

Abstract
Hippocampal CA1 neurons are particularly sensitive to ischemic damage, such as experienced following cardiac arrest and cardiopulmonary resuscitation. In recent years transient receptor potential M2 (TRPM2) channels have been identified as mediators of ischemic damage. We previously demonstrated that neuroprotective strategies targeting TRPM2 channels preferentially protect male cortical neurons from ischemic injury both in vitro and in vivo. It is important to determine the role of TRPM2 in ischemic injury of hippocampal neurons as this population of neurons are particularly sensitive to ischemic injury and are therapeutic targets. Here we report significantly decreased neuronal cell death following in vitro ischemia preferentially in male hippocampal neurons using TRPM2 inhibitors or knockdown of TRPM2 expression. Electrophysiological characterization of sex-stratified cultures shows similar levels of functional TRPM2 channel expression in male and female hippocampal neurons under basal conditions. In contrast, recordings made during reperfusion following in vitro ischemia revealed that TRPM2 channels are activated only in male neurons, resulting in rapid and complete depolarization. These findings provide strong evidence for TRPM2 as a target for protection against cerebral ischemia in male brain and helps define a molecular cell death pathway that is differentially engaged in male and female neurons.
Introduction
Each year in the U.S., approximately 600,000 people suffer from cardiac arrest and receive cardiopulmonary resuscitation (CA/CPR), an event associated with high mortality and poor neurological outcome1. The principal neurological consequences in the central nervous system (CNS) following CA/CPR-induced ischemia are motor and cognitive deficits, particularly memory acquisition and retention2. Consistent with this, transient global ischemia caused by CA/CPR leads to hippocampal CA1 neuronal cell death3. A great deal of research has focused on ion channels and transporters required for maintenance of ionic balance in neurons to explain hippocampal neuron vulnerability to ischemia. The most well studied channels in ischemia research are ionotropic glutamate receptors, N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors. Large numbers of studies have shown that glutamate receptor antagonists prevent excitotoxic neuronal cell death and decrease ischemic injury. However, clinical trials with compounds designed to inhibit these receptors have proven unsuccessful4. The disappointing results with the above mentioned antagonists has led to the understanding of the importance of identifying and characterizing alternative Ca2+ influx pathways involved in neuronal cell death 5. One such family of channels is the transient receptor potential (TRP) superfamily of cation channels. This study focuses on the relatively recently identified oxidative stress-sensitive ion channel TRPM2.
TRPM2 mediated currents were initially described by our group, characterized as NAD+-activated non-selective cation (NSNAD) channels activated by oxidative stress in pancreatic β-cells and striatal neurons6–8. Cloning and molecular analysis of the TRPM2 gene allowed for greater understanding of the structure and function of TRPM2 channels 9,10, confirming that the channel is sensitive to hydrogen peroxide (H2O2)-induced oxidative stress and revealing that the physiologically relevant activator of TRPM2 is adenine dinucleotide phosphate ribose (ADPr)11,12. Subsequently, it has been demonstrated that TRPM2 channels are expressed throughout the brain, as well as immune cells, endocrine cells, endothelial cells and cardiomyocytes (for review see11,12). Functional TRPM2 channels have been demonstrated in numerous neuronal populations, including hippocampus, cortex and striatal neurons. Not surprisingly, TRPM2 channels have been proposed to contribute to neuronal damage in neurodegenerative diseases such as ischemic stroke13,14. We recently demonstrated that pharmacological inhibition of TRPM2 or shRNA knockdown resulted in neuroprotection from transient focal ischemia in mice and oxygen and glucose deprivation (OGD) in cortical cultures15. The current study extended the previous finding in cortical neurons to determine the timing of TRPM2 channel activation following reperfusion and confirm the role of TRPM2 in ischemic injury in the exquisitely sensitive hippocampal neuron population in order to enhance confidence that TRPM2 inhibition represents a viable therapeutic strategy.
