

Abstract
The Human Genome Project (HGP) provided the initial draft of mankind's DNA sequence in 2001. The HGP was produced by 23 collaborating laboratories using Sanger sequencing of mapped regions as well as shotgun sequencing techniques in a process that occupied 13 years at a cost of ~$3 billion. Today, Next Generation Sequencing (NGS) techniques represent the next phase in the evolution of DNA sequencing technology at dramatically reduced cost compared to traditional Sanger sequencing. A single laboratory today can sequence the entire human genome in a few days for a few thousand dollars in reagents and staff time. Routine whole exome or even whole genome sequencing of clinical patients is well within the realm of affordability for many academic institutions across the country. This paper reviews current sequencing technology methods and upcoming advancements in sequencing technology as well as challenges associated with data generation, data manipulation and data storage. Implementation of routine NGS data in cancer genomics is discussed along with potential pitfalls in the interpretation of the NGS data. The overarching importance of bioinformatics in the clinical implementation of NGS is emphasized.[7] We also review the issue of physician education which also is an important consideration for the successful implementation of NGS in the clinical workplace. NGS technologies represent a golden opportunity for the next generation of pathologists to be at the leading edge of the personalized medicine approaches coming our way. Often under-emphasized issues of data access and control as well as potential ethical implications of whole genome NGS sequencing are also discussed. Despite some challenges, it's hard not to be optimistic about the future of personalized genome sequencing and its potential impact on patient care and the advancement of knowledge of human biology and disease in the near future.
Keywords: Bioinformatics, clinical medicine, next generation sequencing, pathology
INTRODUCTION
DNA sequencing techniques have revolutionized our understanding of human biology over the last forty years. DNA sequencing techniques originated in the early 70s due to the pioneering work of Walter Gilbert[1] and Frederick Sanger.[2] Continuous technological improvements in DNA sequencing instrumentation ever since has created an environment in which the Human Genome Project (HGP)[3] could be finally realized in the year 2001 after a decade of work. The HGP was expected to provide mankind with a dramatic advance in our understanding of human health and spawn a revolution in personalized healthcare approaches. While the pace of progress related to personalized healthcare has been frustrating for some, there is much cause for optimism. The HGP catalysed significant developments within both the academic as well as the commercial biotechnology industry. The emergence of Direct-to-Consumer (DTC) genomic industry can be traced back directly to the wealth of information provided by the HGP project. DTC companies such as 23andMe (23andme.com), and Navigenics (http://www.navigenics.com) provide a broad interrogation of an individual's genome using microarray technology. The customer is then provided a wealth of genomic information such as susceptibility to various inherited diseases and past ancestry. The ability to review this data over the internet in an interactive fashion has been cleverly described as “recreational genomics”. The customer can then use this information as a basis for a discussion with their physicians to assess their disease risk and modification of lifestyle patterns. DTC services have the financial backing from large technology companies such as Google and the venture capitalists in Silicon Valley, a strong indication of the potential to monetize knowledge that resides in the human genome. While our current understanding of many fundamental mechanisms of cellular function remains unclear, there is strong interest in areas of practical application such as pharmacogenomics to deliver improved patient outcomes compared to the “trial-and-error” approaches of traditional pharmacology.
NEXT GENERATION SEQUENCING
Sequencing technology has evolved at a fast pace over the past decade, with simultaneous advantages in declining costs-per-base sequenced. The genome that cost around 3 billion dollars for the HGP a decade ago can be now sequenced for a few thousand dollars.[4,5] Several NGS technologies have been developed using diverse approaches since 2001, each with its own distinctive strengths and weaknesses. The major commercial entities which came into existence after the success of the HGP include[6] 454 sequencing (http://www.my454.com/), Solexa/Illumina (http://www.illumina.com), SOLiD (http://www.appliedbiosystems.com), and Polonator (http://www.polonator.org/). Intense competition among these so called “second generation” DNA sequencing entities has driven down the cost per Mb of sequence produced. A common feature of the “second generation” DNA sequencing technologies involves the isolation of DNA followed by the creation of single stranded DNA libraries. The libraries are created by the fragmentation of the sample DNA using various techniques. The key differentiating features specific to each commercial platform are in the subsequent steps. The DNA fragments are modified with the ligation of an adapter and amplified using a unique adapter chemistry proprietary to each individual commercial platform. These modified DNA library molecules are then amplified either on a bead (Emulsion based PCR method - 454 and SOLiD) or a glass slide (Bridge amplification -Illumina). The amplified single DNA strands on the bead or glass slide are then paired with complementary DNA nucleotides in individual flow cycles of ATGC templates. A complementary match unique to the DNA template strand results in the release of a signal detected by the sequencing instrumentation.
The original instrumentation developed for the purpose of DNA sequencing had a research focus primarily. This resulted in the development of instrumentation such as the 454 GS FLX from Roche, SOLiD from Life Technologies and the HiSeq series from Illumina. These instruments are capable of extraordinary amount of DNA sequence output per single run [Table 1]. However, due to the large amount of throughput, a single run of this instrumentation is approximately 10 days per instrument (SOLiD and HiSeq). The long duration of runs are clearly incompatible with a rapid turnaround scenario such as clinical sequencing. In addition, the cost of these instruments represents a considerable investment sum for a mid-sized academic institution. In response to these concerns, companies have introduced “bench-top” DNA sequencing instrumentation such as 454 FLX Jr from Roche, MiSeq from Illumina and IonTorrent from Life technologies. The Roche and Illumina sequencers are smaller sized versions of the larger instruments based on the same DNA sequencing technology. IonTorrent from Life Technologies is based on a completely different sequencing methodology which uses a hydrogen ion sensing semiconductor chip. There is much excitement among clinicians and laboratorians regarding this technology due to (1) The relatively low cost of the technology and (2) The rapid turnaround time. IonTorrent and MiSeq instruments can complete the runs within a few hours instead of the 10 days required by bigger instruments [Table 1]. With the low cost and fast turnaround time, it is now feasible to introduce next generation sequencing technology into the clinical workplace to provide clinical care of the patients. The sequencing throughput of benchtop instrumentation is far less compared to the bigger instruments. However, for targeted clinical DNA sequence applications this is unlikely to be a major impediment. Also, the throughput of these instruments is increasing every few months, making the issue of DNA throughput redundant for clinical applications.
Table 1.
Popular NGS platforms currently available in the market. The table shows the characteristic features of the high-end sequencing platforms and the recent “bench-top” platforms

Beyond the 2nd generation and benchtop sequencing instrumentation, the “third generation” sequencers Helicos Heliscope (http://www.helicosbio.com), Pacific Biosciences SMRT (http://www.pacificbiosciences.com) and Oxford Nanopore (http://www.nanoporetech.com) are being actively developed. The 3rd generation instruments differ from the 2nd generation instruments in that the initial DNA amplification step is unnecessary. The sample DNA strands are sequenced directly at the single-molecule level using engineered protein polymerases. The advantage of these methods is the avoidance of PCR amplification bias. However, it is important to note that none of the 3rd generation technologies are currently in mass use for DNA sequencing. The potential of these technologies remains unproven as of current date. The reader is referred to exhaustive reviews of the next generation technology platforms and their unique distinguishing characteristics[6–8] for more details [Table 1].
