

Abstract

An enantioselective copper-catalyzed hydroamination / cyclization of N-sulfonyl-2-allylanilines for the synthesis of chiral 2-methylindolines is reported. Chiral 2-methylindolines are important subunits in drugs and bioactive compounds. This method provides chiral N-sulfonyl-2-methyl indolines in up to 90% ee.
The enantioselective hydroamination / cyclization of aminoalkenes is an active topic of interest to organic and organometallic chemists.1 The nitrogen heterocyclic products obtained in these reactions are commonly found as components of bioactive natural products and pharmaceutical agents. While a number of highly selective methods for the metal-catalyzed enantioselective hydroamination / cyclization of 4-pentenylamines have been developed,1 few methods report use of 2-allylaniline-based substrates,1f–h and the highest enantioselectivity reported for the synthesis of a 2-methylindoline via alkene hydroamination is 70% ee.1g 2-Methylindolines can be found in bioactive molecules with diverse biological activities (e.g., Figure 1).2
Figure 1.

Bioactive compounds containing the 2-methylindoline core
The synthesis of chiral 2-methylindolines has been actively pursued3 and resolutions are frequently used to obtain these compounds in optically enriched form.3b,3c The enantioselective hydrogenation of indoles has also been reported an a straightforward method for synthesizing chiral indolines, but these methods often suffer from long reaction times, harsh reaction conditions or limited substrate scope.3d,3e Herein is reported a complementary method for the synthesis of variously substituted chiral 2-methylindolines 2 from N-sulfonyl-2-allylanilines 1 with up to 90% ee, the highest selectivity yet reported for the enantioselective alkene hydroamination / cyclization of a 2-allylaniline derivative (Scheme 1, vide infra).
Scheme 1.

Proposed copper-catalyzed enantioselective hydroamination
Reports of copper(I) and copper(II) complexes catalyzing the racemic hydroamination of alkenes have previously appeared.4 Copper(II) triflate-catalyzed intra- and intermolecular hydroamination of alkenes with sulfonamides have been reported by Komeyama, Chan and Hii, respectively, but these reactions are generally thought to be promoted by trace amounts of Bronsted acid generated under the reaction conditions.4a–c A Cu(O-t-Bu)•Xantphos catalyzed hydroamination / cyclization of electron-rich amines has been reported where copper(I) is thought to directly activate the alkene (via aminocupration), but no enantioselective variant of this reaction has yet appeared.4d The enantioselective copper(II)-catalyzed hydroamination / cyclization reaction reported herein is based off of a family of alkene aminofunctionalization reactions we have recently developed.5 In these reactions, addition of the nitrogen and copper(II) across the alkene is thought to occur via a cis aminocupration transition state (Scheme 1).5b,5c The resulting organocopper(II) intermediate is unstable and suffers homolysis under the reaction conditions, generating a primary organic radical and copper(I). We have shown that the carbon radical can be trapped with (2,2,6,6-tetramethylpiperidin-1-yl)oxyl radical (TEMPO) or undergo addition to various alkenes and arenes.5 We reasoned that atom transfer via H-atom abstraction from a good H-atom donor, e.g. 1,4-cyclohexadiene,5d would provide a net hydroamination product (Scheme 1).5c
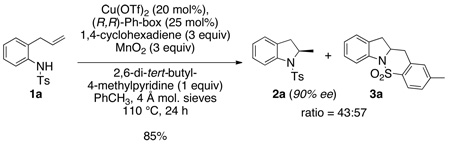
The enantioselectivity would be determined by the cis-aminocupration step, and we have previously shown that the (4R)-(+)-2,2’-isopropylidinebis(4-phenyl-2-oxazoline) [(R,R)-Ph-box] ligand provides very good levels of asymmetric induction in such reactions.5 In the event, N-tosyl-2-allylaniline 1a was subjected to [Cu(R,R)-Ph-box](OTf)2-catalyzed (20 mol%) cyclization in the presence of 1,4-cyclohexadiene (3 equiv) using MnO2 (3 equiv) as the stoichiometric oxidant in toluene at 110 °C (Eq 1). (The MnO2 used in all reactions described herein is activated, ca. 85%, <5 µm, as supplied by commercial vendor.) This reaction provided a 43:57 mixture of the desired hydroamination product 2a along with the intramolecular carboamination product 3a.5e Chiral HPLC analysis of 2-methylindoline 2a indicated it had been formed in 90% ee. Increasing the amount of 1,4-cyclohexadiene to 9 equiv resulted in only a slight decrease in the relative amount of carboamination product 3a (result not shown).
