

Abstract
Host defense against the parasite Toxoplasma gondii requires the cytokine interferon-gamma (IFNγ). However, Toxoplasma inhibits the host cell transcriptional response to IFNγ, which is thought to allow the parasite to establish a chronic infection. It is not known whether all strains of Toxoplasma block IFNγ-responsive transcription equally and whether this inhibition occurs solely through the modulation of STAT1 activity or whether other transcription factors are involved. We find that strains from three North American/European clonal lineages of Toxoplasma, types I, II, and III, can differentially modulate specific aspects of IFNγ signaling through the polymorphic effector proteins ROP16 and GRA15. STAT1 tyrosine phosphorylation is activated in the absence of IFNγ by the Toxoplasma kinase ROP16, but this ROP16-activated STAT1 is not transcriptionally active. Many genes induced by STAT1 can also be controlled by other transcription factors and therefore using these genes as specific readouts to determine Toxoplasma inhibition of STAT1 activity might be inappropriate. Indeed, GRA15 and ROP16 modulate the expression of subsets of IFNγ responsive genes through activation of the NF-κB/IRF1 and STAT3/6 transcription factors, respectively. However, using a stable STAT1-specific reporter cell line we show that strains from the type I, II, and III clonal lineages equally inhibit STAT1 transcriptional activity. Furthermore, all three of the clonal lineages significantly inhibit global IFNγ induced gene expression.
Introduction
The cytokine interferon-gamma (IFNγ) and the transcription factor it activates, signal transducer and activator of transcription (STAT) 1, are critical to host defense against the obligate intracellular parasitic pathogen Toxoplasma gondii; mice deficient in elements of this pathway are acutely susceptible to Toxoplasma infection [1]–[3]. Activated STAT1 induces the expression of genes with gamma activated sequence (GAS) elements in their promoters, including the interferon regulatory factor (IRF) 1 transcription factor. STAT1 and IRF1 together induce a broad transcriptional program including effector mechanisms that mediate pathogen destruction or inhibition of pathogen growth [4].
However, Toxoplasma infection can inhibit IFNγ induced gene expression in host cells, and was first shown to inhibit the basal and IFNγ induced expression of MHC class II molecules, in a variety of cell types [5]–[7]. Since then, Toxoplasma has also been shown to inhibit the expression of IRF1 [8], [9], class II transactivator (CIITA) [7]–[9], inducible nitric oxide synthase (iNOS/NOS2) [10], [11], interferon inducible GTPase 1 (IIGP1) [12], and chemokine (C-X-C motif) ligand 9 (MIG/CXCL9) [12]. This inhibition occurs in a variety of cell types, including human foreskin fibroblasts (HFF), human glioblastoma cells, murine bone marrow-derived macrophages (BMDM), RAW264.7 murine macrophages, murine dendritic cells, and murine microglial cells. Microarray analyses showed that Toxoplasma infection can dysregulate the entire IFNγ induced gene expression program in both HFFs [13] and BMDMs [14].
Toxoplasma infects virtually all warm-blooded animals, including ∼30% of the worldwide human population [15]. Many different strains of Toxoplasma have been isolated from various hosts, and in North America and Europe the majority of Toxoplasma isolates from humans and livestock belong to three main clonal lineages: types I, II, and III [16]. These strains differ in the modulation of multiple host cell signaling pathways through polymorphic effectors secreted into the host cell from rhoptry and dense granule organelles [17]. While all of these strains can inhibit the expression of at least certain IFNγ induced genes, it is unknown whether all of the strains can inhibit global IFNγ induced gene expression and STAT1 transcriptional activity, or whether the degree of inhibition varies between Toxoplasma strains.
Many STAT1 regulated genes can be induced or repressed by other transcription factors, for example NF-κB and STAT3/6, and such genes might not be the best readouts to determine if Toxoplasma specifically inhibits STAT1 activity. Another question that is still unanswered is whether the activation of other transcription factors by Toxoplasma affects the IFNγ response. Specifically, the modulation of STAT3/6 and NF-κB transcription factors through the effector proteins ROP16 [18] and GRA15 [19], respectively, might affect this response.
The polymorphic rhoptry kinase ROP16 from type I and III strains activates the transcription factors STAT3 and STAT6 [18], [20], [21]. In STAT3 deficient cells [22] or cells with STAT6 knocked down [23], increased transcription of STAT1 target genes has been found, suggesting that STAT3 and STAT6 can antagonize STAT1 activity. STAT6 can also compete for promoter sites with STAT1 [24]. It is therefore possible that the activation of STAT3/6 by ROP16 helps to suppress IFNγ induced signaling.
SOCS family proteins are important negative regulators of the IFNγ response and in Socs1 −/− BMDM, Toxoplasma could not inhibit the IFNγ response as well as in wild-type BMDM [12]. ROP16 is a strong activator of SOCS family gene expression; in murine BMDM, Socs1, 2, and 3 are more than 10-fold induced by ROP16 expression [25]. It is therefore possible that ROP16 plays a role in the inhibition of the IFNγ response through the induction of Socs genes. Furthermore, the expression of genes that are co-regulated by both STAT1 and STAT3/6 transcription factors could also be affected by ROP16. If the expression level of such a gene was chosen to measure STAT1 activity, incorrect conclusions might be drawn.
The type II version of the dense granule protein GRA15 activates the host cell NF-κB pathway [19]. NF-κB also co-regulates many of the same genes as STAT1, and NF-κB activation combined with STAT1 activation synergistically induces IRF1 expression and activity [26]. It is therefore possible that strains possessing an active copy of GRA15 do not inhibit IFNγ induced gene expression as well as other strains, or differentially inhibit subsets of IFNγ responsive genes. In fact, a type II Δgra15 strain grows faster in vivo than a type II strain [19], and GRA15 corresponds to a Toxoplasma virulence locus [19], [27].
