

Abstract
Type 2 diabetes and hyperglycemia with the resulting increase of glucose concentrations in the brain impair the outcome of ischemic stroke, and may increase the risk of developing Alzheimer's disease (AD). Reports indicate that glucagon-like peptide-1 (GLP-1) may be neuroprotective in models of AD and stroke: Although the mechanism is unclear, glucose homeostasis appears to be important. We conducted a randomized, double-blinded, placebo-controlled crossover study in nine healthy males. Positron emission tomography was used to determine the effect of GLP-1 on cerebral glucose transport and metabolism during a hyperglycemic clamp with 18fluoro-deoxy-glucose as tracer. Glucagon-like peptide-1 lowered brain glucose (P=0.023) in all regions. The cerebral metabolic rate for glucose was increased everywhere (P=0.039) but not to the same extent in all regions (P=0.022). The unidirectional glucose transfer across the blood–brain barrier remained unchanged (P=0.099) in all regions, while the unidirectional clearance and the phosphorylation rate increased (P=0.013 and 0.017), leading to increased net clearance of the glucose tracer (P=0.006). We show that GLP-1 plays a role in a regulatory mechanism involved in the actions of GLUT1 and glucose metabolism: GLP-1 ensures less fluctuation of brain glucose levels in response to alterations in plasma glucose, which may prove to be neuroprotective during hyperglycemia.
Keywords: blood–brain barrier, 2-deoxy-glucose, diabetes, energy metabolism, GLP-1, glucagon-like peptide-1, glucose, pharmacology
Introduction
Type 2 diabetes is characterized by hyperglycemia. The incidence of type 2 diabetes has increased significantly in the latter years, and patients with diabetes have an increased risk of atherosclerotic macrovascular disease, which may lead to stroke. Indeed, cardiovascular diseases are the main causes of death in this group, accounting for 50% of all diabetes fatalities.1 Type 2 diabetes also increases the risk of developing Alzheimer's disease (AD) 2- to 5-fold; AD shares several pathophysiological features with cardiovascular disease, with a risk that may be related to plasma glucose levels.2, 3
Normal brain function depends on a continuous supply of glucose by the cerebral circulation. However, the search for a regulatory mechanism of blood–brain barrier (BBB) glucose transfer in the brain vasculature has failed. As far as it is known, the only factor that definitively affects BBB glucose transfer is the level of the plasma glucose itself. Hyperglycemia worsens the outcome of stroke, but it remains unclear whether lowering of plasma glucose during stroke actually is neuroprotective.4
The incretin glucagon-like peptide-1 (GLP-1) and artificial GLP-1 receptor agonists are well-established as counter-diabetic medications. The native hormone is mainly secreted by the intestinal L-cells, and is also released as a neuropeptide in different brain regions.5 It has a wide range of physiological actions in glucose metabolism, including stimulation of insulin secretion, inhibition of glucagon secretion, inhibition of gastric emptying, and reduction of appetite and food intake.6, 7 The GLP-1 receptor is abundant throughout the brain,8 and native GLP-1 and GLP-1 agonists have been reported to be neuroprotective in several cell, animal, and human studies (e.g., in AD), to have a neurogenic and antiinflammatory effect in the central nervous system, to enhance synaptic plasticity, to improve learning, and to reduce ischemic damage after stroke. The mechanisms are not clear, but glucose homeostasis appears to be crucial.9, 10
Materials and methods
Ten nonsmoking, healthy, Caucasian males with a mean age of 22.0±2.3 years and a mean body mass index of 24.2±1.5 kg/m2 participated in the study, nine completed the study. They had a normal physical examination, no history of diabetes or cardiovascular disease, and received no medication. The study was conducted in accordance with the Declaration of Helsinki and the protocol was approved by the official Regional Science Ethics Committee of Region Midtjylland. All participants received both oral and written information and signed an approved informed consent form before entering the study. One subject experienced pronounced nausea and withdrew from the study.
In this randomized, double-blinded, placebo-controlled, crossover study, each subject was studied twice in random order of GLP-1 and placebo infusion. Positron emission tomography (PET) sessions were separated by an interval of minimum 4 weeks. Both sessions commenced at 0700 hours after an overnight fast. The subjects did not exercise 24 hours before the sessions. Subjects were placed in bed and two catheters were inserted for infusion of clamp hormones and for the infusion of GLP-1 or placebo. A third catheter was placed in an arterialized (heated) dorsal hand vein for blood sampling. An arterial catheter was placed in the radial artery of the right arm to draw blood samples for measuring input radioactivity during PET.
