

Abstract
E3 ubiquitin ligases, such as the therapeutically relevant VHL, are challenging targets for traditional medicinal chemistry, as their modulation requires targeting protein-protein interactions. We report novel small-molecule inhibitors of the interaction between VHL and its molecular target HIF1α, a transcription factor involved in oxygen sensing.
Keywords: E3 ubiquitin ligases, structure-based design, protein crystallography, protein-protein interactions, von Hippel-Lindau
Protein-protein interactions (PPIs) are vital to most biological processes, yet despite recent advances they remain notoriously difficult to target due their relatively large surfaces lacking the deep pockets of more tractable targets.[1] While targeting these interactions with large alpha-helical mimics[2–5] has been relatively successful, developing drug-like small molecule inhibitors of PPIs remains highly challenging. Recently, some success has resulted from the use of virtual screening[6], fragment based approaches[7] and the targeting of hot-spots,[8] however the hit rates for protein interfaces remain low.[1c]
One class of PPIs with promising therapeutic potential is that of E3 ligases with their substrates. E3 ligases bind to their protein substrates, allowing E2 enzymes to transfer ubiquitin subunits to the target protein. Due to their control of widespread biological systems E3 ligases make highly desirable drug targets.[9] However, since the discovery of the nutlins, the first small molecule E3 ligase inhibitors[10], only a handful of E3 ligases have been successfully targeted.[11–13]
The von-Hippel Lindau protein (VHL) is a component of a multi-subunit E3 ligase that recognizes the prolyl hydroxylated transcription factor HIF1α and tags it for degradation by the proteasome (Figure 1).[14] However, under hypoxic conditions, the prolyl hydroxylase domain enzymes (PHDs) are unable to hydroxylate HIF1α, resulting in the accumulation of HIF1α and subsequent upregulation of the genes involved in the hypoxic response, including GLUT1, VEGF and erythropoietin. HIF1α stabilization, through the use of PHD inhibitors,[15] is being investigated in the clinic as a possible treatment for chronic anemia.[16] Alternatively, the inhibition of the VHL/HIF1α interaction with peptidic inhibitors fused to the tat translocation domain has been shown to stabilize HIF1α,[17] illustrating that inhibition of this interaction is an alternative or complementary strategy to PHD inhibitors for the treatment of anemia.
Figure 1.
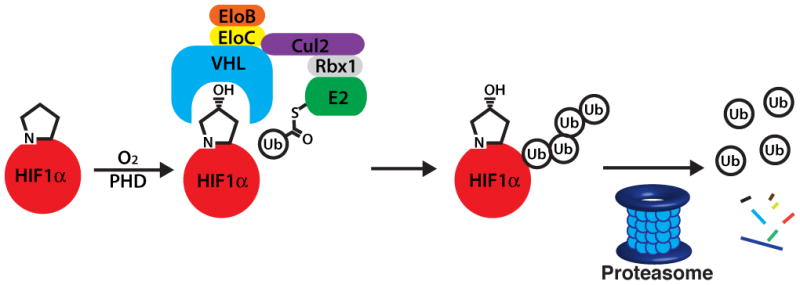
HIF1α is hydroxylated under normoxic conditions, leading to recognition by VHL followed by ubiquitination and degradation by the proteasome.
Recently, we reported a series of VHL ligands, including 1, capable of competitively inhibiting the binding of a fluorescent peptide derived from HIF1α to VHL.[18] These inhibitors contain a hydroxyproline residue, which is crucial for binding to VHL,[19] and an isoxazolylacetamide fragment, which was designed to interact with a water molecule previously identified as an important part of the hydrogen bonding network between VHL and HIF1α.[20] However, these molecules bound with limited potency and only a small number of analogues were made, hindering the ability to draw conclusions about structure-activity relationships (SAR). Herein we report a detailed study of VHL ligand SAR, including the discovery of N-terminal fragments with an alternative binding mode, as shown by X-ray crystallography. The optimization of both the C and N terminal fragments, followed by their combination, yielded our most potent ligand to date, which binds with a submicromolar IC50.
While optimizing the C and N fragments for affinity, we sought to minimize differences in ligand solubility by testing binding affinity in a fluorescence polarization competition assay using 10% DMSO, as opposed to the more physiologically relevant 1% DMSO.[18] While general trends in affinity were similar under both conditions, we found that in cases where solubility was not an issue, ligands had lower IC50 values in 1% DMSO.