 |
(1) |
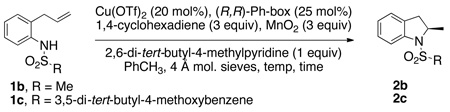
We next explored the hydroamination reactions of N-mesyl-2-allylaniline 1b, which cannot undergo the intramolecular carboamination reaction, and N-(3,5-di-tert-butyl-4-methoxybenzenesulfonyl)-2-allylaniline 1c, where intramolecular carboamination is minimized due to steric hindrance on the arylsulfonyl group (Table 1). The data in Table 1 indicate that the N-mesyl substrate 1b readily converts to the indoline product 2b in good yield (Table 1, entries 1–12). A number of reaction conditions such as temperature, time, amount of 1,4-cyclohexadiene, the presence or absence of 4 Å mol. sieves and catalyst loading were examined. We found that the reaction time could be reduced from 24 h to 8 h (Table 1, compare entries 1 and 2 to entries 3 and 4) and the reaction temperature could be lowered to 90 °C at 20 mol% [Cu(R,R)-Ph-box](OTf)2 loading (Table 1, entries 4–6). We found that flame dried 4 Å mol. sieves were beneficial to the reaction selectivity and reproducibility (Table 1, compare entries 6 and 8 and additional trials not shown), and 2,6-di-tert-butyl-4-methylpyridine was a better base than K2CO3 (Table 1, compare entries 6 and 9). We were able to reduce the catalyst loading to 15 mol% [Cu(R,R)-Ph-box](OTf)2 but the temperature had to be increased to 100 °C for optimal conversion at 8 h (Table 1, compare entries 10 and 11). Thus, the optimal conditions are 15 mol% [Cu(R,R)-Ph-box](OTf)2, 2,6-di-tert-butyl-4-methylpyridine (1 equiv) as base, 3 equiv 1,4-cyclohexadiene at 100 °C for 8 h to give 73% yield of 2-methylindoline 2b in 77% ee (Table 1, entry 11). We attempted to use γ-terpinene as a hydrogen atom source (Table 1, entry 12) under conditions otherwise identical to Table 1, entry 6 and found that this commonly available natural product could transfer a hydrogen atom, but was less efficient than 1,4-cyclohexadiene.
Table 1.
Optimization of the Hydroaminationa
 | |||||
|---|---|---|---|---|---|
| entry | substrate | temp (°C) |
time (h) |
yield (%) |
ee (%) |
| 1b,c | 1b | 110 | 24 | >95d | nd |
| 2c | 1b | 110 | 24 | >95d | nd |
| 3 | 1b | 120 | 8 | >95d | nd |
| 4 | 1b | 110 | 8 | 77 (>95d) |
81 |
| 5 | 1b | 100 | 8 | 77 | 80 |
| 6 | 1b | 90 | 8 | 77 | 77 |
| 7 | 1b | 80 | 8 | 61d | nd |
| 8c | 1b | 90 | 8 | 74 | 73 |
| 9e | 1b | 90 | 8 | 56 (89d) |
76 |
| 10f | 1b | 90 | 8 | 90d | nd |
| 11f | 1b | 100 | 8 | 73 (>95d) |
77 |
| 12g | 1b | 90 | 8 | 50d | nd |
| 13 | 1c | 110 | 8 | >90d (70:30)h |
nd |
| 14 | 1c | 100 | 8 | 80d (81:19)h |
nd |
| 15b | 1c | 110 | 8 | 72 (94:6)h |
86 |
| 16b,f | 1c | 110 | 8 | >90d (77:23)h |
nd |
Conditions: Cu(OTf)2 (20 mol%) and (R,R)-Ph-box (25 mol%) were stirred for 2 h at 60 °C in dry toluene in a sealed tube under Ar. The cooled solution was diluted with dry toluene (0.1 M total concentration with respect to 1) and was treated with sulfonamide 1b (0.237 mmol) or 1c (0.120 mmol), 2,6-di-tert-butyl-4-methylpyridine (1 equiv), MnO2 (3 equiv), 1,4-cyclohexadiene (3 equiv) and 4 Å flame-dried mol. sieves (20 mg/mL) under Ar and was heated in the sealed tube for 8 h unless otherwise noted. Filtration through Celite and flash chromatography on SiO2 afforded the isolated product unless otherwise noted. % ee was determined by chiral HPLC.