A hyperglycemic pancreatic-pituitary clamp was performed according to the principles previously described.11, 12 In brief somatostatin (Ferring GmbH, D-24109 Kiel, Germany) was infused at a rate of 300 μg/h to suppress the endogenous insulin, glucagon, growth hormone, and GLP-1 production. Human glucagon (Glucagen, Novo Nordisk A/S, Copenhagen, Denmark) 0.6 ng per kg per minute, growth hormone (Genotropin Miniquik 0.2 mg, Pfizer ApS, Ballerup, Denmark) 2 ng per kg per minute were infused with the aim of maintaining near basal levels (0 to 360 minutes). Insulin (Actrapid, Novo Nordisk A/S, Copenhagen, Denmark) was infused at a rate of 0.12 mU per kg per minute. Glucose (200 g/L) was infused at a variable rate to clamp plasma glucose at 9 mM (0 to 360 minutes). Glucagon-like peptide-1 or placebo infusion was initiated at time 60 minutes and maintained during the entire session.
Synthetic GLP-1 (7 to 36) amide was purchased from PolyPeptide Laboratories (Wolfenbuttel, Germany), and the same lot number was used in all studies. The peptide was dissolved in sterilized water containing 2% human serum albumin (Human Albumin, guaranteed to be free of hepatitis B surface antigen, hepatitis C virus antibodies, and human immunodeficiency virus antibodies; Statens Serum Institute, Copenhagen, Denmark), and subjected to sterile filtration. The GLP-1 was dispensed into glass ampoules and stored frozen (−20°C) under sterile conditions until the day of the experiment. The peptide was demonstrated to be >97% pure and identical to the natural human peptide by high-performance liquid chromatography mass and sequence analysis. The subjects received either intravenous synthetic GLP-1 (7 to 36 amide) or placebo at a rate of 1.2 pmol per kg per minute (60 to 360 minutes).13 The concentration of GLP-1 (7 to 36 amide) was 20 μg/mL. The test solution consisted of 9 mL GLP-1, 20 mL of human albumin 5% (CSL Behring, King of Prussia, PA, USA) and 0.9% saline solution to yield 100 mL of solution. The placebo solution consisted of the above-mentioned buffer solution containing human albumin and saline. One subject experienced severe nausea during GLP-1 infusion with onset at 300 minutes of infusion and was withdrawn.
Plasma glucose was measured every 5 to 10 minutes until it reached 9.0 mM and then every 10 minutes. Blood for measuring insulin, C-peptide, glucagon, GLP-1 (total and intact), growth hormone, free fatty acids, and cortisol was drawn every 30 minutes. Blood for measuring epinephrine and norepinephrine was drawn at 0, 90, 150, 210 270, 330, and 390 minutes. Arterial blood for measuring input radioactivity was drawn at predetermined intervals during PET (12 × 5 seconds, 8 × 30 seconds, and 8 × 300 seconds).
Assays
Assays were described in details previously.12 Arterial input samples were analyzed on a Packard radioactivity analyzer immediately after end of PET session.
Magnetic Resonance Imaging
A high-resolution T1-weighted MR (magnetic resonance) was acquired for each subject with a 3T GE Signa HDx scanner (GE Medical Systems, Milwaukee, WI, USA) using a 3DIR-fSPGR sequence.
Positron Emission Tomography
[18F]-Fluoro-2-deoxy-glucose (18F-FDG) was used as a tracer for brain glucose uptake and was produced in-house according to the Drug Master File, Danish national marketing authorization number 2165. We used the whole-body PET EXACT HR47 (Siemens Medical, Knoxville, TN, USA) with a 15-cm field of view and an acquisition capacity of 47 transaxial planes with a spatial resolution of 4 to 5 mm at the center of the field of view. All PET data were acquired in 3D mode. A 15-minute transmission scan was performed to correct photon attenuation. Five hours after clamp start and 4 hours after the GLP-1 or placebo infusion was started, a bolus of 175 MBq of 18F-FDG in 10 mL saline was injected intravenously over 10 seconds. Dynamic acquisition commenced at the beginning of tracer injection and continued for 45 minutes to acquire 23 frames (6 × 30 seconds, 7 × 1 minutes, 5 × 2 minutes, and 5 × 5 minutes).
Image Analysis
Positron emission tomography and MR-images were coregistered and entered in Talairach spaces. Regional tissue time–activity curves for 18F-FDG uptake were extracted for total cerebral gray matter, cerebral cortex, thalamus, striatum, cerebellar cortex, brainstem, and white matter.