With the availability of a multitude of platforms and dramatically lower costs of sequencing, NGS technologies are expected to have a major impact on the way we practice medicine in the near future; the $1,000-dollar genome is expected to popularize whole-genome sequencing and is very likely be used in routine clinical diagnostics.[9] Whole-genome sequencing will form the basis of the new field of personalized ‘genomic medicine’[10,11] that aims to integrate the clinical symptoms, personal and family history and the patient's genomic DNA sequence to provide healthcare that is personalized and unique.[12,13]
CANCER AND NEXT GENERATION SEQUENCING
Cancer is a major cause of mortality in the United States, second only to cardiovascular disease. The genomic profile of alterations associated with cancer are complex in nature. Multiple alterations occurring in different types of cancer include mutations, translocations, copy number alterations in the form of gains and losses,[14–17] complex karyotypic rearrangements and epigenetic changes. Using NGS to identify the complete DNA sequence of cancer genomes has the potential to provide major breakthroughs in our understanding of the origin and evolution of cancer.[18] Earlier studies using karyotyping and microarrays provided important insights into structural genomic alterations associated with multiple cancer subtypes. Our knowledge of the molecular mechanisms of cancer began with population based studies of family cohorts susceptible to cancer. Previous epidemiological studies of family cohorts with an increased inherited susceptibility to breast cancer revealed presence of mutations in the BRCA1 and BRCA2 genes.[19] Other similar studies have shown that mutations in the MLH1 and MSH2 genes are associated with a higher risk of colon cancer.[20] The number of genetic mutations associated with cancer is an ever growing list. While useful, epidemiological studies which provide a molecular basis of cancer[21] are limited by the extent of information obtainable from such a study. Traditionally, most of cancer research has focused on single gene and single pathway analysis whereas cancer is well known to be a far more complex entity involving multiple cell signalling pathways. There is a sore need to understand the global levels of gene expression and multiple cellular signalling pathways in the context of cancer. The ability of NGS technology to deliver information on whole genome sequences of different cancers will be an invaluable tool to the future pathologist and clinician. The data obtained from NGS can provide a comprehensive assessment of the genomic landscape associated with the genesis and evolution of different cancers.[14,18,22–24] Large consortia such as the Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium have been formed to sequence thousands of cancers and generate a freely available dataset of DNA sequence changes in different cancer subtypes.
In order to make sense of the large datasets generated by NGS, there is a crucial need for computational algorithms and software capable of performing large scale informational integration. Cross disciplinary tools such as pathway network analysis and graph theory (from computer science and pure mathematics) will be useful to model regulatory networks and interactions associated with cancer tumors.[11,25] Several groups have used principles of network theory to develop software tools and databases capable of displaying complex biological interactions such as (KEGG,[26] Cytoscape,[27] IPA (http://www.ingenuity.com), STRING,[28] GALAXY[25,29]). These interactions are modeled based on data gathered from extensive review of experimental biological data and current scientific literature which are constantly updated to reflect the advancements in the knowledge database. In one such interesting proposal, Friend and Ideker propose a personalized health model based on the integration of multiple sources of data, including clinical, molecular, environmental and social data which are then integrated using tools of graph networks in a personalized healthcare setting.[30] In their model, future clinicians will use multiple data networks to infer a patient's medical condition and choose a personalized treatment approach.[30] Friend and Ideker envision a future visit to the clinic will be based on a seamless integration of biological knowledge obtained from a network analysis of multitude “omics” sources personalized to each patient. The ability of next generation sequencing to provide data regarding the genomic, transcriptomic and epigenetic makeup of a patient in near real time provides the most promising window into such a personalized healthcare future.[30]
Till date, global analysis of cancers using techniques of NGS has revealed the occurrence of hundreds of genomic variants in each different type of cancer.[15–17,24,31–33] Our current understanding of the biological significance of each of these mutational findings is limited. However, we already possess an expanding choice of investigational therapies capable of targeting a particular cell signalling pathway mutation. Such treatments have on occasion been associated with dramatic clinical responses.[34] However, not all mutations are causative and targetable for treatments. Understanding the nature of these “passenger” versus “driver” mutations in specific signaling pathways is an active area of research in cancer genomic biology. NGS is well suited to assess global driver gene expression patterns in tumors by sequencing cDNA and has the potential to provide an enhanced understanding of tumor biology at an individual level. With increasing knowledge of the mechanisms of these driver mutations, clinicians should be able to provide tailored therapeutic choices on a patient by patient basis in the near future.
At the current time, useful but still limited multi-gene biomarker panels such as the 21-gene, OncotypeDX from Genomic Health and the 70 gene, Mammaprint gene expression array from Agendia are available commercially. These panels are used to assess risk for recurrence of cancer and make treatment decisions whether to deliver adjunct chemotherapy in breast cancer patients or not (http://www.agendia.com/pages/mammaprint/21. php). In contrast, data from NGS platforms can provide gene expression information from several thousands of genes simultaneously, increasing the power of prediction exponentially compared to limited gene panel expression assays such as Oncotype DX and Mammaprint. With suitable computational algorithms and computing power, there is the very real chance that NGS will replace many of the current, limited cancer biomarker test panels such as OncotypeDX and Mammaprint in the foreseeable future.
NGS DATA ANALYSIS WORKFLOW
Once the raw sequence data is obtained from the NGS instrument, the computationally intensive step of read mapping is performed. The raw data is usually in the form of a text file which contains “reads” which are short sequences of DNA letters corresponding to the nucleotides incorporated during the process of sequencing. Each data output file contains millions of these “reads” in each data file. The mapping software then attempts to “map” the individual sequence NGS “reads” onto a reference genome sequence available from online genome databases. This process is known as reference mapping. Alternatively, when the reference genome is unknown apriori, the individual “read” fragments are linked to each other by overlapping the common sequences at the ends of each read to form a longer, much complete version of the genome under study. This is known as denovo mapping of DNA sequence. For the purposes of clinical sequencing, reference mapping is performed most of the time due to our pre-existing knowledge of the human reference genome sequence.
The latest version (GRCh37) of the human reference genome is provided by the Genome Reference Consortium (see http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/index.shtml for details). An important factor in the processing and analysis of any large dataset, such as next generation sequencing data, is the presence or absence of inherent parallelism of the data. Data parallelism is a concept that alludes to the self-similar nature of the individual components of the dataset under study. Next generation sequencing data is usually a text (or binary) file in the FASTQ format.[35] Since all of the raw NGS data is a collection of similar FASTQ lines of text in each individual read, we may consider NGS data to be inherently self-similar. It is then possible to use a computing solution to map the NGS reads onto the reference sequence by allocating separate computational processors to deal with different chunks of the self-similar NGS read data. Once the data is mapped onto the reference genome by different processors, the mapped read data is then aggregated by a head computing node to provide the final, mapped genomic sequence desired. The computer processing time required for the read mapping of the data varies depending on the size of the genome that needs to be mapped. More importantly, the computational power required for the mapping varies depending on the type of the mapping performed. In the case of reference mapping, where a pre-existing reference genome sequence is already present, the computational power and time required for the mapping process is not huge. Data generated from a bench-top sequencer can be easily mapped using multicore processor. Most of the bench top sequencer data reads need a reasonably powerful desktop solution (8-12 multicore, 96-256 GB RAM) to perform the mapping of sequence data. The computational requirements for a whole genome are proportionately higher due to the larger size of the whole genome (3GB versus the 40-60 MB for the human exome and a few MB in the case of a routine clinical gene panel). In the case of the whole genome, a parallel processing solution composed of multiple nodes with larger RAM might be adequate to perform reference mapping.
In contrast, there is a vastly increased need for computing power in the case of denovo sequencing where the genomic sequence is unknown apriori. Denovo sequencing also requires longer read lengths and larger computational power to perform the overlapping process efficiently. Denovo read mapping is a much more computationally intensive process compared to the reference mapping process.
With the availability of increased computing power (a cheap commodity nowadays) it is possible to routinely implement NGS technology in a medium sized facility such as an academic centre if the DNA sequencing instrumentation is available. A potential schematic illustrating the NGS workflow process is shown in Figure 1. The goal of clinical NGS is the identification of point mutations and potentially larger structural changes such as translocations, rearrangements, inversions, deletions and duplications either in germline samples or in tumor samples paired with normal genomes for cancer diagnostics. While there are few NGS instruments lending to standardization of some sort, the same cannot be said for the software used to analyse the data. There is a vast ecosystem of bioinformatics software available for the purpose of analysis of DNA sequencing data (for e.g. see http://seqanswers.com/wiki/Software/list). In order to make the implementation of clinical NGS possible, there is a need for standardization of bioinformatics NGS software in the clinical workplace. At present, an array of free and commercial software is used for the purpose of NGS data analysis which is described in the next section.