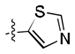
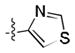
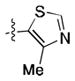
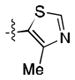
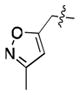
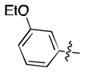
After the discovery of 1,[18] we sought to systematically investigate other 5-membered heteroaromatic substituents (Table 1). After examining various oxazoles (1, 2, 3) and thiazoles (4, 5, 6, 7), we found that the original substitution at the 5 position of the heteroaromatic substituent and at the para position of the aryl ring was optimal.
Table 1.
Optimization of the C-terminal Fragment
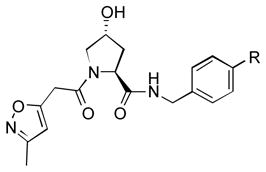
| |||
|---|---|---|---|
| # | R (para) | IC50 (μM) [a] (10%DMSO) | IC50 (μM) [a] (1% DMSO) |
| 1 |
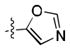
|
7.0 ± 0.5 | 4.1 ± 0.4[b] |
| 2 |
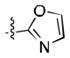
|
11 ± 1 | N.D. |
| 3 |

|
5.1 ± 0.2 | 12.7 ± 0.7 |
| 4 |
|
17 ± 1 | 14.0 ± 0.5 |
| 5 |
|
119 ± 2 | 77 ± 3 |
| 6 |
|
3.8 ± 0.3 | 3.2 ± 0.4 |
| 7 |
 (meta) (meta) |
17.0 ± 0.4 | 19 ± 1 |
| 8 |
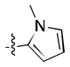 (meta) (meta) |
16.4 ± 0.6 | 32 ± 4 |
| 9 |
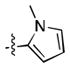
|
17.8 ± 0.3 | 33 ± 9 |
| 10 |
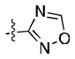
|
36 ± 12 | 19 ± 2 |
| 11 |
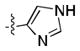
|
270 ± 20 | 180 ± 10 |
| 12 |
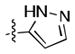
|
12.1 ± 0.6 | 8.97 ± 0.07 |
| 13 |
|
50 ± 10 | 43 ± 2 |
| 14 |
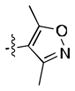
|
120 ± 30 | 70 ± 10 |
| 15 |
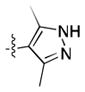
|
18 ± 2 | 32 ± 3 |
IC50 values determined by the displacement of FAM-DEALA-Hyp-YIPD from VCB, with standard error of the mean (SEM) reported.
Literature value.[18]
Preliminary molecular modeling suggested that an N-methyl pyrrole or 4-methyl azole would increase hydrophobic interactions with Pro99 of VHL. While the N-methyl pyrroles had only moderate activity, the 4-methyloxazole, 3, was slightly less potent than 1. However, the 4-methylthiazole, 6, was not only more potent than 4 but also more potent than the oxazole 1.
Given the apparent role of the oxazole C-H donation to the Pro99 carbonyl,[18] we sought to increase the potency of the ligands by using a better H-bond donor. Unfortunately, we were disappointed to find that imidazole 11 had poor binding affinity and that pyrazoles 12 and 13 had moderate affinity but were inferior to 1. However, the direct comparison of pyrazole 15 and isoxazole 14, which lacks the ability to act as a H-bond donor, was informative, showing that the H-bond can lead to a significant increase in potency.
We simultaneously sought to optimize the N-terminal fragment, while keeping the p-chlorobenzylamino fragment of 16 constant due to its small size and availability of the starting material. We originally explored aryl and heteroaryl acetamides. Disappointingly, each was significantly worse than the isoxazole fragment. This included the pyrazole, 22, and imidazoles, 19 and 21, which were designed to interact more strongly with the structural water molecule due to their increased capacity as H-bond acceptors.[21] Testing more diverse fragments led to more success (Table 2), as we found that the chlorobenzamide 24 approached the activity of the corresponding isoxazoleacetamide, 16.
Table 2.
Exploration of Diverse N-terminal Fragments and the Discovery of the Benzamide Fragment
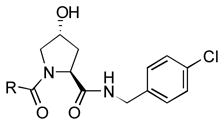
| ||
|---|---|---|
| # | R | IC50 (μM) [a] (10%DMSO) |
| 16 |
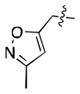
|
38 ± 3 |
| 17 |
|
330 ± 10 |
| 18 |
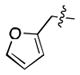
|
>1000 |
| 19 |
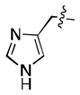
|
>1000 |
| 20 |
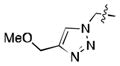
|
280 ± 40 |
| 21 |
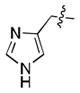
|
>1000 |
| 22 |
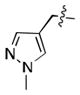
|
372 ± 7 |
| 23 |
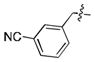
|
300 ± 50 |
| 24 |
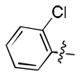
|
52 ± 2 |
IC50 values determined by the displacement of FAM-DEALA-Hyp-YIPD from VCB, with SEM reported.