Kinetic Analysis
For the use of 18F-FDG to trace the phosphorylation rate of glucose, the lumped constant is a necessary isotope conversion factor.14 In the three-compartment model of irreversible accumulation of FDG-6-phosphate, the lumped constant is the ratio between the net clearances of 18F-FDG and glucose, which depends on the relative affinities of transport across the BBB and phosphorylation of both hexoses (FDG and glucose). Then, the net clearance of glucose (K) by definition of the lumped constant is
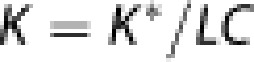 |
where K* is the net clearance of 18F-FDG. With the three-compartment model of irreversible FDG-6-phosphate accumulation, we obtained values of K1* (unidirectional blood–brain clearance, where symbols marked with asterisk indicate 18F-FDG), k2* (efflux rate constant), k3* (phosphorylation rate constant), and Vp (volume of tissue occupied by intravascular blood plasma) for 18F-FDG in each region of interest. The rate of dephosphorylation and the absolute quantity of 18F-FDG were both considered negligible during the tomography sessions period. The net clearance of 18F-FDG can then be described as,15, 16
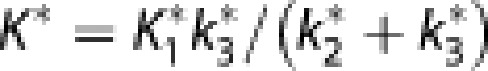 |
In the competition between native 18F-FDG and glucose in the brain, both transport across the BBB (K1*, k2*) and phosphorylation by hexokinase (k3*) obey the Michaelis–Menten equation, which yields fixed ratios of unidirectional blood–brain clearances (τ=K1*/K1) and phosphorylation rates (ϕ=k3*/k3) for the tracer and native glucose.17 By substituting transfer constants of 18F-FDG for those of glucose, using the constants τ and ϕ, we determined the lumped constant directly for each subject in each region of interest as described previously,17 using the values of τ=1.48 and ϕ=0.39.18 As the ratio between the net 18F-FDG and glucose clearances, the lumped constant then equals,12, 17
 |
and the unidirectional glucose flux from blood to brain (Jglc) is
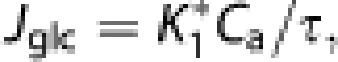 |
where Ca is the arterial steady-state plasma glucose concentration (300 to 360 minutes). The net cerebral metabolic rate of glucose then is
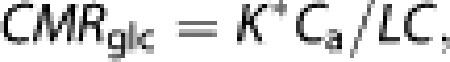 |
the cerebral tissue glucose concentration Ctissue is
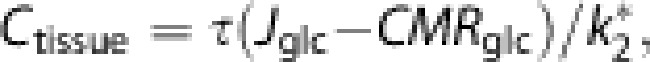 |
and, by virtue of the Michaelis–Menten description of blood–brain and brain–blood glucose transfer, the exchange volume Ve is the same for glucose and 18F-FDG,15, 16, 19 such that,
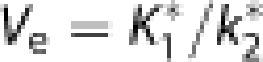 |
Calculations and Statistics
We analyzed the PET data with a linear mixed effects model (repeated measurement analysis of variance) with subject and all interactions involving subjects, including the interaction between subject and brain region and the interaction between subject and treatment, as random effects. Treatment (GLP-1 versus placebo), region, and the interaction between the two were included in the analysis as fixed effects. To compare the two treatment groups' circulating hormones and metabolites, a similar statistical model was used with time instead of region. Significant differences were at significance levels of P<0.05, and we present data as mean±s.d.
Results
The infusion of GLP-1 at a constant rate resulted in high pharmacologically relevant plasma concentrations of the intact hormone12 (Figure 1H). During the PET scan, the Ca concentrations averaged 8.7±0.4 mM (GLP-1) and 9.0±0.4 mM (placebo), respectively, with no significant difference (P=0.17; Figure 1A). The glucose infusion rates rose significantly with GLP-1 infusion (13.5±4.7 mg per kg per minute (GLP-1) versus 2.1±2.3 mg per kg per minute (placebo) P<0.0001) (Figure 1B). The levels of serum insulin and C-peptide were significantly increased with GLP-1 infusion (Figures 1C and 1D). Insulin levels rose to 190±127 pmol/L during scans with GLP-1 infusion compared with 53±13 pmol/L during placebo scans (Figure 1C). Norepinephrine concentration decreased during scans in the placebo group, P=0.02 (Figure 1F). As expected, there were no significant differences among other circulating hormones and metabolites (epinephrine, cortisol, glucagon, growth hormone, and free fatty acids—Figures 1E and 1G) between sessions during the scans.