Figure 1.
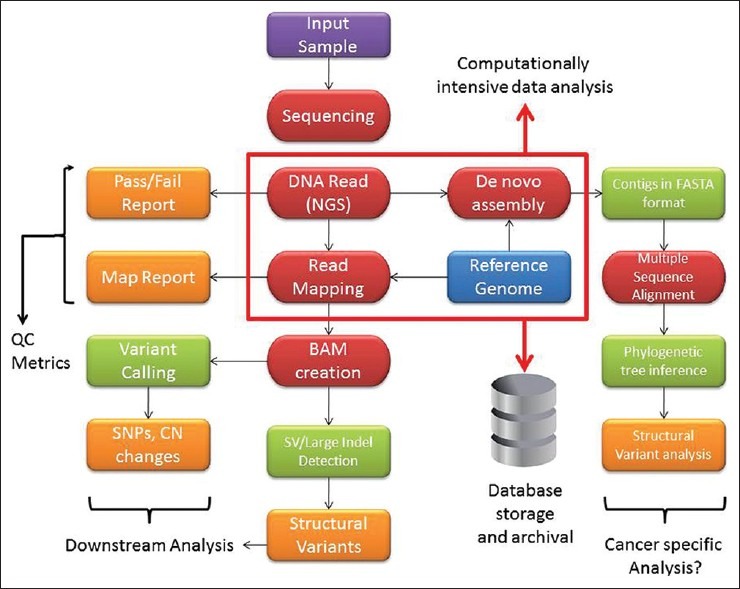
Cancer genome analysis workflow. Various aspects of the workflow start from obtaining the clinical sample to examining the reads for possible variants in the genome
ALIGNMENT AND ASSEMBLY
The first step of NGS data processing is the alignment of reads obtained followed by assembly of the genome of the cancer sample. In cancer NGS, the raw NGS reads must be aligned to a specific location on the various chromosomes to recreate the structural variation within the cancer genome. Currently, there are multiple read mapping software tools available suited for this step. Some of the freely available versions include MAQ,[36] BWA,[37] Bowtie,[38] SOAP,[39] ZOOM,[40] SHRiMP[41] and Novoalign. Instrument vendors such as Illumina and SOLiD provide their own alignment software which may be used for the purpose of read mapping. Commercial, third party vendors such as CLC Genomics provide another avenue for software for the purpose of NGS read mapping. All the different sources have their inherent advantages and disadvantages. Open source software, while free, suffers from the lack of extensive documentation and necessitate the end-user to figure out the available options in the software. Open source tools are often written with the UNIX command line user in mind. Lack of UNIX command skills can be a particularly significant disadvantage in the use of open source NGS tools. Nevertheless, they can provide a significant return if sufficient time is invested in learning the *nix platforms. Commercial vendors provide proprietary algorithms which are optimized to map NGS data. However, the software may be expensive and out for reach for the mid-level academic institutions. It is noteworthy that the open source software have much more frequent updates of algorithms compared to commercial solutions which is a potential advantage in a fast moving field such as NGS. Open source NGS tools such as BWA,[37] Bowtie[38] and SOAP,[39] which are based on the Burrows-Wheeler transformation (BWT) algorithm, perform the mapping process extremely fast which may be an advantage in a situation such as clinical NGS where turnaround time is critical. Softwares based on the BWT algorithm represent the next step in the evolution of the alignment algorithms and can map a human genome in a matter of hours instead of the several days required by other software tools such as MAQ[36] and Novoalign (see Bao et al. for a detailed comparison of mapping software speeds).[42]
An important additional consideration in the analysis of cancer genomes is the need to detect unique rearrangements and accurately map the chromosomal breakpoints within an individual's cancer sample. Software which can perform de novo assembly of cancer genomes is likely to be a much more powerful tool although current algorithms used for de novo assembly are quite slow. These include software such as Velvet,[43,44] EULER-SR,[45] EDENA,[46] QSRA,[47] AbYSS,[48] AllPathsLG (http://www.broadinstitute.org/software/allpaths-lg), Ray and SoapDeNovo (http://soap.genomics.org.cn). However, it is important to remember that most of the software used for the purpose of mapping has its own advantages and disadvantages. The complexity of the assembly pipelines including proper metrics to perform quality assurance are a crucial element in setting up a clinical bioinformatics pipeline. Currently, it is up to the implementing clinical lab to ensure adequate quality of the DNA sequence that is produced and analysed. National organizations such as the College of American Pathologists are in the process of formulating clinical guidelines for NGS, though it is a work in progress at the present moment (personal communication). With improving algorithms for analysis, improved NGS techniques such as paired end mapping and the implementation of strict QA criteria, there is hope for the routine detection of large scale complex structural variation in cancer in the clinical setting.
VARIANT DETECTION
Once the alignment process is completed, downstream bioinformatics analysis is performed to detect the clinically relevant structural genomic alterations. Different software programs are designed to detect different kinds of structural variants. NGS methods can provide a diverse array of information regarding the cancer genome; the primary genomic alterations are described in detail below first followed by the software used for detecting these variants:
Single nucleotide polymorphisms (SNPs) and point mutation discovery
Molecular diagnostics assays for cancer typically have focused on the discovery of mutations in particular pre-identified genes or small panels of genes (for e.g., Ion Ampliseq from IonTorrent). As an example, certain mutations in the epidermal growth factor receptor (EGFR) gene are associated with favorable responses in certain lung cancers treated with targeted therapies like gefitinib compared to lung cancers with wild type EGFR.[49] With NGS technologies, it is possible to scan the entire genome for the presence or absence of mutations in an unbiased fashion. Of all the structural variants associated with a cancer genome, SNPs are the most reliably detected variants in the genome and the most abundant. Additional genomic variants such as indels are equally important which are relatively difficult to identify. Variants such as indels need to be rigorously looked for in the context of clinical NGS. Another impediment to successful and reliable identification of somatic mutations in cancer is the presence of contaminating normal cells. A solution to circumvent this problem is to use laser capture microdissection to obtain DNA from a population highly enriched for cancer cells. Simultaneous analysis of a normal sample from a patient provides a baseline germline sequence to compare against the cancer genome and detect variations.
There are a variety of software tools which are used for the detection of single nucleotide variants in NGS data. Open source tools such as SAM tools,[50] use principles of Bayesian detection to detect the somatic SNP variants in the NGS data analyzed. It is important to remember that a majority of these programs are used for variant discovery in germline DNA and not cancer DNA. Efficient tools which are capable of detecting somatic variation in cancer genotypes are currently in various stages of development. Most of the software packages used for somatic variant detection are based on different statistical models of base calling. Examples of such software include SNVmix,[51] VarScan[52] and SomaticSniper (http://gmt.genome.wustl.edu/somatic-sniper/current/).
Higher order variation in the cancer genome
Cancer genomes are highly unstable leading to diverse chromosomal abnormalities such as large insertions and deletions of chromosomal material. Karyotyping, which was for a long time the standard way to identify the presence of chromosomal abnormalities, suffers from the inability to identify structural abnormalities smaller than ~5 megabases. SNP and oligonucleotide microarrays have revolutionized the field of cytogenetics by providing a high resolution (a few kb) capability to identify large and small copy number variants as well as areas of copy neutral loss of heterozygosity. NGS technologies also can identify structural variations in the genome, although routine alignment tools are ill-equipped to perform such analysis since they cannot identify more than a few nucleotide mismatches. Specialized software for analyzing indels from paired-end reads such as Pindel[53] are being developed which enables identification of structural variants by identifying the flanking end regions of the NGS read data. Another noteworthy tool used for this purpose is the GATK indel genotyper[54] from the Broad Institute, which employs heuristic cutoffs for indel calling. Even then, calling larger amplifications and deletions in cancer chromosomes remains a formidable challenge at the present time using NGS technology. Various algorithms are being developed to identify these larger variations. For example, the circular binary segmentation algorithm of arrays was adopted by Campbell et al.[55] The SegSeq algorithm uses a merging procedure to join localized SNP changes with whole chromosome changes to compare tumor to normal samples.[56] A number of other programs are also available to identify large scale structural variations in the genome, such as BreakDancer[57] which can identify candidate structural variants. While the experimental resolution of NGS technologies is in no doubt and has been successfully utilized to identify variations in different types of cancer such as lung cancer, melanomas and breast cancer at a single nucleotide level,[14,16,18,58,59] significant hurdles still remain in scaling these algorithms to the chromosomal level.
