IL-6 and anti-TNFRII antibodies had no effect (Fig. 5c). These results suggest that IL-1β is the key inflammatory cytokine produced by DCs in response to H. pylori that is responsible for Treg expansion.
Fig. 5.
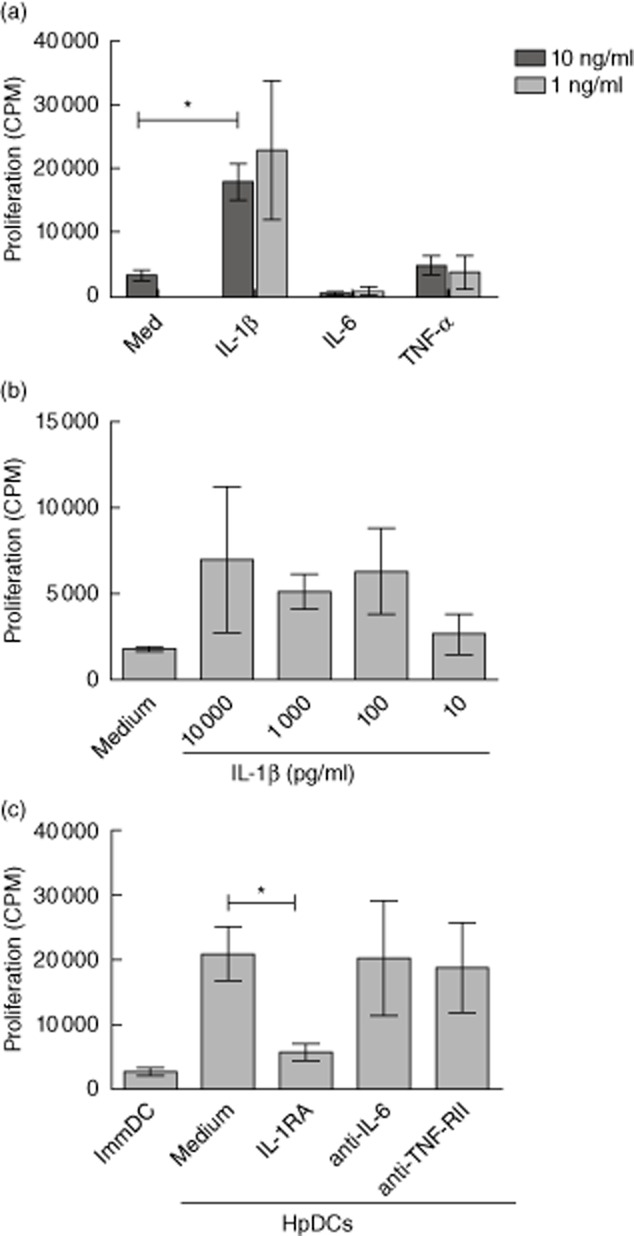
Helicobacter pylori dendritic cells (HpDCs) enhance regulatory T cell (Treg) proliferation through the production of interleukin (IL)-1β. (a) Immature DCs (ImmDCs) were co-cultured with allogeneic Tregs for 5 days in the presence of either 1 or 10 ng/ml of IL-1β, IL-6 or tumour necrosis factor (TNF)-α and their proliferation was determined by [3H]-thymidine incorporation. (a) Tregs were stimulated with CD3/CD28 beads for 5 days in the presence of 10–10 000 or pg/ml of IL-1β and their proliferation was determined by [3H]-thymidine incorporation. (c) Tregs were stimulated by HpDCs in the presence of either IL-RA, anti-IL-6 or anti-TNFRII, and their proliferation was determined after 5 days. Graphs show mean ± standard deviation of cumulative data from three experiments. *P < 0·05.