Literature value.[18]
To elucidate the mode of binding of the chlorobenzamide fragment, a crystal structure of 24 with the pVHL-ElonginB-ElonginC complex (VCB) was obtained (Figure 2). The structure showed that the hydroxyproline core reorients slightly, maintaining many of the previously highlighted interactions.[18] In particular, the hydroxyl group H-bonds to both S111 and H115, while the amide H-bonds to the H110 backbone and Y98 sidechain. Interestingly, the N-terminal fragment clearly binds to VHL in an alternative fashion. While the isoxazole moiety interacted with the structural water previously identified[20] that binds to N67, R69 and H115, the benzamide instead is oriented away from the water pocket, and lays adjacent to the side chain of W88.
Figure 2.

Crystal structure of VCB with 24. pVHL is shown in pale green surface representation, the ligand in grey stick representation. pVHL residues forming the binding pocket are shown in yellow stick representation.
We then synthesized a number of benzamides and found a wide variety of compounds with improved binding potency (Table 3). In particular, we found that the 3-amino-2-methylbenzamide, 28, the m-cyanobenzamide, 33, and the 2-bromo-4-chlorobenzamide, 36, were more potent than the isoxazoleacetamide 16. Additionally, we found that we were able to add large substituents, at the meta position while maintaining potency.
Table 3.
Optimization of the Benzamide N-terminal Fragment
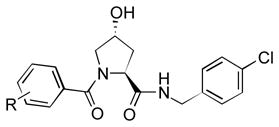
| ||
|---|---|---|
| # | R | IC50 (μM) [a] (10%DMSO) |
| 24 | 2-Cl | 52 ± 2 |
| 25 | H | 110 ± 3 |
| 26 | 3-,4 dimethoxy | 24 ± 2 |
| 27 | 2-NH2 | 38 ± 1 |
| 28 | 3-NH2-2-Me | 10.4 ± 0.8 |
| 29 | 4-NH2 | 29 ± 3 |
| 30 | 4-Cl | 70 ± 10 |
| 31 | 3-F | 38.5 ± 0.5 |
| 32 | 3-Br | 19.6 ± 0.8 |
| 33 | 3-CN | 8.9 ± 0.1 |
| 34 | 3-OMe | 26.3 ± 0.4 |
| 35 | 3-OH | 17 ± 0.6 |
| 36 | 2-Br-4-Cl | 15.5 ± 0.4 |
| 37 | 3-Ph | 33 ± 4 |
| 38 | isonicontinamide | 32 ± 1 |
IC50 values determined by the displacement of FAM-DEALA-Hyp-YIPD from VCB, with SEM reported.
After optimizing both outer fragments independently, we sought to optimize potency of the overall molecule in a combinatorial fashion (Table 4). We found that while in some cases, such as the ethoxybenzamide class, the trend of increasing activity of C-terminal aryl substituents from chloro, to oxazolyl to methylthiazolyl remained similar to the original isoxazolylacetamide, in many cases the oxazolyl compound was worse than both the chloro and methylthiazolyl. In most cases, the methylthiazolyl fragment remained the most potent compound. We were also pleased to find that 51 bound to VHL with an IC50 of 0.9 μM in 1% DMSO, our most potent compound to date, and the only small molecule compound thus far that inhibits the interaction between VHL and the HIF1α peptide below 1 μM.
Table 4.