Figure 1.

Plasma content of hormones and metabolites. (A) Plasma glucose (PG); (B) glucose infusion rate (GIR); (C) insulin; (D) C-peptide; (E) glucagon; (F) Norepinephrine; (G) epinephrine; (H) glucagon-like peptide-1 (GLP-1). Data are represented as mean±s.d. *P<0.05.
Glucagon-like peptide-1 lowered the intracerebral concentration of glucose (Ctissue) significantly by 0.18 mM (P=0.023) (95% confidence interval (CI): 0.02 to 0.32) in all regions (Figure 2A) (P=0.47 for interaction between region and treatment). The cerebral metabolic rate for glucose (CMRglc) was increased significantly overall (P=0.039) but not to the same extent (P=0.022 for interaction between region and treatment); significantly so in thalamus and cerebellum (P=0.001 and 0.049) and with a strong trend in GM and cortex (P=0.05 and 0.06) (Figure 2B). The unidirectional glucose transfer Jglc across the BBB was not changed significantly (P=0.099) in any region (Figure 2C). The net clearance of 18F-FDG (K*) increased 0.002 mL per cm3 per minute (P=0.006) (95% CI: 0.0005 to 0.003) with GLP-1 infusion with the same effect in all regions (Figure 2D). Glucagon-like peptide-1 induced significant increases (P=0.0001 to 0.04) of the net clearance of glucose (K) in GM, cortex, thalamus, striatum, and cerebellum (Figure 2E). There was an overall significant effect of GLP-1 on the unidirectional clearances of 18F-FDG (K1*) and glucose (K1) (P=0.013), which increased by 0.007 mL per cm3 per minute (95% CI: 0.001 to 0.01) (Figure 2F). We found an overall increase of the efflux rate constant of 18F-FDG (k2*) (P=0.046) with interaction between region and treatment (P=0.04). The efflux constant increased in GM, cortex, and thalamus (P=0.002 to 0.04) as a consequence of lowered brain glucose (Figure 2G). For the same reason, the phosphorylation coefficient for 18F-FDG (k3*) increased by 0.005 pr. minute (95% CI: 0.001 to 0.01) in the brain as a whole (P=0.017) (Figure 2H). The lumped constant and volume of tissue occupied by intravascular blood in the brain (Vp) remained constant (P=0.73 and 0.92, respectively), as did the exchange volumes (Ve) between sessions (P=0.46) (data not shown).
Figure 2.

Positron emission tomography (PET) results. (A) Intracerebral glucose concentration (Ctissue); (B) cerebral metabolic rate (CMRglc); (C) cerebral glucose transport (Jglc); (D) net clearance of Fluoro-2-deoxy-glucose (18F-FDG) (K*); (E) net clearance of glucose (Kglc); (F) unidirectional clearance (K1); (G) efflux rate constant (k2*); (H) phosphorylation coefficient (k3*). Data are represented as mean±s.d. *P<0.05, **P=0.05 to 0.06.
Discussion
In the present hyperglycemia study, GLP-1 administration elicited the combined effects of lowered intracerebral glucose content and raised cerebral metabolic rate in specific regions. The net clearance of 18F-FDG and hence of glucose increased, and GLP-1 had an impact on the rates of unidirectional clearance, efflux, and phosphorylation of glucose. Glucagon-like peptide-1 infusion led to endogenous insulin secretion, despite somatostatin administration.
Insulin and Glucose Infusion
Insulin plays a significant role in the metabolism of glucose in peripheral tissues, while the possible roles in the brain metabolism and transport have been subject to debate for decades. Numerous studies confirm that insulin has no effect per se on BBB transport20, 21, 22, 23, 24, 25 or metabolism21, 25, 26, 27 of glucose in the human brain, and hence is unlikely to affect the magnitudes of the transport constants K1*, k2*, and k3*, and a recent study concluded that hyperinsulinemia also failed to increase brain glucose metabolism in subjects with normal glucose tolerance.28
The facilitated and insulin-insensitive diffusion of glucose across the BBB is predominantly dependent on the GLUT1 glucose transporters in the two membranes of the microvascular brain endothelium.16, 29 By contrast, increased insulin levels have a marked effect on the 18F-FDG plasma activity curves, because of rapid clearance of glucose from plasma, due to the insulin-dependent increase in 18F-FDG uptake by skeletal and cardiac muscle tissue,21, 30 and this clearance by peripheral tissues is evident in the additional need for glucose infusion following the GLP-1 infusion.