HpDCs inhibit Treg suppression of Teffs via IL-1β
Suppression of pathogen-responsive Teffs by Tregs at a site of infection is key to determining pathogen persistence/clearance and the degree of tissue injury caused by local inflammation. To determine, therefore, whether H. pylori affects the suppressive capacity of Tregs, ImmDcs and HpDCs were used to stimulate allogeneic Teff in the presence and absence of 1:1 Tregs for 5 days and suppression of proliferation calculated. HpDCs impaired suppression by Tregs when compared to co-cultures stimulated with ImmDCs (Fig. 6a). To rule out the possibility that proliferation of Teff impurities in the Treg population caused an apparent loss of suppression, we repeated the experiments with CD25hi Tregs and CD4+CD25− Teff FACS-sorted to >98% purity. As before, suppression of Teffs was still impaired significantly by HpDCs (Fig. 6b). To determine whether the loss of suppression was mediated by IL-1β, Tregs and Teffs were co-cultured at a 1:1 ratio and activated with HpDCs in the presence of IL-1RA. Antagonism of IL-1β resulted in partial restoration of suppression (Fig. 6c), suggesting that suppression of Teffs by Tregs is abrogated by IL-1β produced by HpDC. To determine the capacity of Tregs to inhibit the effector function of Teffs, we measured proinflammatory cytokine concentrations in supernatants of Teffs, Tregs and 1:1 Treg : Teff co-cultures stimulated by immDCs or HpDCs. IL-17 production was not detectable in this system, and IFN-γ production was not inhibited by Tregs in co-cultures stimulated with HpDCs, whereas ImmDC-stimulated Tregs could suppress IFN-γ production. (Fig. 6d). Taken together, these data demonstrate that the presence of H. pylori instructs DCs to inhibit Treg-mediated suppression of Teffs in an IL-1β-mediated manner.
Fig. 6.
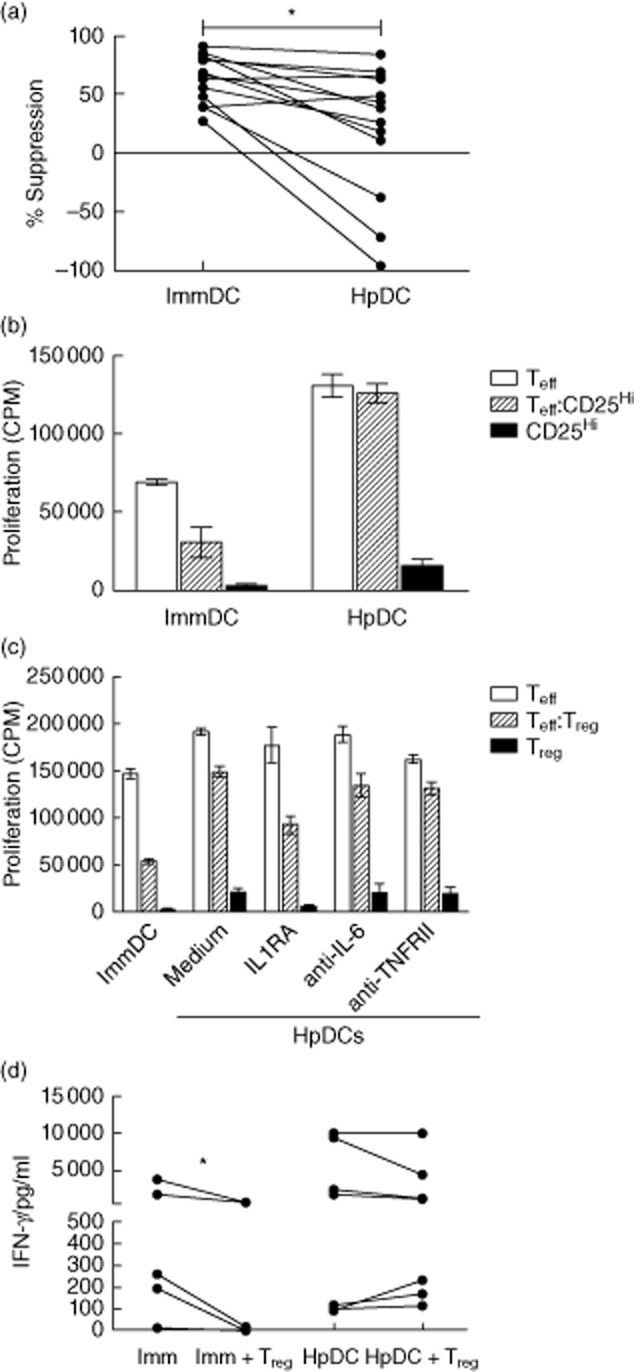
Helicobacter pylori dendritic cells (HpDCs) impair regulatory T cell (Treg) suppression through the production of interleukin (IL)-1β. (a) Effector T cells (Teff) : Treg (1:1) co-cultures were stimulated with HpDCs or immature DCs (ImmDCs) and their suppression at day 5 determined. Graph shows data from 12 independent experiments. (b) Suppression of Teffs by fluorescence activated cell sorted (FACS) CD25hi cells was determined at day 5 following stimulation by ImmDCs or HpDCs. The graph shows one experiment as a representative from two. (c) Suppression of Teff by Tregs in response to HpDCs was determined in the presence of either IL-RA, anti-IL-6 or anti-TNFRII. Graphs show data from a representative example from three independent experiments. (d) IFN-γ production was determined for day 5 Teff : Tregs (1:1) co-cultures stimulated with either HpDCs or ImmDCs. *P < 0·05.
Discussion