5 equiv of 1,4-cyclohexadiene was used.
No molecular sieves were used.
% conversion (NMR) given.
K2CO3 (1 equiv) was used instead of 2,6-di-tert-butyl-4-methylpyridine.
15 mol% Cu(OTf)2 and 19 mol% (R,R)-Ph-box were used.
γ-terpinine was used instead of 1,4-cyclohexadine.
Ratio of hydroamination (2c) to carboamination (3c) as determined by analysis of the crude 1H NMR spectrum. nd = not determined.
The hydroamination reaction of N-arylsulfonyl-2-allylaniline 1c, a hindered analog of N-tosyl-2-allylaniline 1a, was next investigated. The hope was the enantioselectivity could be increased, relative to the reaction of 1b (note hydroamination of 1a occurs with 90% ee, Eq. 1, vide supra) without reducing the hydroamination yield due to competing carboamination. When substrate 1c was subjected to the reaction conditions [110 °C, 20 mol% Cu[(R,R)-Ph-box]OTf2 and 3 equiv 1,4-cyclohexadiene, Table 1, entry 13], >90% conversion was achieved but the hydroamination reaction was accompanied by formation of the corresponding carboamination product 3c (hydroamination : carboamination = 70:30). When the temperature was decreased, the ratio improved but the reaction did not go to completion (Table 1, entry 14). Increasing the amount of 1,4-cyclohexadiene to 5 equiv and running the reaction at 110 °C provided a tolerable hydroamination:carboamination ratio of 94:6 where the reaction went to completion (Table 1, entry 15). This reaction provided the highest isolated yield (72%) of 2c, which was formed in 86% ee. Reduction of the catalyst loading to 15 mol% led to an increased formation of the undesired carboamination product (Table 1, entry 16).
The reaction enantioselectivity as a function of time was briefly examined (not shown) to ascertain if enantioenrichment could be lost due to reversible hydroamination.4c In such a scenario, the reaction enantioselectivity could be higher in the initial hours of the reaction. Running substrate 1b under the same conditions as Table 1, entry 11 but for 3 hours only led to a 30% isolated yield of 2b (the rest being substrate 1b) in 67% ee. This indicates that the reaction is not more selective in its early stages and the observed enantioselectivity is nearly within experimental error of Table 1, entry 11 for the detection method (±5% ee for chiral HPLC). We also ran a control experiment using the same conditions as Table 1, entry 11 but without Cu[(R,R)-Ph-box]OTf2 (not shown). This control reaction gave 8% conversion of 1b to 2b, indicating there is a degree of uncatalyzed background reaction that can lead to racemic product.