Methods
Experimental Animals
All experimental protocols were approved by the Institutional Animal Care and Use Committee and conformed to the National Institutes of Health guidelines for the care and use of animals in research. Culture experiments performed on embryos obtained from pregnant C57Bl/6 mice on embryonic day 17.
Primary Cell culture
Experiments were performed on sex-stratified mouse hippocampal neuronal cultures. Hippocampi were dissected from embryonic day 17 (E 17) C57BL/6 mice and embryos sexed as described previously15. Briefly, E17 embryos were rapidly removed from timed pregnant mice and sex of each embryo was identified by exploratory laparotomy to inspect gonads and internal organs. The isolated hippocampi were then digested with papain (20 μg/ml: (Worthington Biochemical, Lakewood, NJ, USA)), halted by addition of trypsin inhibitor (Sigma-Aldrich, St. Louis, MO) and triturated and filtered through cell sorting nylon mesh. Cells were plated at a concentration of 2.5 × 105 cells per well (24-well plate) coated with poly-D-lysine and grown at 37°C. On day 3 in vitro, 1.5 μM AraC (Cytosine-1-β-D-arabino furanoside, Sigma-Aldrich, St. Louis, MO, USA) was added to each well to inhibit the growth of astrocytes in the culture (>95% pure neurons). Half the medium (neurobasal without Phenol red + B27) was replaced with fresh medium every 3–4 days.
Oxygen-Glucose Deprivation and oxidative stress
In vitro ischemia (OGD) was induced by transferring cells to glucose-free neurobasal-A medium and placing them into an anaerobic incubator for 2 h. The anaerobic incubator contained an oxygen reacting catalyst to ensure the presence of < 5 ppm O2, kept at 37°C with 5% CO2 and 95% N2 (Coy Laboratory Products, Grass Lake, MI, USA). Oxidative stress was induced by treating cells with H2O2 (50 μM) for 2 hrs. Re-oxygenation was initiated by transferring cells to culture media in aerobic incubator or glucose containing saline for electrophysiology recordings (see below). Cells were exposed to TRPM2 inhibitors for 15 min. prior to OGD and inhibitor maintained throughout OGD and re-oxygenation. Cell viability was determined 24 h after initiation of re-oxygenation using the MTT assay. The MTT assay is a standard colorimetric assay for measuring the activity of mitochondrial enzymes that reduce MTT to formazan, producing a purple color16. After OGD, MTT was added to cells for 1 h and after 1 h incubation, formazan accumulated in living cells was dissolved in 200 μl DMSO. Formazan was quantified by measuring optical density at 540 nm with microplate reader (Victor3, Perkin Elmer, Waltham, MA, USA). Survival ratio treatment group/oxygenated control was expressed as percent, with control being 100%.
Electrophysiology
Cells were transferred to a recording chamber mounted on an inverted Leica DM IRB microscope (Leica, Houston, TX, USA) with continuous gravity fed saline perfusion (in mM): 140 NaCl, 2.5 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES and 10 Glucose, pH 7.35 with NaOH. Patch-clamp recordings were obtained from the soma of hippocampal neurons using an Axopatch 200B (Molecular Devices, Union City, CA) amplifier interfaced to a computer (Dell, Round Rock, TX, USA). Data was collected at a frequency of 10–20 kHz and analyzed using pClamp10, Clampfit (Molecular Devices, Sunnyvale, CA, USA), and IgorPro6 software (Wavemetrics, Lake Oswego, OR, USA). A Flaming Brown electrode puller (Sutter 97; Sutter Instruments, Novato, CA, USA) was used to pull microelectrodes with a resistance of 2–3 MΩ when polished. Internal pipette solution consisting of (in mM): 140 K-Gluconate, 0.05 EGTA, 10 HEPES, 1 MgCl2, 5 MgATP, 0.5 NaGTP, pH of 7.3 with KOH. Whole-cell voltage clamp recordings of TRPM2 channel activity were performed using an internal pipette solution consisting (in mM): 140 K-Gluconate, 10 HEPES, 1 MgCl2, 1 EGTA, 0.3 ADPR, 1.05 CaCl2 to produce 1 μM free Ca2+ (pH 7.3). Whole-cell current-clamp experiments were performed to monitor membrane potential passively, with no bias current applied. Whole-cell capacitance and series resistance was electronically compensated to 60–80% for voltage-clamp experiments. Adequate whole cell access (Ra<20 MΩ) was achieved prior to experimentation and verified at end of recordings.