BEYOND SEQUENCING THE GENOME
The initial enthusiasm associated with sequencing the human genome in 2001 has not surprisingly become tempered with the realization that the underlying cell biology is not solely dependent on the genome sequence alone. The ENCODE project is one effort that is aimed at understanding these multiple layers of biologic complexity.[60–63] In this context, NGS platforms offer additional versatility and application in transcriptomic profiling (as demonstrated by Mortazavi et al[64]), chromatin immunoprecipitation, small RNA's and epigenomics studies (discussed below). Transcriptomics via NGS can also be employed to probe alternate splicing, the process by which multiple RNA isoforms (and hence proteins) can arise from a single gene. These isoforms contribute to the specificity of individual cell types and most likely play a significant role in the specificity of cancerous cell types. Identification of novel splicing variants is important for understanding biological specificity in the context of normal and abnormal cellular function. Software tools such as TOPHAT,[65] facilitate de novo discovery of splicing variants.
In recent years, the role played by small RNAs (18- 35 bp) in the regulation of gene expression has become increasingly recognized. Small RNAs play an important role in the regulation of expression and translation of mRNAs, and thus, the functionality of cells where they are expressed. NGS instruments can perform deep sequencing of small RNA species for discovery and analysis. There is a specific advantage to short read platforms such as Illumina and SOLiD in small RNA discovery due to the short nature of small RNAs. There are many small RNA databases along with bioinformatics tools such as MirCat[66] and mirDeep[67,68] which facilitate the identification and discovery of small RNAs. For a comprehensive listing of noncoding RNAs, the reader is referred to online database of NONCODE (http://www.noncode.org/NONCODERv3/).
Epigenomics deals with the chemical modifications (e.g., 5′ methylation) of DNA and RNA and the impacts of such changes on levels of gene expression. The epigenomic status of individual genes determines the overall tumor prognosis.[69] The traditional method of assessing methylation status of a gene is to use bisulfite treatment which converts unmethylated (but not methylated) cytosines to uracil which are then identified using methods such sequencing or restriction endonuclease analysis. A pitfall associated with these methods is the labor intensive process required to identify epigenetic changes on an individual gene basis. NGS offers the potential to explore broad changes in DNA methylation pattern across the entire genome as a part of a single experiment. It should thus be possible to capture epigenetic information from multiple genes at once and, in theory, provide enhanced information content in the prognostication of the tumor methylation status.
The versatility in NGS platforms to examine a variety of cellular properties coupled with its rapidly falling costs, thus offers an integrated and efficient platform beyond determination of genomic sequence alone.
INSTITUTIONAL LEVEL CHALLENGES FOR THE IMPLEMENTATION OF CLINICAL NGS SEQUENCING
At the present, there are multiple commercial NGS operations in existence which perform clinical NGS as a service, such as Illumina clinical genome service, Complete Genomics, Seqwright and Beijing Genomics Institute to name a few. In addition, there is a fast developing interest in academic centres to establish NGS facilities for clinical next generation sequencing. However, establishing a clinical NGS facility at a mid-sized academic centre is not a trivial undertaking. Some of the main challenges involved include: (i) NGS technology overhead - space, power, tools, infrastructure for NGS. (ii) Need for computational architecture for data analysis. (iii) Data archival and retrieval facilities. This is an important issue for clinical NGS due to the turnaround of the number of patients and samples involved. (iv) Computational tools for data processing and management, (v) Identification and training of technical and bioinformatics personnel, and vi) Building a pipeline for acquisition and management of properly consented samples.
Bioinformatics is the single largest bottleneck in the routine implementation of next generation sequencing in clinical practice at the current time. A general guideline is that every dollar spent on sequencing hardware must be matched by a comparable investment in informatics.[70] Some of the considerations in implementing a clinical NGS facility from scratch are described below in detail.
NGS hardware implementation requires substantial in-house investment for the necessary infrastructure, or alternatively can be outsourced to a third party vendor. However, this is an issue which is becoming less crucial with the advent of bench top NGS instrumentation. A mid-sized hospital can obtain a Illumina MiSeq for $125,000 or an IonTorrent for $50,000 at the present time. One advantage of having an in-house clinical NGS sequencing capability is that it enables greater control over the nature of data generated. This is critical for applications such as clinical sequencing, where process quality control is the utmost priority for medico-legal reasons. Outsourcing to a commercial vendor can make it more difficult to understand the data produced.
Availability of computational resources required for NGS data analysis- Availability of in-house, institutional computer clusters in a university centre greatly facilitates the NGS data analysis step. While the availability of such resources is crucial for purposes of whole genome sequencing in the context of research, they are not as critical for facilities using a bench-top sequencer for purposes of clinical NGS. Most of the data generated from a bench-top sequencer can be easily analyzed with a high end desktop server. Institutional computer clusters have the advantage of scheduled maintenance, professional back-up facilities, and dedicated and shared nodes for research. However, the use of such facilities for the purpose of clinical NGS where there are issues of HIPAA violation of patient information, it is preferable to use an in-house computing facility. Most academic centres have existing centralized computing resources which can be readily leveraged for the purpose of in-house NGS analysis. Figure 1 shows a schematic of a routine bioinformatics workflow required for analysis of NGS data.
-
Long-term storage of clinical NGS data – The rate of growth of the sequencing data generation has outstripped the commonly used Moore's law paradigm for measuring the rate of growth of computational hardware speed [Figure 2]. Moore's law states that the rate of growth of computers doubles every 18 months which is particularly relevant to the field of NGS due to needs for computational power to collect, analyze and manage NGS data.
The amount of data generated by NGS for purposes of whole genome sequencing for research is normally in the range of terabytes,[71] and is dependent on the scientific workflow. In a research setting, it is not unreasonable to imagine a core facility producing many terabytes and potentially a petabyte of data over the course of a year at a national level centre. However, the cost per MB of hard drive space has been falling dramatically over the last decade, providing cheap storage options for long-term storage of DNA sequencing data [Figure 3]. Data management of this magnitude requires a well-defined policy of efficient NGS data management. Assessment of data storage needs is complicated by variability of data formats and the lack of industry wide standardization for data output from different NGS platforms. Multiple groups have attempted to come up with unified solutions such as the SAM and BAM data formats.[50] SAM stands for Sequence Alignment/Map format, which is a text format for storing sequence data in a series of tab delimited ASCII columns. SAM is typically generated as a human readable version of the BAM (binary alignment map) format, which stores the same data in a compressed, indexed, binary form. The BAM data file format (binary text based format) is reasonably efficient for storage.
For clinical genomic data using NGS, the issues of data storage and management are not as acute due to the small size of data generated using bench-top sequencers. However, with the ever lowering costs of generating NGS data, it is not unrealistic to reach point where a bench-top sequencer may produce data sufficient for whole genomic analysis in the future. Clinical NGS facilities need to plan for such a development and be nimble enough to implement a data storage solution for those needs. One may also foresee a time in the future when it is much cheaper to resequence the patient DNA “on-demand” and circumvent the need for long-term storage. An additional concern related to clinical NGS is that privacy concerns must be addressed to ensure proper storage of NGS data in a HIPAA-compliant manner. In addition, sustained operation of a clinical NGS facility over the long-term requires regular attention to service contracts, equipment turnover, data storage contracts, continuing grant support, as well as potential institutional or charitable support.