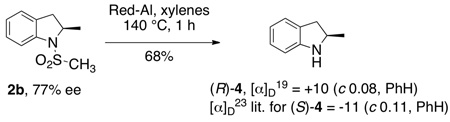
The absolute stereochemistry of N-mesyl-2-methylindoline 2b (77% ee), initially assigned R by analogy to other Cu[(R,R)-Ph-box]OTf2 catalyzed reactions we have reported with substrates 1,5 was confirmed by removal of the mesyl group by treatment with sodium bisaluminumhydride (Red-Al) in refluxing in xylenes (Eq 2). (Removal of the arylsulfonyl group of 2c is best performed as previously described.5f) The optical rotation of the 2-methylindoline was compared to the known value for (S)-2-methylindoline (4)3b and found to be of the opposite sign, establishing the configuration of our major product as R. 2-Methylindoline (4) is a key intermediate in the synthesis of the clinically used diuretic indapamide, thus methods for the enantioselective synthesis of this compound and analogs thereof are useful.2a
 |
(2) |
The hydroamination reactions of various N-sulfonyl-2-allylanilines 1 were examined under the optimal reaction conditions (Table 1, entry 11 for N-mesyl substrates 1d and 1e and Table 1, entry 15 for N-arylsulfonyl substrates 1f–j and N-SES substrate 1k) as illustrated in Scheme 2. Yields of the isolated hydroamination products are given along with the hydroamination (HA) to carboamination (CA) ratios (based on analysis of the crude 1H NMR) of substrates with N-arylsulfonyl groups. The hydroaminations of substrates 1f and 1g were run for 24 h and the reaction of 1g was run with 3 equiv of 1,4-cyclohexadiene (reactions performed prior to reaction time optimization). The N-mesyl-2-allylanilines 1d and 1e gave the respective indolines in 76% and 73% ee while the N-SES substrate 1k gave indoline 2k in 72% ee. The N-arylsulfonyl-2-allylanilines 1c and 1f–1j gave higher levels of enantioselectivity but the yields were somewhat eroded by formation of the competing carboamination products 3. We observed that anilines 1c (Table 1) and 1i (containing H and Me aniline substituents, respectively) gave higher ratios of hydroamination to carboamination (3, Fig. 2) than did the anilines 2f, 2g, 2h and 2j (functionalized with MeO, Cl, Br, CN aniline substituents). Nevertheless, moderately good yields of the hydroamination products can be obtained in all cases and the enantioselectivities of the N-arylsulfonyl hydroamination products range from 84–89%. Indoline products 2c and 2g-2j were isolated as solids, so further enantiomeric enrichment of these compounds via recrystallization can in principle be achieved.5g,5i
Scheme 2.

Scope of the Enantioselective Indoline Synthesis
Figure 2.

Carboamination side products
Ar = 3,5-di-tert-butyl-4-methoxybenzene, Ms = methane sulfonyl, SES = 2-trimethylsilylethylsulfonyl. % Yield refers to amount of pure isolated indoline 2. HA = hydroamination product (2); CA = carboamination product (3); ratio obtained by analysis of the crude 1H NMR spectrum. Reactions of R2NMs substrates were performed at 100 °C with 15 mol% [Cu]; reactions of R2NSO2Ar and R2NSES substrates were performed at 110 °C with 20 mol% [Cu]. nd = not determined. Enantiomers could not be separated by chiral HPLC.
In summary, we have developed the first copper-catalyzed enantioselective alkene hydroamination/cyclization. The method provides an expedient route to enantioenriched 2-methylindolines, compounds of value in medicinal chemistry and drug design. This chiral 2-methylindoline synthesis method is competitive with other reported methods.3 The N-sulfonyl-2-allylaniline substrates 1 can be synthesized in 2-3 steps5 and the hydroamination reaction uses a commercially available, amino-acid derived chiral ligand and a widely available copper(II) salt. The hydrogen atom source, 1,4-cyclohexadiene, is low molecular weight, relatively inexpensive and produces a volatile by-product. This enantioselective intramolecular alkene hydroamination provides the highest enantioselectivity yet reported for the hydroamination of 2-allylaniline-derived substrates and several of the products are crystalline, so the enantiomeric excesses of such products can in principle be even further enhanced. Further reaction optimization and expansion of the substrate scope are underway and will be reported in due course.
Supplementary Material
ACKNOWLEDGMENT
We thank the National Institutes of Health (GM078383) for support of this work.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Full experimental details, characterization data and NMR spectra for all new compounds, chiral HPLC traces for all chiral products. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
REFERENCES
- 1.Reviews: Mueller TE, Hultzsch KC, Yus M, Foubelo F, Tada M. Chem. Rev. 2008:3795–3892. doi: 10.1021/cr0306788. Zi GF. J. Organomet. Chem. 2011;696:68–75. Chemler SR. Org. Biomol. Chem. 2009;7:3009–3019. doi: 10.1039/B907743J. Allaud I, Collin J, Hannedouche J, Schulz E. Dalton Trans. 2007:5105–5118. doi: 10.1039/b711126f. Hultzsch KC. Org. Biomol. Chem. 2005;3:1819–1824. doi: 10.1039/b418521h. Enantioselective hydroamination of 2-allylanilines Knight PD, Munslow I, O’Shaughnessy PN, Scott P. Chem. Commun. 2004:894–895. doi: 10.1039/b401493f. Watson DA, Chiu M, Bergman RG. Organometallics. 2006;25:4731–4733. doi: 10.1021/om0606791. Shen X, Buchwald SL. Angew. Chem. Int. Ed. 2010;49:564–567. doi: 10.1002/anie.200905402.