Solutions and Drugs
ACA: N-(p-Amylcinnamoyl)anthranilic acid was obtained from EMD Bioscience (NJ, USA). 2-APB: 2-Aminoethoxy diphenyl borate, CTZ: clotrimazole, and FFA: flufenamic acid were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Generation of pseudo-type Lentivirus
To generate the 3rd generation lentiviral transfer vectors, DNA encoding shRNA sequences targeted to TRPM2 (shRNA 89) were cloned into the lentivirus vector FUGW, expressing eGFP driven by ubiquitin promoter15. The lentiviral vectors used were pCMVΔR8.9 (viral core packaging), pHCMV-g (VSV-g envelope protein) and the FUGW transfer vector, as previously described15. Probe sequence for shRNA89: TTTGGCTCATGGATTCCCGAGAATACTCGAGTATTCTCGGGAATCCATGAGCTTTTTG
Statistical Analysis
All data is presented as mean ± SEM. Each n represents an individual cell for electrophysiology experiments or an individual culture for in vitro experiments. Statistical significance was determined using students t-test (unpaired, 2-tailed, if p<0.05) or one way analysis of variance (ANOVA) with Neuman-Keuls post hoc analysis, p<0.05. Significance of neuroprotection from H2O2-induced oxidative stress with CTZ was assessed using repeated measures two-way-ANOVA, p<0.05.
Results
TRPM2 inhibitors protect male but not female neurons against OGD-induced cell damage
Significant cell death was observed in male and female hippocampal neurons exposed to in vitro ischemia induced by oxygen-glucose deprivation (OGD). Cell death measured 24 hr after exposure to 2 hr OGD was not different in male and female neurons, exhibiting 50.3 ± 6.9% (n=12) and 48.3 ± 8.9% (n=12) cell death, respectively. We used four different compounds demonstrated to potently inhibit TRPM2 channel activity, specifically: ACA, 2-APB, CTZ and FFA12. Each inhibitor significantly decreased neuronal cell death following OGD in male hippocampal neurons (Figure 1a), decreasing male hippocampal neuronal cell death from 50.3 ± 6.9% (n = 12) in vehicle treated neurons to 24.5 ± 7.8% in 100 μM 2-APB (n = 5; P < 0.05), 10.8 ± 2.2% in 30 μM FFA (n = 4; P < 0.05), 15.8 ± 6.6% in 10 μM ACA (n = 4; P<0.05) and 10.0 ± 4.0% in 20 μM CTZ (n = 4; P < 0.05). In contrast, none of the inhibitors altered survival of female hippocampal neurons following OGD, having 48.3 ± 8.9% (n = 12) cell death in vehicle treated neurons and 43.7 ± 9.3% in 100 μM 2-APB (n = 5), 41.8 ± 4.8% in 30 μM FFA (n = 4), 35.8 ± 17.4% in 10 μM ACA (n = 4) and 41.6 ± 2.4% 20 μM CTZ (n = 4). To determine if TRPM2 inhibition is protective when administered after the ischemic event, CTZ, the most effective and well characterized inhibitor was applied only during re-oxygenation. CTZ, applied 15 minutes after re-oxygenation, provided significant protection, decreasing cell death to 16.7 ± 4.5% (n = 5; P < 0.05) in male neurons (Figure 1). These data support previous results obtained in cortical neurons and demonstrate the efficacy of TRPM2 inhibition to protect when administered post-ischemia.