-
Cloud computing solutions as an alternative to in-house computational infrastructure - In the last 3-4 years, online computer clusters have become available commercially for public use through the web. This “on-demand” access to supercomputing is referred to as a “cloud computing” solution. Large technology companies such as Amazon, Google and Microsoft have adopted centralization of supercomputing facilities made possible mainly by a technology known as virtualization of software.[72]
Virtualization refers to the process where a user can access an “image” of the operating system (Linux or Windows) residing on a server of a company hosting the cloud. This interface image connects the user's desktop with the company server. This OS “image” is indistinguishable from an ordinary desktop interface with the only difference being that the virtual operating system is hosted on a remote server [Figure 4] and not locally on the user's desktop. The major advantage of using the cloud solution is the availability of supercomputing power without having to install and maintain expensive supercomputing hardware. The fee schedule for these elastic compute cloud services is competitive and affordable for an average user, with pay-as-you-go pricing. The National Science Foundation has initiated a recent program to provide funding and computing cloud access to individual research groups in collaboration with Microsoft. Private vendors such as Amazon S3 also provide long-term storage of datasets using networked storage facilities. This is a critically important given the scale of NGS datasets (often running into petabytes).
There are some disadvantages with cloud services, a major one being, privacy of patient data. Cloud services also necessitate the transfer of patient data over an Internet network which is potentially vulnerable to hacking. Another disadvantage is the need for high capacity networking access. Genomic datasets are often large (in the range of terabytes), making efficient data transfer a challenge. A potential solution is the use of postal mail to send NGS data which is then uploaded to the server by the cloud service provider. For clinical genomic data, an unknown variable is the data policy of the companies running the compute cloud cluster. What happens to this data over the long-term? Would the company retain rights to such data indefinitely? Would it be possible to erase the data upon patient request? Currently, clear guidelines are lacking, though cloud services have begun to address these issues. Since most cloud computing services charge on the basis of data transmitted and computational time, these services may become expensive. This is the reason large centers prefer to develop their own computational clusters and storage facilities.
Analysis and interpretation of NGS data – Routine analysis of NGS data requires multidisciplinary teams of clinical and biomedical bioinformaticians, computational biologists, molecular pathologists, programmers, statisticians, biologists, as well as clinicians. Most facilities are unlikely to have the financial resources to recruit an experienced bioinformatician, so it is important to organically build a group of people trained in-house, including graduate students and postdocs from various backgrounds and with different levels of expertise. Particularly crucial personnel include specialists with core skills in computing systems, programming, biology, and statistics to create an environment that encourages collaboration of ideas for data analysis.
-
Other Considerations - While there are several polished commercial solutions for NGS analysis, as well as several other not-so-polished open-source solutions, some research labs prefer to create custom-solutions for their requirements.[70] These data filters and file converters take information from one format, process it and generate an output in another format. They also guide raw sequence data through other processing steps such as data cleanup, collection of quality metrics, alignment to a reference genome, and others. The challenge with locally-developed solutions is the lack of compatibility with other NGS lab solutions. Also, in the case of clinical NGS, the lack of standardization of data analysis is a major hurdle in the implementation of the technique.
As NGS technology is applied to a wider range of biological and clinical problems, it becomes critical to standardize the quality metrics for the NGS data generated.[7] These include validation and comparison among platforms, data reliability, robustness and reproducibility, and quality of assemblers. Guidelines for standardization of NGS protocols, along the lines of- efforts initiated by the FDA: the microarray quality control (MAQC) project[73] and the sequencing quality control (SEQC) project (http://www.fda.gov/MicroArrayQC/), will be particularly important for NGS going forward.
Figure 2.
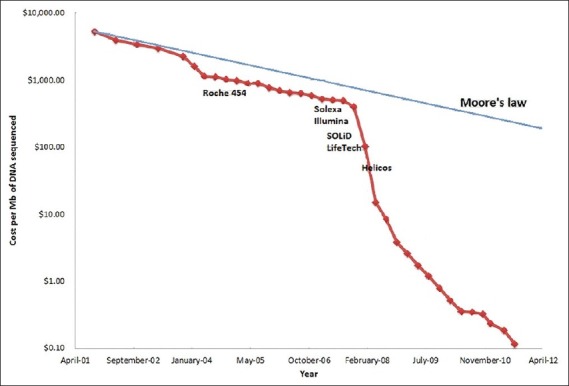
Cost per megabase of DNA sequenced in the last decade. The semi-log plot shows a dramatic reduction in the cost per megabase of DNA sequenced in the last decade. Also shown are the approximate dates of introduction of different NGS instruments by commercial vendors into the market. The costs have fallen dramatically since 2007 due to competition from multiple vendors. Data source – http://www.genome.gov/sequencingcosts/
Figure 3.

Storage and computational processor trends over time. Note the semi-log scale on the y-axis. The linearity of the semi-log plot is in concordance with the Moore's law over time. This is in contrast to the costs of DNA sequencing showing a dramatic reduction in costs [Figure 1]
Figure 4.
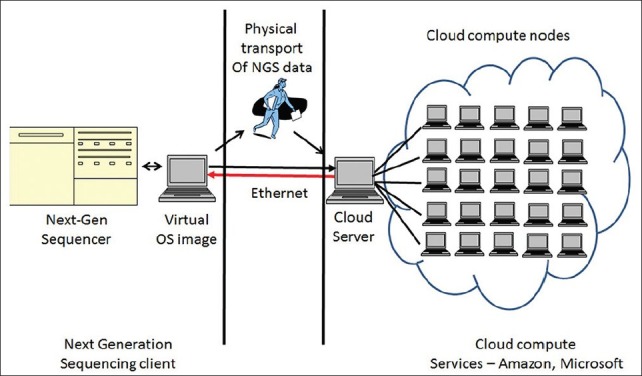
A schematic illustrating the organization of a cloud computing solution for analysis of NGS data
IMPACT OF NGS IN PHARMACOGENOMICS
A frequent criticism of the use of NGS technology in the clinical workplace has been the lack of definitive evidence regarding the utility of NGS data to alter patient outcomes. Much work still needs to be done to establish NGS in a clinical diagnostic and prognostic role. However, one immediate area of application with the potential to alter clinical outcomes favourably would be Pharmacogenomics. The arsenal of potential therapeutic agents available to the clinician has been steadily increasing over the past fifteen years. In parallel, we now have a better understanding of the genes involved in the metabolism of drugs. Pharmacogenomics is the area of study that uses the knowledge of specific genetic variations to provide a “personalized” approach to treatment and dosing of patients with cancer and other clinical disorders. Previously, the main stay of cancer treatment was a combination of ‘one size fits all’ modality of surgery, chemotherapy and radiotherapy. We now know that different drugs are metabolized at different rates depending on the polymorphic genetic variation in individuals leading to uneven eradication of cancer cells. Chemotherapy has significant side effects due to the toxicity of the drugs, whose metabolism is influenced by the patient's genetic differences. Cancer cells are prone to develop resistance to chemotherapy due to somatic mutations and could potentially benefit from a targeted therapeutic agent or regimen.
A paradigm of pharmacogenomics is in the treatment of breast cancer in women with a targeted molecular approach. A subset of breast cancers is known to overexpress a protein, Her2/Neu. Such patients respond favourably to Trastuzumab, a drug targeted towards the Her2/Neu receptor. Pharmacogenomic evaluation of Her2/ Neu status before treatment can stratify subsets of breast cancer patients who are likely to respond to Trastuzumab versus patients who are unlikely to do so. Other cancers for which targeted therapies are available based on genetic variations in the tumor or germline include chronic myeloid leukaemia (treated with Imatinib mesylate as well as second generation tyrosine kinase inhibitors), colorectal cancer (treated with an array of therapies including Irinotecan, Cetuximab, Panitumab) and lung cancer (treated with Erlotinib, which is directed towards EGFR over expression in a subset of lung cancers). Companion diagnostics to identify subsets of patients likely to respond to treatment are becoming commonplace.