Norepinephrine. was constant during sessions but lower in the placebo group than in the GLP-1 group, due to a decline during the tomography. The difference was not reflected in other catecholamines that remained almost constant during the tomography and norepinephrine is not known to affect CMRglc.31
Ve
Estimates of K1*/k2* ratios are expressions of the exchange volume (Ve) for dissolved tracers in models with multiple compartments for tracer molecules and metabolites, as is the case for 18F-FDG and its metabolites. In the brain, the GLUT1 proteins in the capillary endothelium are the main sites of facilitated diffusion of glucose and glucose analogs from the circulation to the tissue. The exchange volume indicates the magnitude of the joint extra- and intracellular pools of solvent available for solution of intact tracer 18F-FDG, relative to the solvent in which the tracer concentrations in the circulation are measured, that is, arterial plasma in this case. The values remained unchanged between sessions with values averaging 0.5 mL/cm3 with minor differences among regions. This value implies that 0.5 mL fluid of a total brain tissue quantity of 1 cm3 is available for joint extra- and intracellular solution of 18F-FDG. It is a real physical volume and as such should not undergo dynamic or short-term changes. The total distribution volume of unchanged 18F-FDG in the brain tissue then consists of the plasma and the extracellular and intracellular volumes of solvent.
Positron Emission Tomography
We previously linked GLP-1, cerebral glucose transport, and neuroprotection during normoglycemia.12 Thus, GLP-1 provides a possible regulatory mechanism for the link between plasma glucose and brain glucose, and the reported neuroprotective effects may be linked to the lowering of glucose fluctuations in the brain when plasma glucose is increased.9 Hyperglycemia with concomitantly increased glucose concentration in the brain tissue accelerates ischemic damage and worsens the outcome after ischemic stroke.4 Glucagon-like peptide-1 is reported to counter the effects of stroke by unknown means,10 but the present results indicate a mechanism by means of which GLP-1 lowers the Ctissue because of increased net clearance of glucose (and CMRglc). The continuous metabolism of glucose transferred across the BBB maintains Ctissue levels that are lower than the concentration of glucose in plasma.32 Thus, the Ctissue depends on the BBB glucose transfer as well as on the glucose phosphorylation rate. In contrast to the hypothesis, however, the net glucose flux Jglc was not changed significantly during the sessions, although a trend emerged in all regions. The values of CMRglc were increased in specific regions, but the rates are not held to depend on the tissue glucose concentration because the low Michaelis half-saturation constant (KM 0.05 mM) ensures that brain hexokinase is fully saturated at all but the lowest glucose concentrations. The increased CMRglc is reflected in the increased net clearance, and the unidirectional clearance, the efflux coefficient, and the phosphorylation coefficients all relate descriptively to the net clearance.
The results show that transport across the BBB is affected by the acute infusion of GLP-1. These end points are not independent and hence do not yield independent evidence of the direction of an effect of GLP-1 but do throw light on different aspects of the kinetics of blood–brain transport and brain metabolism. In the three-compartment tracer analysis, K1 represents the ‘unidirectional' clearance of glucose across the BBB from blood to the combined extra- and intracellular spaces, k2* represents the rate of efflux of 18F-FDG across the BBB to the circulation, and k3* represents the rate of phosphorylation of 18F-FDG. Therefore, the K1* and k2* values reflect GLUT1 activity and k3* reflects the activity of hexokinase,33, 34 all of which were increased.
With the three-compartment model of irreversible FDG-6-phosphate accumulation, the apparent magnitude of k3* is affected by both the magnitude of the inherent phosphorylation rate and the effect of loss of FDG-6-phosphate from the tissue due to dephosphorylation, expressed by the rate constant k4* in the three-compartment model of reversible FDG-6-phosphate accumulation. If the magnitude of k4* decreased during GLP-1 infusion, then statement of an increase in the rate of phosphorylation could be inaccurate. However, there are two reasons why this is an unlikely outcome. First, insulin increased during GLP-1, and hyperinsulinemia appears, if anything, to raise the magnitude of k4*,21 which supports the observation that k3* actually did increase. Second, the magnitude of the lumped constant is likewise affected by changes of the magnitude of k4* and hence counteracts the effect.17 The absence of a change of the lumped constant further indicates that no change of the relationship between the two rate constants occurred.