Figure 1.
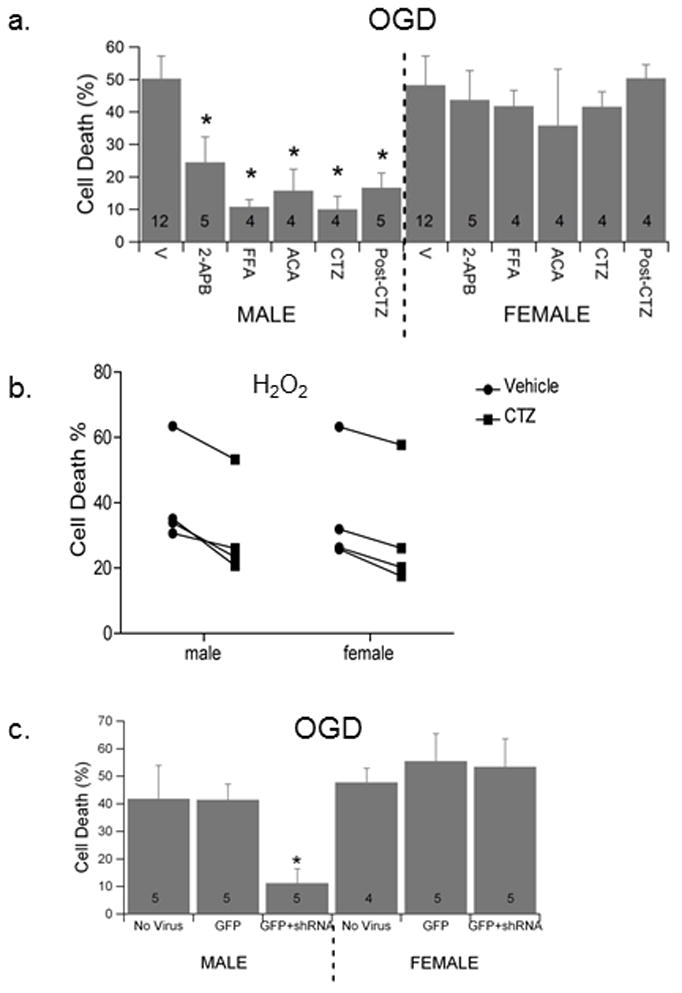
TRPM2 inhibition decrease OGD-induced hippocampal cell death in males preferentially. (a) Quantification of cell death 24 hr after 2 hr OGD in male and female hippocampal cultures. Neurons exposed to each inhibitor 15 min prior to OGD and inhibitor maintained throughout OGD and re-oxygenation, with the exception of bar labeled Post-CTZ in which CTZ was added during re-oxygenation only. (b) Cultures were treated with 50 μM H2O2 in the presence of vehicle or CTZ for 2hrs. Quantification of cell death was performed 24 hrs after return to normal media. Each data point represents an individual culture. Repeated measures 2-way ANOVA was used to assess significance of treatment and sex (p < 0.05). (c) Quantification of cell death 24 hr after 2 hr OGD in male and female hippocampal cultures exposed to control lentivirus and lentivirus expressing shRNA89. Data presented is mean ± SEM. The numbers in each bar represent number of experiments and symbol * indicates P<0.05 compared to vehicle treated cultures of same sex.
Hippocampal cultures were treated with H2O2 (50 μM) for 2 hrs followed by 24 hr incubation in normal media. Similar neuronal cell death was observed in male cultures (40.8±7.6) and female cultures (36±8.9). Remarkably, figure 1b illustrates that CTZ treatment reduced cell death in all cultures tested, with males decreasing to 30.8±7.5 (n = 4, P < 0.05) and females to 30.4±9.3 (n = 4, p < 0.05). Thus, unlike what is seen with OGD, CTZ treatment is capable of protecting both male and female neurons in response to oxidative stress.