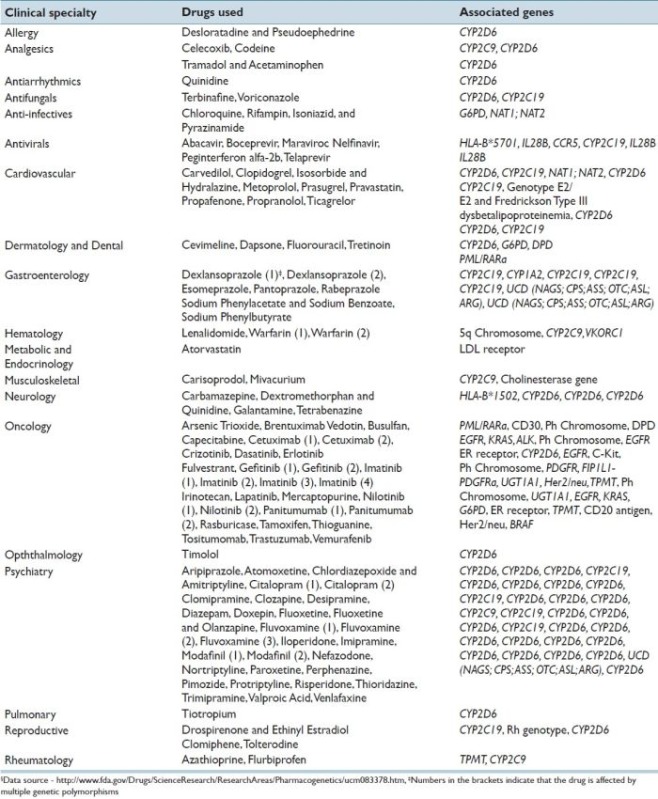
There is great excitement over the prospect of using NGS technology to identify somatic variants to direct changes in therapy early on, as resistant tumor clones begin to emerge. NGS also offers broad potential for pharmacogenetic use in a wide variety of non-neoplastic conditions. Warfarin is an important anticoagulant for patients at risk for venous thrombosis. It has long been known that patients can clinically respond differently to a standard dose of Warfarin. Patients who are sensitive to low doses of Warfarin due to genetic variation are at the risk of catastrophic bleeding which can lead to death. In contrast, there are patients with genetic variants who may be resistant to Warfarin and are at an increased risk of developing clots. The major component of sensitivity to Warfarin has been traced to genetic polymorphisms of the VKORC1 and CYP2C9 genes.[74] NGS technology has the potential to classify the patient's sensitivity status to Warfarin and prevent catastrophic complications. These are just a few examples of the potential role NGS technology can play in the future during the implementation of the personalized medicine. A detailed list of different drugs and their associated genes for currently approved by FDA pharmacogenomic evaluation is provided in Appendix A.
TRAINING AND IMPLEMENTATION OF NGS IN THE CLINICAL WORKPLACE
The traditional model of clinical medicine has relied on bedside interaction of trainees with ‘tutor’ physicians and a hands-on approach to learning medicine. NGS technology is a quantum leap in our ability to provide a comprehensive, molecular scale view of the human genome. These massive datasets contain vital clues that can explain the basis for human disease and provide useful tools for its prognostication. However, we are at the very beginning of obtaining clinical expertise and knowledge to curate and interpret NGS data on a routine basis in the hospital. Barriers hindering the routine implementation of NGS technology in the clinical workplace include lack of correlative data, a lack of physician-friendly computational data analysis tools, and structured training programs and curricula to train physicians in the use and interpretation of NGS and genomic data.
In the near term future, there is going to be a pressing need for such physicians skilled with the interpretation of NGS data. The pressure for such skillsets and training is bound to increase due to the availability of direct-to-consumer (DTC) genetic tests. Once could easily imagine a scenario where a patient walks into a clinic with a report from a DTC firm demanding an explanation of the findings. At present, medical school training is inadequate to deal with complex genomic issues due to the lack of a formal curriculum. The problem is even more acute in the practicing physician who has neither the time nor the resources to develop an understanding of genomics. At present, there are approximately 1,000 medical geneticists and 3,000 genetic counsellors in the United States. These numbers are grossly inadequate to deal with the explosive growth of genomics testing. One feasible solution is to form strategic collaborations between disciplines to deal with genomics.[75] At the present time, there are over 17,000 pathologists in the United States. Pathologists are perfectly suited to take on the role of dealing with the complexities of genomic data interpretation due to their broad training in anatomic pathology and laboratory medicine and their ability to integrate knowledge of large datasets with clinical findings.[75]
The need to train pathologists in the use of computational analysis tools is necessary to manage large datasets such as those encountered in the NGS. There is a critical need to create a subspecialty of “Computational Pathology” to train pathologists with the ability to manage and interpret high-throughput biological data. Computational pathologists would not only understand the basis of molecular testing, but also possess skills for data manipulation, analysis and interpretation and create lab workflows suitable for NGS data analysis. Skills to manage and explore large datasets would be invaluable not only in genomic analysis, but also in high throughput proteomic and metabolomics analysis.
REIMBURSEMENT ISSUES RELATED TO NGS IN THE CLINICAL LABORATORY
An important consideration in the widespread implementation of NGS in the clinical workplace is related to issues of reimbursement. Appropriate reimbursement will be necessary for the widespread adoption of clinical next generation sequencing, so it's notable that the Centre for Medicare and Medicaid Services (CMS) Coverage and Analysis Officer commented in October 2011 “I hope people realize that whole genome sequencing itself is probably something that CMS would never cover.”[76] CMS and other payers are increasingly looking for evidence of clinical utility for services which contribute to patient management and outcomes. The AMA CPT Editorial Panel also expects laboratory procedures seeking a category 1 CPT code to be performed widely and have publications supporting their importance in patient care.[77] Over the past two years the CPT Editorial Panel has been revising billing codes to make molecular pathology services more transparent; it's interesting that next generation sequencing is mentioned in the description of service for one assay (Long QT Syndrome) which assesses a medium-sized panel of genes.
The charge for the clinical and laboratory community seems straightforward: demonstrate that next generation sequencing assays contribute meaningfully to patient care through diagnoses or improved therapeutic decisions that cannot currently be made and/or savings compared to existing test strategies to include the retirement of other tests no longer needed. Published next generation experiences till date have been impressive case reports, but series of consecutive patients in whom actionable variations can be consistently identified for specific indications will likely be needed to change the mind of the CMS Coverage and Analysis Officer.
An interesting concept is the use of whole genome or exome sequencing as a one-time ‘universal’ and ‘reusable’ laboratory test. A relevant subset of genomic information would be useful for an initial episode of care; other subsets could be interpreted for subsequent medical issues without again performing the technical portion of the assay. The interpretation of next generation sequence data is complex and requires a trained professional, the reason most professional organizations have recommended that CPT codes for next generation sequencing be placed on the Physician Fee Schedule.
OWNERSHIP AND PRIVACY CONCERNS RELATED TO NGS
An important issue related to the clinical implementation of NGS is ownership of genomic data. Questions include, does a patient exclusively own his/her digital data? Does the facility, either a hospital or a commercial lab, own this DNA data? If so, what rights do they possess over the data? Can a facility commercially “lease” the sequencing data to third-party data miners? Is it legally possible to expunge the sequencing data on concerns of abuse of genomic data? Would an institutional review board (IRB) kind of a model work to govern the proper usage of these data? What rights do physicians have regarding the exploration of such data? Is clinical data exploration limited only to diseases and subsets for which the patient presents for evaluation and management? Is the pathologist obligated to report findings elsewhere in the genome unrelated to the current focus of investigation? Like many other areas of science, NGS technology is far ahead of the law. We need to carefully consider the social and societal implications of such a revolutionary technology in greater detail with proper oversight.