- 2.(a) Temperini C, Cecchi A, Scozzafava A, Supuran CT. J. Med. Chem. 2009;52:322–328. doi: 10.1021/jm801386n. [DOI] [PubMed] [Google Scholar]; (b) Khan PM, Correa RG, Divlianska DB, Peddibhotla S, Sessions EH, Magnuson G, Brown B, Suyama E, Yuan H, Mangravita-Novo A, Vicchiarelli M, Su Y, Vasile S, Smith LH, Diaz PW, Reed JC, Roth GP. ACS Med. Chem. Lett. 2011;2:780–785. doi: 10.1021/ml200158b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yoon SJ, Chung YH, Lee MS, Choi DR, Lee JA, Lee HS, Yun HR, Lee DK, Moon EY, Hwang HS, Choi CH, Jung SH. Arylsulfonylimidazoline derivatives as a antitumor agent. 5,932,742. U.S. Patent. 1999 Aug 3;
- 3.(a) Anas S, Kagan HB. Tetrahedron Asym. 2009:2193–2199. [Google Scholar]; (b) Arp FO, Fu GC. J. Am. Chem. Soc. 2006;128:14264–14265. doi: 10.1021/ja0657859. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hou XL, Zheng BH. Org. Lett. 2009;111:1789–1791. doi: 10.1021/ol9002543. [DOI] [PubMed] [Google Scholar]; (d) Xiao YC, Wang C, Yao Y, Sun J, Chen YC. Angew. Chem. Int. Ed. 2011;50:10661–10664. doi: 10.1002/anie.201105341. [DOI] [PubMed] [Google Scholar]; (e) Wang DS, Chen QA, Li W, Yu CB, Zhou YG, Zhang X. J. Am. Chem. Soc. 2010;132:8909–8911. doi: 10.1021/ja103668q. [DOI] [PubMed] [Google Scholar]
- 4.Copper-catalyzed hydroaminations: Komeyama K, Morimoto T, Takaki K. Angew. Chem. Int. Ed. 2006;45:2938–2941. doi: 10.1002/anie.200503789. Rao W, Kothandaraman P, Koh CB, Chan PWH. Adv. Synth. Catal. 2010;352:2521–2530. Taylor JG, Whittall N, Hii KK. Org. Lett. 2006;8:3561–3564. doi: 10.1021/ol061355b. Ohmiya H, Moriya T, Sawamura M. Org. Lett. 2009;11:2145–2147. doi: 10.1021/ol9007712. Munro-Leighton C, Delp SA, Alsop NM, Blue ED, Gunnoe TB. Chem. Commun. 2008:111–113. doi: 10.1039/b715507g.
- 5.(a) Chemler SR. J. Organomet. Chem. 2011;696:150–158. doi: 10.1016/j.jorganchem.2010.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Paderes MC, Belding L, Fanovic B, Dudding T, Keister JB, Chemler SR. Chem.-Eur. J. 2012;18:1711–1726. doi: 10.1002/chem.201101703. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sherman ES, Fuller PH, Kasi D, Chemler SR. J. Org. Chem. 2007;72:3896–3905. doi: 10.1021/jo070321u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Miller Y, Miao L, Hosseini AS, Chemler SR. J. Am. Chem. Soc. 2012 doi: 10.1021/ja3034075. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zeng W, Chemler SR. J. Am. Chem. Soc. 2007;129:12948–12949. doi: 10.1021/ja0762240. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Fuller PH, Kim JW, Chemler SR. J. Am. Chem. Soc. 2008;130:17638–17639. doi: 10.1021/ja806585m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Bovino MT, Chemler SR. Angew. Chem. Int. Ed. 2012;51:3923–3927. doi: 10.1002/anie.201109044. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Liwosz TW, Chemler SR. J. Am. Chem. Soc. 2012;134:2020–2023. doi: 10.1021/ja211272v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Sequeira FC, Bovino MT, Chipre AJ, Chemler SR. Synthesis. 2012;44:1481–1484. doi: 10.1055/s-0031-1289762. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.