Persistence of H. pylori is the result of both resistance against the local gastric microenvironment and immunological evasion [32]. Despite making physical contact with immune cells in the lamina propria [33], H. pylori evades immune clearance through a variety of mechanisms including its unique site of colonization, modulation of adhesion and alteration of the host immune response [34]. H. pylori colonization results in recruitment of inflammatory immune cells and chronic increase in proinflammatory cytokines locally, including IL-1β, IL-6, IL-8, IL-17 and TNF-α [35,36]. Among the others, IL-1 has been shown to be a key cytokine in initiating and amplifying the inflammatory responses against H. pylori [37–39]. Very recently, IL-1β present in the gastric mucosa has been shown to play an important role in H. pylori-induced epigenetic changes linking inflammation to carcinogenesis [40]. Finally, H. pylori virulence and IL-1B genes contribute to peptic ulcers and intestinal metaplasia [41].
Elevation of Tregs at the site of infection and H. pylori-specific Tregs in the circulation [20,21] has been suggested as a mechanism of pathogen persistence, on the assumption that Tregs are differentiated cells with professional suppressive function. In this study we show for the first time that H. pylori interacts with human Tregs indirectly via DCs and modifies their function. Our data show that H. pylori-treated DCs stimulate Treg proliferation, diminish their suppressive function and that DC-derived IL-1β drives this process. Biopsy data from in-vivo H. pylori-infected antrum corroborated these findings, showing that a significant portion of Tregs found in infected gastric biopsies are actively undergoing mitosis. The persistence of H. pylori in the gastric mucosa may allow continual restimulation of the Treg population. This restimulation may allow for expansion of the Treg population beyond the 3-day peak observed in vitro. In this model it is not the presence of Tregs that promote the persistence of infection, but rather the persistence of infection that expands the Treg population in an attempt to limit the damage caused by a prolonged and excessive inflammatory response.
Demonstrations that suppressive function of Tregs can be undermined by pathogens have been shown previously in the context of L. major and H. hepaticus infections, limiting inflammation while hindering pathogen clearance [18,19]. Although pathogens can influence Treg function directly, such as through engagement of TLR-2, -4 and -8 [42–44], we found that H. pylori had no direct effect on Tregs and that the changes induced in Treg behaviour could be explained by cytokine production from DCs. We have found that IL-1β plays a central role in mediating the effects of H. pylori on Tregs. This is of particular interest, as virulent strains of H. pylori expressing cagPAI are associated with elevated levels of IL-1β [13,45]. As a result, the influence of H. pylori DCs on Tregs may be enhanced by the local microenvironment. In addition, IL-1β has a significant inhibitory effect on gastric acid production [46], which encourages H. pylori colonization to spread and downstream pathological events (gastritis and gastric cancer). As IL-1β appears to have a central role in H. pylori biology and its mechanisms of immune evasion and chronic inflammation, it may be revealing to study the relationship between polymorphisms in IL-1β and interactions between H. pylori and the host [47,48].