We suggest that GLP-1 plays a role in the regulatory mechanism that is implicit in the actions of GLUT1 and glucose metabolism. The role is exerted in the brain vasculature in support of the findings reported by Lerche et al.12 Hence, GLP-1 assures less fluctuation of brain glucose levels in response to alterations in plasma glucose, which may prove to be neuroprotective during hyperglycemia. Cerebral blood flow was not measured in this study, but as described earlier,12 clearance in principle is a measure that includes a blood flow effect.
Type 2 diabetes and hyperglycemia increases the risk for AD2, 3 and PET studies demonstrate a reduced glucose uptake in some brain regions of AD patients,35 opposite to the trend in the present study. However, this reduction is possibly due to lower functional demands for glucose rather than a direct effect of the pathophysiology of AD. If so, effects that lower brain glucose may be beneficial. Glucagon-like peptide-1 affects the symptoms in animal models of AD,9 a claim examined in ongoing human studies. Furthermore, hyperglycemia and hence increased glucose content in the brain markedly affects cognition and memory,36 and thus support the claim of neuroprotective effects of GLP-1. Studies show that excess glucose from the increased glucose levels in the brain can be converted into sorbitol and fructose, which influence several intracellular cascades, including associations with critical AGE- and ROS-formation.37
If the metabolic effect was due to an increase of the maximum phosphorylation rate (Vmax), it would presumably be evident also in hypoglycemia, a hypothesis in need of testing. This is important because the global glucose metabolic rate is higher in aware subjects during hypoglycemia, compared with the unaware subjects.38
The authors declare no conflict of interest.
References
- Morrish NJ, Wang SL, Stevens LK, Fuller JH, Keen H. Mortality and causes of death in the WHO Multinational Study of vascular disease in diabetes. Diabetologia. 2001;44 (Suppl 2:S14–S21. doi: 10.1007/pl00002934. [DOI] [PubMed] [Google Scholar]
- Luchsinger JA, Reitz C, Honig LS, Tang MX, Shea S, Mayeux R. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology. 2005;65:545–551. doi: 10.1212/01.wnl.0000172914.08967.dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu WL, von SE, Qiu CX, Winblad B, Fratiglioni L. Uncontrolled diabetes increases the risk of Alzheimer's disease: a population-based cohort study. Diabetologia. 2009;52:1031–1039. doi: 10.1007/s00125-009-1323-x. [DOI] [PubMed] [Google Scholar]
- Bellolio MF, Gilmore RM, Stead LG. Insulin for glycaemic control in acute ischaemic stroke. Cochrane Database Syst Rev. 2011;9:CD005346. doi: 10.1002/14651858.CD005346.pub3. [DOI] [PubMed] [Google Scholar]
- Brubaker PL, Drucker DJ. Minireview: glucagon-like peptides regulate cell proliferation and apoptosis in the pancreas, gut, and central nervous system. Endocrinology. 2004;145:2653–2659. doi: 10.1210/en.2004-0015. [DOI] [PubMed] [Google Scholar]
- Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368:1696–1705. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- Holst JJ. On the physiology of GIP and GLP-1. Horm Metab Res. 2004;36:747–754. doi: 10.1055/s-2004-826158. [DOI] [PubMed] [Google Scholar]
- Hamilton A, Holscher C. Receptors for the incretin glucagon-like peptide-1 are expressed on neurons in the central nervous system. Neuroreport. 2009;20:1161–1166. doi: 10.1097/WNR.0b013e32832fbf14. [DOI] [PubMed] [Google Scholar]
- Bak AM, Egefjord L, Gejl M, Steffensen C, Stecher CW, Smidt K, Brock B, Rungby J. Targeting amyloid-beta by glucagon-like peptide -1 (GLP-1) in Alzheimer's disease and diabetes. Expert Opin Ther Targets. 2011;15:1153–1162. doi: 10.1517/14728222.2011.600691. [DOI] [PubMed] [Google Scholar]