VHL Ligands with Optimized C- and N-terminal Fragments
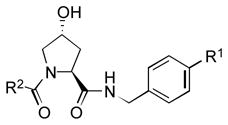
| |||||
|---|---|---|---|---|---|
| # | R1 | R2[b] | IC50 (μM) [a] (10% DMSO) | IC50 (μM)[a] (1% DMSO) | Ki [b] (μM) |
| 16 | Cl |
|
37.8 ± 3.1 | 20.5 ± 1.9[c] | 15.2 |
| 1 | 5-oxazole | 7.0 ± 0.5 | 4.1 ± 0.4[c] | 3.0 | |
| 6 | 5-(4-methyl) thiazole | 3.8 ± 0.3 | 3.2 ± 0.4 | 2.4 | |
|
| |||||
| 39 | Cl |
|
62.9 ± 2.0 | N.D. | N.D. |
| 40 | 5-oxazole | 27.0 ± 0.7 | 19.5 ± 1.0 | 14.5 | |
| 41 | 5-(4-methyl) thiazole | 14.1 ± 0.3 | 11.2 ± 0.9 | 8.3 | |
|
| |||||
| 32 | Cl |
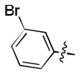
|
19.6 ± 0.8 | N.D. | N.D. |
| 42 | 5-oxazole | 39.5 ± 3.7 | 31.5 ± 1.9 | 23.3 | |
| 43 | 5-(4-methyl) thiazole | 7.7 ± 0.4 | 26.8 ± 1.8 | 19.9 | |
|
| |||||
| 36 | Cl |
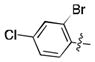
|
17.0 ± 0.6 | N.D. | N.D. |
| 44 | 5-oxazole | 21.2 ± 0.4 | 24.1 ± 0.6 | 17.8 | |
| 45 | 5-(4-methyl) thiazole | 4.9 ± 0.2 | 11.9 ± 0.7 | 8.8 | |
|
| |||||
| 33 | Cl |
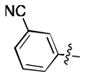
|
8.0 ± 0.1 | N.D. | N.D. |
| 46 | 5-oxazole | 40.3 ± 3.9 | 47.3 ± 1.3 | 35.1 | |
| 47 | 5-(4-methyl) thiazole | 12.0 ± 0.5 | 11.0 ± 0.8 | 8.1 | |
|
| |||||
| 35 | Cl |
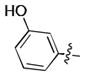
|
17.0 ± 0.6 | N.D. | N.D. |
| 48 | 5-oxazole | 23.4 ± 0.4 | 18.0 ± 0.4 | 13.3 | |
| 49 | 5-(4-methyl) thiazole | 3.5 ± 0.2 | 3.71 ± 0.03 | 2.8 | |
|
| |||||
| 28 | Cl |
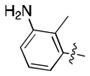
|
10.4 ± 0.8 | N.D. | N.D. |
| 50 | 5-oxazole | 18.9 ± 2.3 | 14.0 ± 0.7 | 10.4 | |
| 51 | 5-(4-methyl) thiazole | 1.76 ± 0.06 | 0.90 ± 0.03 | 0.67 | |
We next obtained a crystal structure of the most potent ligand, 51, in complex with VCB (Figure 3). The optimized methylthiazolyl moiety of the C-terminal fragment binds in a similar conformation to the oxazolyl fragment of 1 that had been previously crystallized.[18] While the methyl group is solvent exposed, it is possible that the larger and more hydrophobic aromatic sulfur atom is better able to fill the small hydrophobic pocket underneath P99 than the corresponding oxygen in 1. The aryl ring of the N-terminal fragment is oriented similarly to 24, but we were surprised at the orientation of the aniline. Rather than interacting with the structural water in the pocket bound by N67, R69 and H115 (as the earlier isoxazole fragment does), the aniline appears to make a completely novel water-mediated H-bond to the side chain of Q96.
Figure 3.

Crystal structure of VCB with 51. pVHL is shown in pale green surface representation, the ligand in orange stick representation. pVHL residues forming the binding pocket are shown in yellow stick representation.
In summary, we report the structure-based design and synthesis of novel VHL ligands, illustrating structure-activity relationships and improving potency. We individually optimized two fragments of the ligands, arriving at a new class of N-terminal benzamide fragments. The interactions made by this new class were then elucidated through x-ray crystallography of the protein:ligand complex. Structural studies of this ligand class revealed both a new binding modality as well as a novel water-mediated hydrogen bond. Finally, using these optimized fragments, we assemble new VHL ligands in a combinatorial fashion, arriving at 51, the first small molecule submicromolar inhibitor of the interaction between VHL and a peptide derived from HIF1α. While no cell-based activity has yet been observed, we hope to take advantage of the interactions highlighted by the crystal structure of 51 to design more drug-like inhibitors of VHL with improved cell permeability. These ligands and the novel interactions and binding modes highlighted have the potential to guide the development of future chemical probes that can target the E3 ligase VHL or stabilize HIF1α proteins.