Others have suggested that Treg function can be modulated by the local cytokine microenvironment, in murine models inhibition of suppression by lipopolysaccharide (LPS)-treated DCs can be reversed by the addition of IL-6 neutralizing antibody [25]. We did not observe a role for IL-6 in the biological effects of H. pylori on Tregs, which is at variance with both the publication of Pasare and descriptions of IL-6R expression by Tregs in inflammatory environments [49]. This can be explained by suggestions that IL-6 is incapable of blocking suppression on its own and requires co-operative action with IL-1 to do so [26], whereas IL-1β has no obligate requirement for IL-6 and can break suppression of T cell proliferation on its own [24]. Alternatively, the variance could reflect differences between murine and human cells. Others have also suggested that IL-12 (but not IL-23) may also be capable of reversing suppression [28], but this result may not be of significance in H. pylori infections, as we have demonstrated previously that H. pylori-stimulated DCs are poor producers of IL-12 [10,13]. We also failed to find a role for TNF-α in the effect of H. pylori on Tregs. Although there is evidence in patients with rheumatoid arthritis that anti-TNF therapy reverses a defect in Tregs [27,50] we postulate that, in similar fashion to IL-6, this effect may be mediated through modification of other cytokines, such as IL-1, that may act in co-operation with TNF.
Finally, it has often been assumed that the presence of Tregs in inflamed sites indicates active T cell suppression. Our observations that H. pylori-stimulated DCs, as well as IL-1β, can subvert Treg suppression suggests that we should be cautious in this assumption. Equally, emerging data suggest that Tregs, or a subset of Tregs, retain the capacity to convert to the Th17 lineage when stimulated appropriately in the context of inflammation, in particular (for human Tregs) by IL-1β [51]. Such IL-17-producing, or ‘plastic’, Tregs have been described previously in lesional sites of Crohn's disease [52]. We have shown previously that DCs infected with H. pylori stimulate autologous CD4+ T cells to produce IL-17 and that this cytokine is expressed in gastric biopsies of patients with H. pylori infection [13]. Infection with H. pylori might not only inhibit Treg-mediated suppression but also differentiate subsets of Tregs to proinflammatory lineages, such as Th17. While, in this study, we looked for Th17 conversion of Tregs by HpDCs in vitro, we were unable to demonstrate Th17 conversion (data not shown), suggesting that Th17 conversion, if it occurs in response to H. pylori, is restricted to the in-vivo setting, where other components may be involved.
Very recently, a different role for H. pylori infection of DCs has been published. Oertli et al. have demonstrated in a murine model that H. pylori infection reprograms DCs towards a semi-mature phenotype, with the capacity to induce Tregs via the secretion of IL-18 [53]. This finding was supported partially in man by showing that DCs in H. pylori infected human gastric biopsies have a semi-mature phenotype and expressed DC-specific intercellular adhesion molecule-3-grabbing non-integrin (SIGN) [53]. In addition to this, the virulence factor vacuolating cytotoxin has also been shown to regulate DC maturation negatively [54], suggesting that the modulation of DC maturation plays an important role in H. pylori's subversion of the immune response. The study presented here has focused on the effect of H. pylori-infected DCs on naturally occurring Tregs, and whether or not infected DCs are able to produce IL-18 and induce de-novo Tregs has not been investigated. However, many reports published in the last few years have confirmed that H. pylori infection induced DC maturation and the release of IL-23 [10,13,55–57].