- Darsalia V, Mansouri S, Ortsater H, Olverling A, Nozadze N, Kappe C, Iverfeldt K, Tracy LM, Grankvist N, Sjoholm A, Patrone C. Glucagon-like peptide-1 receptor activation reduces ischaemic brain damage following stroke in Type 2 diabetic rats. Clin Sci (Lond) 2012;122:473–483. doi: 10.1042/CS20110374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degn KB, Brock B, Juhl CB, Djurhuus CB, Grubert J, Kim D, Han J, Taylor K, Fineman M, Schmitz O. Effect of intravenous infusion of exenatide (synthetic exendin-4) on glucose-dependent insulin secretion and counterregulation during hypoglycemia. Diabetes. 2004;53:2397–2403. doi: 10.2337/diabetes.53.9.2397. [DOI] [PubMed] [Google Scholar]
- Lerche S, Brock B, Rungby J, Botker HE, Moller N, Rodell A, Bibby BM, Holst JJ, Schmitz O, Gjedde A. Glucagon-like peptide-1 inhibits blood-brain glucose transfer in humans. Diabetes. 2008;57:325–331. doi: 10.2337/db07-1162. [DOI] [PubMed] [Google Scholar]
- Toft-Nielsen MB, Madsbad S, Holst JJ. Continuous subcutaneous infusion of glucagon-like peptide 1 lowers plasma glucose and reduces appetite in type 2 diabetic patients. Diabetes Care. 1999;22:1137–1143. doi: 10.2337/diacare.22.7.1137. [DOI] [PubMed] [Google Scholar]
- Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD, Sakurada O, Shinohara M. The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem. 1977;28:897–916. doi: 10.1111/j.1471-4159.1977.tb10649.x. [DOI] [PubMed] [Google Scholar]
- Gjedde A. Calculation of cerebral glucose phosphorylation from brain uptake of glucose analogs in vivo: a re-examination. Brain Res. 1982;257:237–274. doi: 10.1016/0165-0173(82)90018-2. [DOI] [PubMed] [Google Scholar]
- Gjedde A, Christensen O. Estimates of Michaelis-Menten constants for the two membranes of the brain endothelium. J Cereb Blood Flow Metab. 1984;4:241–249. doi: 10.1038/jcbfm.1984.33. [DOI] [PubMed] [Google Scholar]
- Kuwabara H, Evans AC, Gjedde A. Michaelis-Menten constraints improved cerebral glucose metabolism and regional lumped constant measurements with [18F]fluorodeoxyglucose. J Cereb Blood Flow Metab. 1990;10:180–189. doi: 10.1038/jcbfm.1990.33. [DOI] [PubMed] [Google Scholar]
- Hasselbalch SG, Madsen PL, Knudsen GM, Holm S, Paulson OB. Calculation of the FDG lumped constant by simultaneous measurements of global glucose and FDG metabolism in humans. J Cereb Blood Flow Metab. 1998;18:154–160. doi: 10.1097/00004647-199802000-00005. [DOI] [PubMed] [Google Scholar]
- Gjedde A, Wienhard K, Heiss WD, Kloster G, Diemer NH, Herholz K, Pawlik G. Comparative regional analysis of 2-fluorodeoxyglucose and methylglucose uptake in brain of four stroke patients. With special reference to the regional estimation of the lumped constant. J Cereb Blood Flow Metab. 1985;5:163–178. doi: 10.1038/jcbfm.1985.23. [DOI] [PubMed] [Google Scholar]
- Crone C. Facilitated transfer of glucose from blood into brain tissue. J Physiol. 1965;181:103–113. doi: 10.1113/jphysiol.1965.sp007748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselbalch SG, Knudsen GM, Videbaek C, Pinborg LH, Schmidt JF, Holm S, Paulson OB. No effect of insulin on glucose blood-brain barrier transport and cerebral metabolism in humans. Diabetes. 1999;48:1915–1921. doi: 10.2337/diabetes.48.10.1915. [DOI] [PubMed] [Google Scholar]
- Hom FG, Goodner CJ, Berrie MA. A [3H]2-deoxyglucose method for comparing rates of glucose metabolism and insulin responses among rat tissues in vivo. Validation of the model and the absence of an insulin effect on brain. Diabetes. 1984;33:141–152. doi: 10.2337/diab.33.2.141. [DOI] [PubMed] [Google Scholar]
- McCall AL, Gould JB, Ruderman NB. Diabetes-induced alterations of glucose metabolism in rat cerebral microvessels. Am J Physiol. 1984;247:E462–E467. doi: 10.1152/ajpendo.1984.247.4.E462. [DOI] [PubMed] [Google Scholar]
- Namba H, Lucignani G, Nehlig A, Patlak C, Pettigrew K, Kennedy C, Sokoloff L. Effects of insulin on hexose transport across blood-brain barrier in normoglycemia. Am J Physiol. 1987;252:E299–E303. doi: 10.1152/ajpendo.1987.252.3.E299. [DOI] [PubMed] [Google Scholar]