TRPM2 knockdown protects male hippocampal neurons from OGD-induced cell death
We have previously reported TRPM2 knockdown and neuroprotection in cortical neurons in vitro and in vivo using shRNA8915. Male and female hippocampal neurons were infected with lentivirus (GFP or GFP+shRNA89) 4 days prior to exposure to 2 hr OGD and 24 hr reoxygenation. Figure 1c illustrates that infection with shRNA89 producing lentivirus significantly reduced male hippocampal neuronal cell death, decreasing cell death from 41.4 ± 5.3% (n = 4) in control cells exposed to GFP expressing virus (empty virus) to 11.1 ± 5.8% (n = 4; P < 0.05) in cells treated with GFP+shRNA89. In contrast, infection with GFP+shRNA89 had no effect on survival of female hippocampal neurons following OGD, having 53.4 ± 10.2% (n = 4) and 55.4 ± 10.0% (n = 4) cell death in GFP and GFP+shRNA89, respectively. Control GFP expressing virus has no effect on neuronal viability or sensitivity to OGD, compared to uninfected neurons (No Virus; Figure 1b).
TRPM2 channels are activated during re-oxygenation
Whole-cell voltage-clamp recordings were performed to confirm the presence of functional TRPM2 channels in male and female hippocampal neurons. Experiments performed in the presence of 300 μM ADPribose (ADPr) and 10 μM free calcium in the patch pipette resulted in the generation of inward current that was completely inhibited by the extracellular application of 20 μM clotrimazole (Figure 2a). The magnitude of ADPr-activated current was not significantly different in male and female hippocampal neurons, observing 4.3 ± 1.1 pA/pF (n = 5) and 3.5 ± 0.8 pA/pF (n = 7) of current at −40 mV in male and female neurons, respectively. Recordings made in the absence of internal ADPr did not exhibit inward currents (Fig 2b). These data confirm previous findings demonstrating the expression of TRPM2 channels in hippocampal neurons17 and further show that male and female neurons express similar levels of functional channels.
Figure 2.

Male and female hippocampal neurons express similar levels of functional TRPM2 channels. (a) Representative whole-cell voltage clamp recording from cultured hippocampal neuron demonstrating ADPribose-activated current (300 μM ADPr in patch pipette). Current was completely abolished by extracellular application of 30 μM CTZ. (b) Representative recording in the absence of ADPribose. (c) Quantification of current density of ADPribose-activated current in male and female hippocampal neurons. Data presented is mean ± SEM. The numbers in each bar represent number of experiments.
To determine whether OGD results in activation of TRPM2 currents, whole-cell current clamp recordings were obtained from hippocampal neurons within the first 60 min. of reoxygenation. Male neurons exhibited rapid and complete depolarization in all 19 cells recorded (Fig 3a). Time to onset of depolarization varied between 15 and 74 min after reoxygenation averaging 37.4±3.7 min (n = 19). Kinetics of depolarization were remarkably similar in all 19 cells, depolarizing completely within 2 minutes, similar to previous observations following oxidative stress (H2O2)-induced TRPM2 depolarization6,7. OGD had no effect on any of the biophysical properties measured at the onset of recordings (data not shown). Application of TRPM2 inhibitors immediately upon initiation of depolarization caused reversal of the process and hyperpolarization towards the initial resting membrane potential, in 6 out of 9 experimental cells (Figure 3b). In contrast, we did not observe rapid depolarization in any female hippocampal neuron after OGD (Figure 3c; n = 7). Finally, we recorded from male hippocampal neurons infected with GFP+sh89 virus after OGD and observed action potential activity similar to female neurons (Figure 3d), but importantly, no membrane depolarization (n = 4).
Figure 3.