In conclusion, we have found that H. pylori expands Tregs in vitro and in vivo and subverts their suppressive function through the production of IL-1β from DCs. These findings question the role of Tregs at H. pylori-infected sites and provide mechanistic and therapeutic insights into the mechanisms of H. pylori-associated chronic gastritis and potential targets for the local treatment of inflammation associated with H. pylori in patients who do not respond to standard eradication therapy.
Acknowledgments
The authors acknowledge financial support from the Department of Health via the National Institute for Health Research (NIHR) comprehensive Biomedical Research Centre award to Guy's & St Thomas' NHS Foundation Trust in partnership with King's College London and King's College Hospital NHS Foundation Trust. The authors acknowledge the support of the MRC Centre for Transplantation.
Funding
This work was funded by grants from the Medical Research Council (to B.A., P.M. and R.I.L.), the British Heart Foundation and Guy's and St Thomas' Charity Trust (R.I.L. and G.L.).
Disclosure
The authors of this manuscript have no conflicts of interest to disclose.
References
- 1.Logan R, Walker M. ABC of the upper gastrointestinal tract. Epidemiology and diagnosis of Helicobacter pylori infection. BMJ. 2001;323:920–922. doi: 10.1136/bmj.323.7318.920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murray LJ, Bamford KB, O'Reilly DP, McCrum EE, Evans AE. Helicobacter pylori infection: relation with cardiovascular risk factors, ischaemic heart disease, and social class. Br Heart J. 1995;74:497–501. doi: 10.1136/hrt.74.5.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dhalla F, da Silva SP, Lucas M, Travis S, Chapel H. Review of gastric cancer risk factors in patients with common variable immunodeficiency disorders, resulting in a proposal for a surveillance programme. Clin Exp Immunol. 2011;165:1–7. doi: 10.1111/j.1365-2249.2011.04384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris A, Misiewicz JJ. ABC of the upper gastrointestinal tract. Management of Helicobacter pylori infection. BMJ. 2001;323:1047–1050. doi: 10.1136/bmj.323.7320.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med. 2002;347:1175–1186. doi: 10.1056/NEJMra020542. [DOI] [PubMed] [Google Scholar]
- 6.Schmausser B, Andrulis M, Endrich S, et al. Expression and subcellular distribution of Toll-like receptors TLR-4, TLR-5 and TLR-9 on the gastric epithelium in Helicobacter pylori infection. Clin Exp Immunol. 2004;136:521–526. doi: 10.1111/j.1365-2249.2004.02464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maeda S, Akanuma M, Mitsuno Y, et al. Distinct mechanism of Helicobacter pylori-mediated NF-kappa B activation between gastric cancer cells and monocytic cells. J Biol Chem. 2001;276:44856–44864. doi: 10.1074/jbc.M105381200. [DOI] [PubMed] [Google Scholar]
- 8.Nishi T, Okazaki K, Kawasaki K, et al. Involvement of myeloid dendritic cells in the development of gastric secondary lymphoid follicles in Helicobacter pylori-infected neonatally thymectomized BALB/c mice. Infect Immun. 2003;71:2153–2162. doi: 10.1128/IAI.71.4.2153-2162.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ninomiya T, Matsui H, Akbar SM, Murakami H, Onji M. Localization and characterisation of antigen-presenting dendritic cells in the gastric mucosa of murine and human autoimmune gastritis. Eur J Clin Invest. 2000;30:350–358. doi: 10.1046/j.1365-2362.2000.00629.x. [DOI] [PubMed] [Google Scholar]