The Health Insurance Portability and Accountability Act (HIPAA) enacted by the U.S. Congress in 1996 deals with privacy concerns related to patient data. Title II explicitly deals with issues related to establishment of national standards of electronic health care for providers, insurance companies and employers. The “privacy rule” establishes guidelines and regulations for the use and disclosure of protected health information (PHI). In the context of genomics, all of the regulations of HIPAA apply to currently used Sanger sequencing. However, one could easily imagine practical HIPAA hurdles related to NGS data. Clinical NGS datasets are extremely large and may require transfer to sites beyond an institutional firewall into the cloud for further processing. The potential for misuse of NGS data by commercial entities is a real concern if adequate safeguards are not in place. Commercial vendors of computational services are cognizant of the ethical implications of patient sensitive data, and governmental guidelines regulating the storage and transfer of NGS data by commercial and academic users are sorely required. A temporary solution to ensure compliance with HIPAA regulations would be to anonymize NGS data using identifier numbers and remove all patient identifiers. However, this is a temporary fix, and permanent solutions need to be researched to solve this problem.
FUTURE OF PERSONALIZED GENOMIC MEDICINE
The future of genomic sequencing is very bright. The ability of NGS to integrate a diverse set of data analysis techniques into a single platform (i.e., DNA-seq, RNA-seq, transcriptome analysis, methylation analysis) is a revolutionary development in the field of biology. As an analogy, electronic signal processing moved from the analog into the “digital” domain when the personal computer was invented. Digitization of signals obviated the need for specialized analog processing hardware for different kinds of instruments. The “brains” of the instrument moved to a single platform (i.e., personal computer) capable of controlling a variety of physical instruments. The current situation in biology and medicine is somewhat similar. Traditional biological research was and is mostly performed in a linear fashion. Techniques of molecular biology study cellular physiology on a gene-by-gene basis to understand cell signal transduction. With the advent of microarrays, studies of cell biology have become more parallelized, albeit still limited by the number and type of probes available on the array. NGS overcomes the limitations of microarray studies and represents the next step in the advancement of biological discovery which is “independent” of the hardware (molecular biology). NGS technology provides biological knowledge discovery in a digitized, massively parallel fashion. We are at the beginning of a revolution in high throughput, massively parallel biological data discovery.
The advent of large dataset biological data has necessitated the need for development of computational tools required to handle and understand this data. Progress is being slowly made on that front using techniques of systems biology. Our ability to mine this vast array of digitized biologic data and correlate it with the accumulated datasets of other individuals provides an enormous potential to create models of “personalized” genomic approaches to patient care and management. In the specific case of cancer, it is important to remember that the significance of genomic sequence data is only one component of the biologic behaviour of individual cancers. There is a need to create specific protocols to analyse correlations across individual cancer subtypes and across populations of cancer types to unlock the true potential of NGS technology approaches.
NGS presents a huge opportunity for interdisciplinary teams of scientists, physicians, computer scientists, statisticians, bioinformaticians, mathematicians, and biologists to formulate personalized approaches to the treatment of cancer and revolutionize the way we practice clinical medicine. Wet-bench biology provides the feedback loop to NGS data enabling predictive modeling and an understanding of the fundamental aspects of cancer. As bioinformatics tools become increasingly sophisticated and user friendly, computation assisted medicine is bound to become a reality for the future physician. In the near future (~ next 5-10 years) we will witness a dramatic revolution in our knowledge of biology due to NGS technologies and its’ impact on the way clinical medicine is practiced.
APPENDIX A
Appendix A: List of drugs currently approved by the Food and Drug Administration (FDA) with associated pharmacogenomic information. Specific nucleic sequence variants (genetic polymorphisms) in genes lead to varying metabolism and/or distribution of individual drugs§.
ACKNOWLEDGEMENTS
The authors would like to acknowledge the following individuals of the Pittsburgh NGS task force for their role in the planning and execution of the 25th UPCI Cancer symposium- William Bigbee, Michelle Broido, Uma Chandran, Tianjiao Chu, Nancy Davidson, Maryann Donovan, Bino John, Bill LaFramboise, Adrian Lee, Cecelia Lo, Xinghua Lu, George Michalopoulos, Patrick Moore, David Moses, David Peters, Marjorie Romkes, Ralph Roskies, Yoel Sadovsky, Irtisha Singh, Ashok Srinivasan, Scot Stevens, and Paul Wood.
In memoriam
One of the contributors of the current article, Dr. Jeffrey A Kant, passed away due to a sudden illness in the month of September while the current article was in review. This article is dedicated to his memory and his lasting accomplishments in the field of molecular pathology.
Footnotes
Available FREE in open access from: http://www.jpathinformatics.org/text.asp?2012/3/1/40/103013
REFERENCES
- 1.Maxam AM, Gilbert W. A new method for sequencing DNA. Proc Natl Acad Sci U S A. 1977;74:560–4. doi: 10.1073/pnas.74.2.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanger F, Coulson AR. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol. 1975;94:441–8. doi: 10.1016/0022-2836(75)90213-2. [DOI] [PubMed] [Google Scholar]
- 3.Watson JD. The human genome project: Past, present, and future. Science. 1990;248:44–9. doi: 10.1126/science.2181665. [DOI] [PubMed] [Google Scholar]
- 4.Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science. 2010;327:78–81. doi: 10.1126/science.1181498. [DOI] [PubMed] [Google Scholar]
- 5.Zhou X, Ren L, Li Y, Zhang M, Yu Y, Yu J. The next-generation sequencing technology: A technology review and future perspective. Sci China Life Sci. 2010;53:44–57. doi: 10.1007/s11427-010-0023-6. [DOI] [PubMed] [Google Scholar]
- 6.Mardis ER. Next-Generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- 7.Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–45. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 8.Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 9.Service RF. Gene sequencing. The race for the $1000 genome. Science. 2006;311:1544–6. doi: 10.1126/science.311.5767.1544. [DOI] [PubMed] [Google Scholar]
- 10.Kahvejian A, Quackenbush J, Thompson JF. What would you do if you could sequence everything? Nat Biotechnol. 2008;26:1125–33. doi: 10.1038/nbt1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boguski MS, Arnaout R, Hill C. Customized care 2020: How medical sequencing and network biology will enable personalized medicine. F1000 Biol Rep. 2009;1:5. doi: 10.3410/B1-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bryer S. The case for personalized medicine. Personalized Medicine Coalition. 2009:1–24. [Google Scholar]
- 13.Diamandis M, White NM, Yousef GM. Personalized medicine: Marking a new epoch in cancer patient management. Mol Cancer Res. 2010;8:1175–87. doi: 10.1158/1541-7786.MCR-10-0264. [DOI] [PubMed] [Google Scholar]
- 14.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–60. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–20. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A, et al. Mutational evolution in a lobular breast tumor profiled at single nucleotide resolution. Nature. 2009;461:809–13. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- 19.Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet. 1998;62:676–89. doi: 10.1086/301749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gille JJ, Hogervorst FB, Pals G, Wijnen JT, van Schooten RJ, Dommering CJ, et al. Genomic deletions of MSH2 and MLH1 in colorectal cancer families detected by a novel mutation detection approach. Br J Cancer. 2002;87:892–7. doi: 10.1038/sj.bjc.6600565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seng KC, Seng CK. The success of the genome-wide association approach: A brief story of a long struggle. Eur J Hum Genet. 2008;16:554–64. doi: 10.1038/ejhg.2008.12. [DOI] [PubMed] [Google Scholar]