Supplementary Material
Acknowledgments
This research was partially supported by the NIH AI084140, GM032136, BBSRC BB/G023123/1 and EC PIEF-GA-2010-275683. J.L.G. thanks the NIH for a postdoctoral fellowship (F32GM10052101). A.G.R. is a Leopoldina-Nationale Aka- demie der Wissenschaften Postdoctoral Fellow. We are grateful to the beamline scientists of the Proxima-1 beamline at the Soleil synchrotron facility for their assistance.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Dennis L. Buckley, Departments of Chemistry, Molecular, Cellular & Developmental Biology and Pharmacology, and Center for Molecular Discovery, Yale University, New Haven, Connecticut 06511, United States
Dr. Jeffrey L. Gustafson, Departments of Chemistry, Molecular, Cellular & Developmental Biology and Pharmacology, and Center for Molecular Discovery, Yale University, New Haven, Connecticut 06511, United States
Dr. Inge Van Molle, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, United Kingdom
Dr. Anke G. Roth, Departments of Chemistry, Molecular, Cellular & Developmental Biology and Pharmacology, and Center for Molecular Discovery, Yale University, New Haven, Connecticut 06511, United States
Dr. Hyun Seop Tae, Departments of Chemistry, Molecular, Cellular & Developmental Biology and Pharmacology, and Center for Molecular Discovery, Yale University, New Haven, Connecticut 06511, United States
Dr. Peter C. Gareiss, Departments of Chemistry, Molecular, Cellular & Developmental Biology and Pharmacology, and Center for Molecular Discovery, Yale University, New Haven, Connecticut 06511, United States
Prof. William L. Jorgensen, Departments of Chemistry, Molecular, Cellular & Developmental Biology and Pharmacology, and Center for Molecular Discovery, Yale University, New Haven, Connecticut 06511, United States
Dr. Alessio Ciulli, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, United Kingdom
Prof. Craig M. Crews, Email: craig.crews@yale.edu, Departments of Chemistry, Molecular, Cellular & Developmental Biology and Pharmacology, and Center for Molecular Discovery, Yale University, New Haven, Connecticut 06511, United States
References
- 1.a) Arkin MR, Wells JA. Nat Rev Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]; b) Chène P. Chem Med Chem. 2006;1:400–411. doi: 10.1002/cmdc.200600004. [DOI] [PubMed] [Google Scholar]; c) Wells JA, Mcclendon CL. Nature. 2007;450:1001. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 2.Liskamp RMJ, Rijkers DTS, Kruijtzer JAW, Kemmink J. Chem Bio Chem. 2011;12:1626. [Google Scholar]
- 3.a) Baek S, Kutchukian PS, Verdine GL, Huber R, Holak TA, Lee KW, Popowicz GM. J Am Chem Soc. 2012;134:103. doi: 10.1021/ja2090367. [DOI] [PubMed] [Google Scholar]; b) Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL. J Am Chem Soc. 2007;129:2456. doi: 10.1021/ja0693587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Udugamasooriya DG, Dineen SP, Brekken RA, Kodadek T. J Am Chem Soc. 2008;130:5744. doi: 10.1021/ja711193x. [DOI] [PubMed] [Google Scholar]
- 5.Boersma MD, Haase HS, Peterson-Kaufman KJ, Lee EF, Clarke OB, Colman PM, Smith BJ, Horne WS, Fairlie WD, Gellman SH. J Am Chem Soc. 2012;134:315. doi: 10.1021/ja207148m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geppert T, Bauer S, Hiss JA, Conrad E, Reutlinger M, Schneider P, Weisel M, Pfeiffer B, Altmann KH, Waibler Z, Schneider G. Angew Chem Int Ed. 2012;51:258. doi: 10.1002/anie.201105901. [DOI] [PubMed] [Google Scholar]