- 10.Mitchell P, Germain C, Fiori PL, Khamri W, et al. Chronic exposure to Helicobacter pylori impairs dendritic cell function and inhibits Th1 development. Infect Immun. 2007;75:810–819. doi: 10.1128/IAI.00228-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bamford KB, Fan X, Crowe SE, et al. Lymphocytes in the human gastric mucosa during Helicobacter pylori have a T helper cell 1 phenotype. Gastroenterology. 1998;114:482–492. doi: 10.1016/s0016-5085(98)70531-1. [DOI] [PubMed] [Google Scholar]
- 12.Karttunen R, Karttunen T, Ekre HP, MacDonald TT. Interferon gamma and interleukin 4 secreting cells in the gastric antrum in Helicobacter pylori positive and negative gastritis. Gut. 1995;36:482–492. doi: 10.1136/gut.36.3.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khamri W, Walker MM, Clark P, et al. Helicobacter pylori stimulates dendritic cells to induce interleukin-17 expression from CD4+ T lymphocytes. Infect Immun. 2010;78:845–853. doi: 10.1128/IAI.00524-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mizuno T, Ando T, Nobata K, et al. Interleukin-17 levels in Helicobacter pylori-infected gastric mucosa and pathologic sequelae of colonization. World J Gastroenterol. 2005;11:6305–6311. doi: 10.3748/wjg.v11.i40.6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sakaguch IS, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10:490–500. doi: 10.1038/nri2785. [DOI] [PubMed] [Google Scholar]
- 16.Coffer PJ, Burgering BM. Forkhead-box transcription factors and their role in the immune system. Nat Rev Immunol. 2004;11:889–899. doi: 10.1038/nri1488. [DOI] [PubMed] [Google Scholar]
- 17.Belkaid Y, Rouse B. Natural regulatory T cells in infectious disease. Nat Immunol. 2005;6:353–360. doi: 10.1038/ni1181. [DOI] [PubMed] [Google Scholar]
- 18.Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420:502–507. doi: 10.1038/nature01152. [DOI] [PubMed] [Google Scholar]
- 19.Maloy KJ, Salaun L, Cahill R, Dougan G, Saunders NJ, Powrie F. CD4+CD25+ T(R) cells suppress innate immune pathology through cytokine-dependent mechanisms. J Exp Med. 2003;197:111–119. doi: 10.1084/jem.20021345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lundgren A, Suri-Payer E, Enarsson K, Svennerholm AM, Lundin BS. Helicobacter pylori-specific CD4+ CD25high regulatory T cells suppress memory T-cell responses to H. pylori in infected individuals. Infect Immun. 2003;71:1755–1762. doi: 10.1128/IAI.71.4.1755-1762.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lundgren A, Strömberg E, Sjöling A, et al. Mucosal FOXP3-expressing CD4+ CD25high regulatory T cells in Helicobacter pylori-infected patients. Infect Immun. 2005;73:523–531. doi: 10.1128/IAI.73.1.523-531.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamazaki S, Iyoda T, Tarbell K, et al. Direct expansion of functional CD25+ CD4+ regulatory T cells by antigen-processing dendritic cells. J Exp Med. 2003;198:235–247. doi: 10.1084/jem.20030422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng Y, Manzotti CN, Liu M, Burke F, Mead KI, Sansom DM. CD86 and CD80 differentially modulate the suppressive function of human regulatory T cells. J Immunol. 2004;172:2778–2784. doi: 10.4049/jimmunol.172.5.2778. [DOI] [PubMed] [Google Scholar]
- 24.O'Sullivan BJ, Thomas HE, Pai S, et al. IL-1 beta breaks tolerance through expansion of CD25+ effector T cells. J Immunol. 2006;176:7278–7287. doi: 10.4049/jimmunol.176.12.7278. [DOI] [PubMed] [Google Scholar]
- 25.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 26.Kubo T, Hatton RD, Oliver J, Liu X, Elson CO, Weaver CT. Regulatory T cell suppression and anergy are differentially regulated by proinflammatory cytokines produced by TLR-activated dendritic cells. J Immunol. 2004;173:7249–7258. doi: 10.4049/jimmunol.173.12.7249. [DOI] [PubMed] [Google Scholar]