- Seaquist ER, Damberg GS, Tkac I, Gruetter R. The effect of insulin on in vivo cerebral glucose concentrations and rates of glucose transport/metabolism in humans. Diabetes. 2001;50:2203–2209. doi: 10.2337/diabetes.50.10.2203. [DOI] [PubMed] [Google Scholar]
- Hertz MM, Paulson OB, Barry DI, Christiansen JS, Svendsen PA. Insulin increases glucose transfer across the blood-brain barrier in man. J Clin Invest. 1981;67:597–604. doi: 10.1172/JCI110073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro ET, Cooper M, Chen CT, Given BD, Polonsky KS. Change in hexose distribution volume and fractional utilization of [18F]-2-deoxy-2-fluoro-D-glucose in brain during acute hypoglycemia in humans. Diabetes. 1990;39:175–180. doi: 10.2337/diab.39.2.175. [DOI] [PubMed] [Google Scholar]
- Hirvonen J, Virtanen KA, Nummenmaa L, Hannukainen JC, Honka MJ, Bucci M, Nesterov SV, Parkkola R, Rinne J, Iozzo P, Nuutila P. Effects of insulin on brain glucose metabolism in impaired glucose tolerance. Diabetes. 2011;60:443–447. doi: 10.2337/db10-0940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall AL. Cerebral glucose metabolism in diabetes mellitus. Eur J Pharmacol. 2004;490:147–158. doi: 10.1016/j.ejphar.2004.02.052. [DOI] [PubMed] [Google Scholar]
- Huitink JM, Visser FC, van Leeuwen GR, Van LA, Bax JJ, Heine RJ, Teule GJ, Visser CA. Influence of high and low plasma insulin levels on the uptake of fluorine-18 fluorodeoxyglucose in myocardium and femoral muscle, assessed by planar imaging. Eur J Nucl Med. 1995;22:1141–1148. doi: 10.1007/BF00800596. [DOI] [PubMed] [Google Scholar]
- Moller K, Qvist T, Tofteng F, Sahl C, Sonderkaer S, Dethloff T, Knudsen GM, Larsen FS. Cerebral blood flow and metabolism during infusion of norepinephrine and propofol in patients with bacterial meningitis. Stroke. 2004;35:1333–1339. doi: 10.1161/01.STR.0000128418.17312.0e. [DOI] [PubMed] [Google Scholar]
- Leybaert L. Neurobarrier coupling in the brain: a partner of neurovascular and neurometabolic coupling. J Cereb Blood Flow Metab. 2005;25:2–16. doi: 10.1038/sj.jcbfm.9600001. [DOI] [PubMed] [Google Scholar]
- Gejl M, Sondergaard HM, Stecher C, Bibby BM, Moller N, Botker HE, Hansen SB, Gjedde A, Rungby J, Brock B. Exenatide alters myocardial glucose transport and uptake depending on insulin resistance and increases myocardial blood flow in patients with type 2 diabetes. J Clin Endocrinol Metab. 2012;97:E1165–E1169. doi: 10.1210/jc.2011-3456. [DOI] [PubMed] [Google Scholar]
- Morita K, Katoh C, Yoshinaga K, Noriyasu K, Mabuchi M, Tsukamoto T, Kageyama H, Shiga T, Kuge Y, Tamaki N. Quantitative analysis of myocardial glucose utilization in patients with left ventricular dysfunction by means of 18F-FDG dynamic positron tomography and three-compartment analysis. Eur J Nucl Med Mol Imaging. 2005;32:806–812. doi: 10.1007/s00259-004-1743-2. [DOI] [PubMed] [Google Scholar]
- Johnson KA, Fox NC, Sperling RA, Klunk WE. Brain imaging in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006213. doi: 10.1101/cshperspect.a006213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brands AM, Kessels RP, de Haan EH, Kappelle LJ, Biessels GJ. Cerebral dysfunction in type 1 diabetes: effects of insulin, vascular risk factors and blood-glucose levels. Eur J Pharmacol. 2004;490:159–168. doi: 10.1016/j.ejphar.2004.02.053. [DOI] [PubMed] [Google Scholar]
- Humpel C. Chronic mild cerebrovascular dysfunction as a cause for Alzheimer's disease. Exp Gerontol. 2011;46:225–232. doi: 10.1016/j.exger.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingham EM, Dunn JT, Smith D, Sutcliffe-Goulden J, Reed LJ, Marsden PK, Amiel SA. Differential changes in brain glucose metabolism during hypoglycaemia accompany loss of hypoglycaemia awareness in men with type 1 diabetes mellitus. An [11C]-3-O-methyl-D-glucose PET study. Diabetologia. 2005;48:2080–2089. doi: 10.1007/s00125-005-1900-6. [DOI] [PubMed] [Google Scholar]