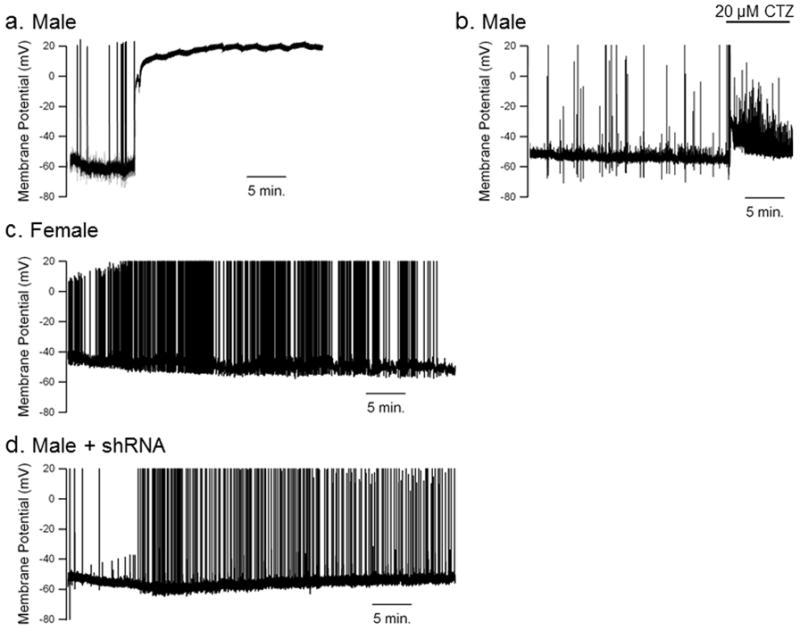
OGD-induced depolarization of hippocampal neurons involves activation of TRPM2 channels. (a) Representative current clamp recording of male hippocampal neuron during re-oxygenation following exposure to 2 hr OGD. Note delayed, rapid and complete depolarization. (b) Representative current clamp recording of male hippocampal neuron during re-oxygenation following exposure to 2 hr OGD. 20 μM clotrimazole (CTZ) is rapidly applied to the extracellular recording solution at the initiation of depolarization. Note the repolarization and lack of complete depolarization. (c) Representative current clamp recording of female hippocampal neuron during re-oxygenation following OGD. Female cells exhibited increase excitability without rapid depolarization. (d) Representative recording of male hippocampal neuron 4 days after infection with shRNA expressing lentivirus. Note lack of depolarization observed in infected male neurons.
Discussion
A role for TRPM2 in cerebral ischemia has been proposed in multiple review articles13,14,18,19, due to their sensitivity to oxidative stress. Indeed, our previous work identified TRPM2 as a contributor to damage in cortex and striatum in males only following experimental stroke induced by middle cerebral artery occlusion15. This study supports a gender specific role for TRPM2 in hippocampal neurons and demonstrates a male specific activation of the channel following in vitro ischemia. Interestingly, our data with oxidative stress-induced cell death show that TRPM2 inhibition is capable of providing protection to both males and females, suggesting that TRPM2 is activated equally in this injury model. Voltage-clamp data obtained in male and female hippocampal neurons demonstrates TRPM2 can be activated to similar levels when ADPribose is present. Thus, sexual dimorphism observed with OGD, but not H2O2, likely involves preferential engagement of pathways upstream of TRPM2 which generate high levels of intracellular ADPribose in males but not females. In contrast, H2O2 is a strong activator of oxidative stress pathways that can cause TRPM2 activation in both sexes. While oxidative stress clearly contributes to ischemia-induced neuronal injury, our data provides strong evidence for distinct cellular processes engaged using the more complex and relevant insult of OGD.