- 27.Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood. 2006;108:253–261. doi: 10.1182/blood-2005-11-4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.King IL, Segal BM. Cutting edge: IL-12 induces CD4+CD25– T cell activation in the presence of T regulatory cells. J Immunol. 2005;175:641–645. doi: 10.4049/jimmunol.175.2.641. [DOI] [PubMed] [Google Scholar]
- 29.Scott K, Manunta M, Germain C, et al. Qualitatively distinct patterns of cytokines are released by human dendritic cells in response to different pathogens. Immunology. 2005;116:245–254. doi: 10.1111/j.1365-2567.2005.02218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Veerapathran A, Pidala J, Beato F, Yu X, Anasetti C. Ex vivo expansion of human Tregs specific for alloantigens presented. Blood. 2011;118:5671–5680. doi: 10.1182/blood-2011-02-337097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schulz O, Edwards AD, Schito M, et al. CD40 triggering of heterodimeric IL-12 p70 production by dendritic cells in vivo requires a microbial priming signal. Immunity. 2000;4:453–462. doi: 10.1016/s1074-7613(00)00045-5. [DOI] [PubMed] [Google Scholar]
- 32.Wilson KT, Crabtree JE. Immunology of Helicobacter pylori: insights into the failure of the immune response and perspectives on vaccine studies. Gastroenterology. 2007;133:288–308. doi: 10.1053/j.gastro.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 33.Necchi V, Candusso ME, Tava F, et al. Intracellular, intercellular, and stromal invasion of gastric mucosa, preneoplastic lesions, and cancer by Helicobacter pylori. Gastroenterology. 2007;132:1009–1023. doi: 10.1053/j.gastro.2007.01.049. [DOI] [PubMed] [Google Scholar]
- 34.Cooke CL, Huff JL, Solnick JV. The role of genome diversity and immune evasion in persistent infection with Helicobacter pylori. FEMS Immunol Med Microbiol. 2005;45:11–23. doi: 10.1016/j.femsim.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 35.Algood HM, Cover TL. Helicobacter pylori persistence: an overview of interactions between H. pylori and host immune defenses. Clin Microbiol Rev. 2006;19:597–613. doi: 10.1128/CMR.00006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kabir S. The role of interleukin-17 in the Helicobacter pylori induced infection and immunity. Helicobacter. 2011;16:1–8. doi: 10.1111/j.1523-5378.2010.00812.x. [DOI] [PubMed] [Google Scholar]
- 37.Baldwin AS. Series introduction: the transcription factor NF-kappaB and human disease. J Clin Invest. 2001;107:3–6. doi: 10.1172/JCI11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wallace JL, Cucala M, Mugridge K, Parente L. Secretagogue-specific effects of interleukin-1 on gastric acid secretion. Am J Physiol. 1991;261:559–564. doi: 10.1152/ajpgi.1991.261.4.G559. [DOI] [PubMed] [Google Scholar]
- 39.Kondo S, Shinomura Y, Kanayama S, et al. Interleukin-1 beta inhibits gastric histamine secretion and synthesis in the rat. Am J Physiol. 1994;267:966–971. doi: 10.1152/ajpgi.1994.267.6.G966. [DOI] [PubMed] [Google Scholar]
- 40.Huang FY, Chan AO, Rashid A, Wong DK, Cho CH, Yuen MF. Helicobacter pylori induces promoter methylation of E-cadherin via interleukin-1β activation of nitric oxide production in gastric cancer cells. Cancer. 2012 doi: 10.1002/cncr.27519. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 41.Zambon CF, Basso D, Navaglia F, et al. Helicobacter pylori virulence genes and host IL-1RN and IL-1beta genes interplay in favouring the development of peptic ulcer and intestinal metaplasia. Cytokine. 2002;18:242–251. doi: 10.1006/cyto.2002.0891. [DOI] [PubMed] [Google Scholar]
- 42.Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, Demengeot J. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J Exp Med. 2003;197:403–411. doi: 10.1084/jem.20021633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peng G, Guo Z, Kiniwa Y, et al. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science. 2005;309:1380–1384. doi: 10.1126/science.1113401. [DOI] [PubMed] [Google Scholar]