- 7.a) Maurer T, Garrenton LS, Oh A, Pitts K, Anderson DJ, Skelton NJ, Fauber BP, Pan B, Malek S, Stokoe D, Ludlam MJC, Bowman KK, Wu J, Giannetti AM, Starovasnik MA, Mellman I, Jackson PK, Rudolph J, Wang W, Fang G. PNAS. 2012;109:5299–5304. doi: 10.1073/pnas.1116510109. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sun Q, Burke JP, Phan J, Burns MC, Olejniczak ET, Waterson AG, Lee T, Rossanese OW, Fesik SW. Angew Chem Int Ed. 2012;51:6140. doi: 10.1002/anie.201201358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kozakov D, Hall DR, Chuang GY, Cencic R, Brenke R, Grove LE, Beglov D, Pelletier J, Whitty A, Vajda S. PNAS. 2011;108:13528. doi: 10.1073/pnas.1101835108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Bedford L, Lowe J, Dick LR, Mayer RJ, Brownell JE. Nat Rev Drug Discov. 2011;10:29. doi: 10.1038/nrd3321. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cohen P, Tcherpakov M. Cell. 2010;143:686. doi: 10.1016/j.cell.2010.11.016. [DOI] [PubMed] [Google Scholar]
- 10.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. Science. 2004;303:844. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 11.a) Oost TK, Sun C, Armstrong RC, Al-Assaad AS, Betz SF, Deckwerth TL, Ding H, Elmore SW, Meadows RP, Olejniczak ET, Oleksijew A, Oltersdorf T, Rosenberg SH, Shoemaker AR, Tomaselli KJ, Zou H, Fesik SW. J Med Chem. 2004;47:4417. doi: 10.1021/jm040037k. [DOI] [PubMed] [Google Scholar]; b) Sun H, Nikolovska-Coleska Z, Yang CY, Xu L, Liu M, Tomita Y, Pan H, Yoshioka Y, Krajewski K, Roller PP, Wang S. J Am Chem Soc. 2004;126:16686. doi: 10.1021/ja047438+. [DOI] [PubMed] [Google Scholar]
- 12.Aghajan M, Jonai N, Flick K, Fu F, Luo M, Cai X, Ouni I, Pierce NW, Tang X, Lomenick B, Damoiseaux R, Hao R, Del Moral PM, Verma R, Li Y, Li C, Houk KN, Jung ME, Zheng N, Huang L, Deshaies RJ, Kaiser P, Huang J. Nat Biotechnol. 2010;28:738. doi: 10.1038/nbt.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orlicky S, Tang X, Neduva V, Elowe N, Brown ED, Sicheri F, Tyers M. Nat Biotechnol. 2010;28:733. doi: 10.1038/nbt.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Kaelin WG. Nat Rev Cancer. 2008;8:865–873. doi: 10.1038/nrc2502. [DOI] [PubMed] [Google Scholar]; b) Semenza GL. Trends Mol Med. 2001;7:345. doi: 10.1016/s1471-4914(01)02090-1. [DOI] [PubMed] [Google Scholar]
- 15.a) Rotili D, Altun M, Kawamura A, Wolf A, Fischer R, Leung IKH, Mackeen MM, Tian YM, Ratcliffe PJ, Mai A, Kessler BM, Schofield CJ. Chemistry & Biology. 2011;18:642. doi: 10.1016/j.chembiol.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tian YM, Yeoh KK, Lee MK, Eriksson T, Kessler BM, Kramer HB, Edelmann MJ, Willam C, Pugh CW, Schofield CJ, Ratcliffe PJ. J Biol Chem. 2011;286:13041. doi: 10.1074/jbc.M110.211110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muchnik E, Kaplan J. Expert Opin Investig Drugs. 2011;20:645. doi: 10.1517/13543784.2011.566861. [DOI] [PubMed] [Google Scholar]
- 17.Willam C, Masson N, Tian YM, Mahmood SA, Wilson MI, Bicknell R, Eckardt KU, Maxwell PH, Ratcliffe PJ, Pugh CW. PNAS. 2002;99:10423. doi: 10.1073/pnas.162119399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Buckley DL, Van Molle I, Gareiss PC, Tae HS, Michel J, Noblin DJ, Jorgensen WL, Ciulli A, Crews CM. J Am Chem Soc. 2012;134:4465. doi: 10.1021/ja209924v. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Van Molle I, Thomann A, Buckley DL, So EC, Lang S, Crews CM, Ciulli A. Chemistry & Biology. 2012:19. doi: 10.1016/j.chembiol.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loenarz C, Mecinovic J, Chowdhury R, McNeill LA, Flashman E, Schofield CJ. Angew Chem Int Ed. 2009;48:1784. doi: 10.1002/anie.200805427. [DOI] [PubMed] [Google Scholar]
- 20.Hon WC, Wilson MI, Harlos K, Claridge TDW, Schofield CJ, Pugh CW, Maxwell PH, Ratcliffe PJ, Stuart DI, Jones EY. Nature. 2002;417:975. doi: 10.1038/nature00767. [DOI] [PubMed] [Google Scholar]
- 21.Meanwell NA. J Med Chem. 2011;54:2529. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 22.Huang X. Journal of Biomolecular Screening. 2003;8:34. doi: 10.1177/1087057102239666. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.