Recordings obtained from male neurons during re-oxygenation revealed a rapid and complete depolarization consistent with activation of non-selective cation channels. This data is in strong agreement with previous work demonstrating H2O2-induced activation of TRPM2 channels, membrane depolarization and excessive Ca2+ influx6,20. While current-clamp recordings are indirect evidence for activation of TRPM2 channels, the lack of depolarization observed in male neurons following TRPM2 knockdown, and female neurons, provides evidence strengthening the link between TRPM2 and OGD-induced depolarization in male neurons. This is somewhat surprising considering we demonstrate no significant differences in level of expression of TRPM2 channels in male and female neurons, supporting the notion that sex-specific regulation of TRPM2 channel activity likely involves differential activation of regulators upstream of channel activation. Our functional data indicates that TRPM2 channels are activated during the initial 30–90 minutes of re-oxygenation in male neurons. This data, combined with the CTZ protection observed during re-oxygenation, support TRPM2 inhibition as a target for hippocampal protection when inhibitor is administered within a relatively narrow window after reperfusion. Therefore, the current study provides strong evidence for the possible clinical utility of TRPM2 inhibitors following global cerebral ischemia as experienced following cardiac arrest and CPR, which has a shorter therapeutic window than ischemic stroke and injury is particularly evident in the hippocampus.
There is increasing evidence that calcium entry via non-excitotoxic pathways contributes to the brain response to ischemia and represents new targets for therapeutic intervention (For review see18). In particular, inhibition of the acid sensing ion channel ASIC1a, a calcium and sodium permeable cation channel activated by acidosis, protects the brain with a time window of up to five hours following cerebral ischemia21,22. Similarly, knockdown of TRPM7 channels protects cultures neurons from anoxic damage23 and prevents delayed hippocampal CA1 neuronal cell death following global cerebral ischemia24. Our data adds to this growing list, strongly indicating that inhibition of TRPM2 channels is a novel, and potentially effective, strategy for minimizing ischemic brain damage. In support of this idea, we observed a similar phenomenon using a mouse model of focal cerebral ischemia, in which TRPM2 inhibition reduced neuronal damage after middle cerebral artery occlusion in male brain exclusively, providing no benefit in female mice15. It is of particular interest to examine a role for TRPM2 following cardiac arrest and cardiopulmonary resuscitation which is especially damaging to hippocampal neurons. This congruence between in vitro and in vivo data lends support to the notion that TRPM2 is a compelling new target for neuroprotection that should be considered for the treatment of men exclusively.
Highlights.
TRPM2 inhibition decrease OGD-induced hippocampal cell death in male preferentially
No sex difference in TRPM2 contribution to H2O2-induced hippocampal cell death
Male and female neurons express similar levels of functional TRPM2 channels
TRPM2 channels activated during reperfusion in male neurons
Acknowledgments
Work funded by NIH grant NS058792.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Roger VL, et al. Heart Disease and Stroke Statistics--2012 Update: A Report From the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lim C, Alexander MP, LaFleche G, Schnyer DM, Verfaellie M. The neurological and cognitive sequelae of cardiac arrest. Neurology. 2004;63:1774–1778. doi: 10.1212/01.wnl.0000144189.83077.8e. [DOI] [PubMed] [Google Scholar]
- 3.Horn M, Schlote W. Delayed neuronal death and delayed neuronal recovery in the human brain following global ischemia. Acta Neuropathol(Berl) 1992;85:79–87. doi: 10.1007/BF00304636. [DOI] [PubMed] [Google Scholar]