- 22.Welch JS, Westervelt P, Ding L, Larson DE, Klco JM, Kulkarni S, et al. Use of whole-genome sequencing to diagnose a cryptic fusion oncogene. JAMA. 2011;305:1577–84. doi: 10.1001/jama.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schweiger MR, Kerick M, Timmermann B, Isau M. The power of NGS technologies to delineate the genome organization in cancer: From mutations to structural variations and epigenetic alterations. Cancer Metastasis Rev. 2011;30:199–210. doi: 10.1007/s10555-011-9278-z. [DOI] [PubMed] [Google Scholar]
- 24.Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–72. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hawkins RD, Hon GC, Ren B. Next-generation genomics: An integrative approach. Nat Rev Genet. 2010;11:476–86. doi: 10.1038/nrg2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–60. doi: 10.1093/nar/gkp896. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–8. doi: 10.1093/nar/gkq973. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, et al. Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 2005;15:1451–5. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Friend SH, Ideker T. POINT: Are we prepared for the future doctor visit? Nat Biotechnol. 2011;29:215–8. doi: 10.1038/nbt.1794. [DOI] [PubMed] [Google Scholar]
- 31.Bass AJ, Lawrence MS, Brace LE, Ramos AH, Drier Y, Cibulskis K, et al. Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A-TCF7L2 fusion. Nat Genet. 2011;43:964–8. doi: 10.1038/ng.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Timmermann B, Kerick M, Roehr C, Fischer A, Isau M, Boerno ST, et al. Somatic mutation profiles of MSI and MSS colorectal cancer identified by whole exome next generation sequencing and bioinformatics analysis. PLoS One. 2010;5:e15661. doi: 10.1371/journal.pone.0015661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones SJ, Laskin J, Li YY, Griffith OL, An J, Bilenky M, et al. Evolution of an adenocarcinoma in response to selection by targeted kinase inhibitors. Genome Biol. 2010;11:R82. doi: 10.1186/gb-2010-11-8-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cock PJ, Fields CJ, Goto N, Heuer ML, Rice PM. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010;38:1767–71. doi: 10.1093/nar/gkp1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–8. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li R, Li Y, Kristiansen K, Wang J. SOAP: Short oligonucleotide alignment program. Bioinformatics. 2008;24:713–4. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- 40.Lin H, Zhang Z, Zhang MQ, Ma B, Li M. ZOOM! Zillions of oligos mapped. Bioinformatics. 2008;24:2431–7. doi: 10.1093/bioinformatics/btn416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rumble SM, Lacroute P, Dalca AV, Fiume M, Sidow A, Brudno M. SHRiMP: Accurate mapping of short color-space reads. PLoS Comput Biol. 2009;5:e1000386. doi: 10.1371/journal.pcbi.1000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bao S, Jiang R, Kwan W, Wang B, Ma X, Song YQ. Evaluation of next-generation sequencing software in mapping and assembly. J Hum Genet. 2011;56:406–14. doi: 10.1038/jhg.2011.43. [DOI] [PubMed] [Google Scholar]
- 43.Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zerbino DR. Using the Velvet de novo assembler for short-read sequencing technologies. Curr Protoc Bioinformatics. 2010 doi: 10.1002/0471250953.bi1105s31. Chapter 11:Unit 11.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chaisson MJ, Pevzner PA. Short read fragment assembly of bacterial genomes. Genome Res. 2008;18:324–30. doi: 10.1101/gr.7088808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hernandez D, Francois P, Farinelli L, Osteras M, Schrenzel J. De novo bacterial genome sequencing: Millions of very short reads assembled on a desktop computer. Genome Res. 2008;18:802–9. doi: 10.1101/gr.072033.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bryant DW, Jr, Wong WK, Mockler TC. QSRA: A quality-value guided de novo short read assembler. BMC Bioinformatics. 2009;10:69. doi: 10.1186/1471-2105-10-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. ABySS: A parallel assembler for short read sequence data. Genome Res. 2009;19:1117–23. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 50.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goya R, Sun MG, Morin RD, Leung G, Ha G, Wiegand KC, et al. SNVMix: Predicting single nucleotide variants from next-generation sequencing of tumors. Bioinformatics. 2010;26:730–6. doi: 10.1093/bioinformatics/btq040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, et al. VarScan: Variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25:2283–5. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–71. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109–13. doi: 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chiang DY, Getz G, Jaffe DB, O’Kelly MJ, Zhao X, Carter SL, et al. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Methods. 2009;6:99–103. doi: 10.1038/nmeth.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, et al. BreakDancer: An algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6:677–81. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Berger MF, Levin JZ, Vijayendran K, Sivachenko A, Adiconis X, Maguire J, et al. Integrative analysis of the melanoma transcriptome. Genome Res. 2010;20:413–27. doi: 10.1101/gr.103697.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–8. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–40. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 61.Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guigo R, Flicek P, Abril JF, Reymond A, Lagarde J, Denoeud F, et al. EGASP: The human ENCODE genome annotation assessment project. Genome Biol. 2006;7(Suppl 1):S21–31. doi: 10.1186/gb-2006-7-s1-s2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thomas DJ, Rosenbloom KR, Clawson H, Hinrichs AS, Trumbower H, Raney BJ, et al. The ENCODE Project at UC Santa Cruz. Nucleic Acids Res. 2007;35:D663–7. doi: 10.1093/nar/gkl1017. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mortazavi A, Schwarz EM, Williams B, Schaeffer L, Antoshechkin I, Wold BJ, et al. Scaffolding a caenorhabditis nematode genome with RNA-seq. Genome Res. 2010;20:1740–7. doi: 10.1101/gr.111021.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Trapnell C, Pachter L, Salzberg SL. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moxon S, Schwach F, Dalmay T, Maclean D, Studholme DJ, Moulton V. A toolkit for analysing large-scale plant small RNA datasets. Bioinformatics. 2008;24:2252–3. doi: 10.1093/bioinformatics/btn428. [DOI] [PubMed] [Google Scholar]
- 67.Friedlander MR, Chen W, Adamidi C, Maaskola J, Einspanier R, Knespel S, et al. Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol. 2008;26:407–15. doi: 10.1038/nbt1394. [DOI] [PubMed] [Google Scholar]
- 68.Yang X, Li L. miRDeep-P: A computational tool for analyzing the microRNA transcriptome in plants. Bioinformatics. 2011;27:2614–5. doi: 10.1093/bioinformatics/btr430. [DOI] [PubMed] [Google Scholar]
- 69.Etcheverry A, Aubry M, de Tayrac M, Vauleon E, Boniface R, Guenot F, et al. DNA methylation in glioblastoma: Impact on gene expression and clinical outcome. BMC Genomics. 2010;11:701. doi: 10.1186/1471-2164-11-701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Perkel JM. Sequence Analysis 101: A newbie's guide to crunching next-generation sequencing data. The Scientist. 2011;25:60. [Google Scholar]
- 71.Kahn SD. On the future of genomic data. Science. 2011;331:728–9. doi: 10.1126/science.1197891. [DOI] [PubMed] [Google Scholar]
- 72.Fusaro VA, Patil P, Gafni E, Wall DP, Tonellato PJ. Biomedical cloud computing with Amazon Web Services. PLoS Comput Biol. 2011;7(8):e1002147. doi: 10.1371/journal.pcbi.1002147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shi L, Reid LH, Jones WD, Shippy R, Warrington JA, Baker SC, et al. The MicroArray quality control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat Biotechnol. 2006;24:1151–61. doi: 10.1038/nbt1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ashley EA, Butte AJ, Wheeler MT, Chen R, Klein TE, Dewey FE, et al. Clinical assessment incorporating a personal genome. Lancet. 2010;375:1525–35. doi: 10.1016/S0140-6736(10)60452-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Haspel RL, Arnaout R, Briere L, Kantarci S, Marchand K, Tonellato P, et al. A call to action: Training pathology residents in genomics and personalized medicine. Am J Clin Pathol. 2010;133:832–4. doi: 10.1309/AJCPN6Q1QKCLYKXM. [DOI] [PubMed] [Google Scholar]
- 76.Pathologists CoA. STATLINE Archive. Northfield: College of American Pathologists. [Last accessed on 2011 Oct 13]. Available from: http://www.cap.org/apps/cap.portal .
- 77.Association AM. Chicago: American Medical Association; [Last accessed on 2009 Jul 29]. Applying for CPT® Codes. Available from: http://www.ama-assn.org/ama/pub/physicianresources/solutions-managing-your-practice/coding-billing-insurance/cpt/applying-cpt-codes.page . [Google Scholar]