- 44.Liu H, Komai-Koma M, Xu D, Liew FY. Toll-like receptor 2 signaling modulates the functions of CD4+ CD25+ regulatory T cells. Proc Natl Acad Sci U S A. 2006;103:7048–7053. doi: 10.1073/pnas.0601554103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peek RM, Jr, Miller GG, Tham KT, et al. Heightened inflammatory response and cytokine expression in vivo to cagA+ Helicobacter pylori strains. Lab Invest. 1995;73:760–770. [PubMed] [Google Scholar]
- 46.Kondo S, Shinomura Y, Kanayama S, et al. Interleukin-1 beta inhibits gastric histamine secretion and synthesis in the rat. Am J Physiol. 1994;267:G966–971. doi: 10.1152/ajpgi.1994.267.6.G966. [DOI] [PubMed] [Google Scholar]
- 47.Persson C, Canedo P, Machado JC, El-Omar EM, Forman D. Polymorphisms in inflammatory response genes and their association with gastric cancer: a HuGE systematic review and meta-analyses. Am J Epidemiol. 2011;173:259–270. doi: 10.1093/aje/kwq370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sugimoto M, Furuta T, Yamaoka Y. Influence of inflammatory cytokine polymorphisms on eradication rates of Helicobacter pylori. J Gastroenterol Hepatol. 2009;24:1725–1732. doi: 10.1111/j.1440-1746.2009.06047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Doganci A, Eigenbrod T, Krug N, et al. The IL-6R alpha chain controls lung CD4+CD25+ Treg development and function during allergic airway inflammation in vivo. J Clin Invest. 2005;115:313–325. doi: 10.1172/JCI22433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nadkarni S, Mauri C, Ehrenstein MR, Anti TN. F-alpha therapy induces a distinct regulatory T cell population in patients with rheumatoid arthritis via TGF-beta. J Exp Med. 2007;204:33–39. doi: 10.1084/jem.20061531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Afzali B, Mitchell P, Lechler RI, John S, Lombardi G. Translational mini-review series on Th17 cells: induction of interleukin-17 production by regulatory T cells. Clin Exp Immunol. 2010;159:120–130. doi: 10.1111/j.1365-2249.2009.04038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hovhannisyan Z, Treatman J, Littman DR, Mayer L. Characterization of interleukin-17-producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology. 2011;140:957–965. doi: 10.1053/j.gastro.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oertli M, Sundquist M, Hitzler I, et al. DC-derived IL-18 drives Treg differentiation, murine Helicobacter pylori-specific immune tolerance, and asthma protection. J Clin Invest. 2012;112:1082–1096. doi: 10.1172/JCI61029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim JM, Kim JS, Yoo DY, et al. Stimulation of dendritic cells with Helicobacter pylori vacuolating cytotoxin negatively regulates their maturation via the restoration of E2F1. Clin Exp Immunol. 2011;166:34–45. doi: 10.1111/j.1365-2249.2011.04447.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Drakes ML, Czinn SJ, Blanchard TG. Regulation of murine dendritic cell immune responses by Helicobacter felis antigen. Infect Immun. 2006;74:4624–4633. doi: 10.1128/IAI.00289-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kranzer K, Eckhardt A, Aigner M, et al. Induction of maturation and cytokine release of human dendritic cells by Helicobacter pylori. Infect Immun. 2004;72:4416–4423. doi: 10.1128/IAI.72.8.4416-4423.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nishi T, Okazaki K, Kawasaki K, et al. Involvement of myeloid dendritic cells in the development of gastric secondary lymphoid follicles in Helicobacter pylori-infected neonatally thymectomized BALB/c mice. Infect Immun. 2003;71:2153–2162. doi: 10.1128/IAI.71.4.2153-2162.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]