- 4.Ginsberg MD. Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology. 2008;55:363–389. doi: 10.1016/j.neuropharm.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Besancon E, Guo S, Lok J, Tymianski M, Lo EH. Beyond NMDA and AMPA glutamate receptors: emerging mechanisms for ionic imbalance and cell death in stroke. Trends Pharmacol Sci. 2008;29:268–275. doi: 10.1016/j.tips.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 6.Herson PS, Ashford ML. Activation of a novel non-selective cation channel by alloxan and H2O2 in the rat insulin-secreting cell line CRI-G1. J Physiol. 1997;501 (Pt 1):59–66. doi: 10.1111/j.1469-7793.1997.059bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith MA, Herson PS, Lee K, Pinnock RD, Ashford ML. Hydrogen-peroxide-induced toxicity of rat striatal neurones involves activation of a non-selective cation channel. J Physiol. 2003;547:417–425. doi: 10.1113/jphysiol.2002.034561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herson PS, Dulock KA, Ashford ML. Characterization of a nicotinamide-adenine dinucleotide-dependent cation channel in the CRI-G1 rat insulinoma cell line. J Physiol. 1997;505 (Pt 1):65–76. doi: 10.1111/j.1469-7793.1997.065bc.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hara Y, et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell. 2002;9:163–173. doi: 10.1016/s1097-2765(01)00438-5. [DOI] [PubMed] [Google Scholar]
- 10.Nagamine K, et al. Molecular cloning of a novel putative Ca2+ channel protein (TRPC7) highly expressed in brain. Genomics. 1998;54:124–131. doi: 10.1006/geno.1998.5551. [DOI] [PubMed] [Google Scholar]
- 11.Eisfeld J, Luckhoff A. TRPM2. Handb Exp Pharmacol. 2007:237–252. doi: 10.1007/978-3-540-34891-7_14. [DOI] [PubMed] [Google Scholar]
- 12.Wu LJ, Sweet TB, Clapham DE. International Union of Basic and Clinical Pharmacology. LXXVI Current progress in the mammalian TRP ion channel family. Pharmacological reviews. 2010;62:381–404. doi: 10.1124/pr.110.002725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.MacDonald JF, Jackson MF. Transient receptor potential channels of the melastatin family and ischemic responses of central neurons. Stroke. 2007;38:665–669. doi: 10.1161/01.STR.0000251671.77351.e2. [DOI] [PubMed] [Google Scholar]
- 14.Simard JM, Tarasov KV, Gerzanich V. Non-selective cation channels, transient receptor potential channels and ischemic stroke. Biochim Biophys Acta. 2007;1772:947–957. doi: 10.1016/j.bbadis.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jia J, et al. Sex differences in neuroprotection provided by inhibition of TRPM2 channels following experimental stroke. J Cereb Blood Flow Metab. 2011 doi: 10.1038/jcbfm.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 17.Olah ME, et al. Ca2+-dependent induction of TRPM2 currents in hippocampal neurons. J Physiol. 2009;587:965–979. doi: 10.1113/jphysiol.2008.162289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium. 2010;47:122–129. doi: 10.1016/j.ceca.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 19.Forder JP, Tymianski M. Postsynaptic mechanisms of excitotoxicity: Involvement of postsynaptic density proteins, radicals, and oxidant molecules. Neuroscience. 2009;158:293–300. doi: 10.1016/j.neuroscience.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 20.Herson PS, Lee K, Pinnock RD, Hughes J, Ashford ML. Hydrogen peroxide induces intracellular calcium overload by activation of a non-selective cation channel in an insulin-secreting cell line. J Biol Chem. 1999;274:833–841. doi: 10.1074/jbc.274.2.833. [DOI] [PubMed] [Google Scholar]
- 21.Xiong ZG, et al. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–698. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 22.Pignataro G, Simon RP, Xiong ZG. Prolonged activation of ASIC1a and the time window for neuroprotection in cerebral ischaemia. Brain. 2007;130:151–158. doi: 10.1093/brain/awl325. [DOI] [PubMed] [Google Scholar]
- 23.Aarts M, et al. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115:863–877. doi: 10.1016/s0092-8674(03)01017-1. [DOI] [PubMed] [Google Scholar]
- 24.Sun HS, et al. Suppression of hippocampal TRPM7 protein prevents delayed neuronal death in brain ischemia. Nat Neurosci. 2009;12:1300–1307. doi: 10.1038/nn.2395. [DOI] [PubMed] [Google Scholar]