In this report we show that the polymorphic effectors GRA15 and ROP16 do contribute to strain differences in the modulation of IFNγ-STAT1 signaling. Type II GRA15 induces the expression of IRF1, which can induce a subset of IFNγ responsive genes. ROP16 induces the tyrosine phosphorylation and nuclear translocation of STAT1 but this STAT1 is not transcriptionally active. In spite of these differences, type I, II, and III parasites can all inhibit global IFNγ induced transcription as determined by microarray analysis. Because many STAT1-regulated genes can be controlled by other transcription factors we directly measured STAT1 activity with a stable STAT1-specific reporter cell line and find that neither GRA15 nor ROP16 affects the ability of Toxoplasma to inhibit STAT1 transcriptional activity.
Materials and Methods
Parasites and Cells
Parasites were maintained in vitro by serial passage on monolayers of human foreskin fibroblasts (HFFs), as described previously [19]. RH or GT1 were used as representative type I strains, Pru or ME49 as representative type II strains and CEP or VEG as representative type III strains. A Pru strain engineered to express firefly luciferase and GFP (Pru Δhxgprt A7) [28], and CEP and RH strains engineered to express clickbeetle luciferase and GFP (CEP hxgprt − C22 and RH 1–1) [29] have been described previously. An RH Δrop16 strain (provided by John Boothroyd, Stanford University) [21], an RH Δrop16 strain expressing firely luciferase and GFP (clone 1A2) [25], a PruA7 ROP16I strain [25], and Pru Δgra15, PruA7 Δgra15, and RH GRA15II strains [19] have been described previously. HFFs (provided by John Boothroyd, Stanford University) and RAW264.7 (ATCC) cells were grown as described previously [19], [25]. 293FT and HEK293 cells were grown with additional 10 mM HEPES. U3A STAT1-null cells [30], [31] (provided by George Stark, Cleveland Clinic Foundation Research Institute, Ohio) were grown with 10 mM HEPES, 1 mM sodium pyruvate, and MEM non-essential amino acids. All parasite strains and cell lines were routinely checked for Mycoplasma contamination and it was never detected.
Reagents
Antibodies against IRF1 (BD Biosciences #612046), phospho-STAT1Tyr701 58D6 (Cell Signaling #9167), phospho-STAT1Ser727 (Cell Signaling #9177), total STAT1α p91 (C-24) (Santa Cruz #345), GAPDH (6C5) (Santa Cruz #32233), and Toxoplasma surface antigen (SAG)-1 (kindly provided by John Boothroyd, Stanford University) were used in immunofluorescence and Western blot assays. Secondary antibodies coupled with either Alexa Fluor 488 or Alexa Fluor 594 (Molecular Probes) for immunofluorescence assay or conjugated to peroxidase (Kirkegaard & Perry Laboratories) for Western blots were used. Recombinant human IFNγ (100 U/ml, AbD serotec) and murine IFNγ (100 U/ml, Calbiochem) were used to stimulate cells.
Immunofluorescence Assay
Immunofluorescence assay was performed as described previously [19]. Briefly, cells were fixed with 3% formaldehyde, permeabilized with 100% ethanol and/or 0.2% Triton-X 100, and blocked with 3% BSA and 5% goat serum. Coverslips were incubated with primary antibody at 4°C, and fluorescent secondary antibodies and Hoechst dye were used for antigen and DNA visualization, respectively. Photographs were taken using NIS-Elements software (Nikon) and a digital camera (Coolsnap EZ; Roper Scientific) connected to an inverted fluorescence microscope (model eclipse Ti-S; Nikon). Quantification of nuclear signal was performed by randomly selecting cells in each condition and measuring the average signal intensity per nucleus using the NIS-Elements software and Hoechst dye to define nuclei. The minimum number of cells measured is indicated in the figure legends for each experiment.
Western Blot
Western blots were performed as described previously [19]. Briefly, HFFs were left uninfected or infected with RH Δhxgprt, RH 1–1, RH Δrop16, RH Δrop16 1A2, Pru Δhxgprt A7, or CEP hxgprt - C22 parasites for three hours. Samples were subsequently stimulated with human IFNγ for one hour, or left unstimulated, and then lysed in buffer containing sodium dodecyl sulfate (SDS) and either β-mercaptoethanol (βME) or dithiothreitol (DTT). After immunoblotting, membranes were stripped with boiling 2% SDS and 0.7% βME and reprobed.
Reporter Cell Line Construction
A GAS (TR027PA-1, 5′-AGTTTCATATTACTCTAAATC -3′) pGreenFire1 (pGF1) lentiviral reporter vector containing a Neo selection cassette and a minimal CMV promoter followed by four tandem consensus GAS sites driving the expression of Firefly luciferase was purchased from System Biosciences. The vector was co-transfected into 293FT cells with vectors containing gag, pol, and VSV-G proteins using FuGENE reagent (Roche) according to the manufacturer’s protocol. Supernatant containing virus was collected two and three days after transfection, filtered with a 0.45 µm surfactant-free cellulose acetate filter (Nalgene), and added to HEK293 cells (ATCC) with 8 µg/ml polybrene (Sigma). HEK293 cells containing the pGF1 construct were then selected with 750 µg/ml Geneticin (Invitrogen). Cells were cloned by limiting dilution and were confirmed to be responsive to IFNγ but not to IFNβ, TNFα, or IL4.
Luciferase Assay
HEK293 pGF1-GAS cells were plated in 96-well plates, 3.5–4×104 cells/well, and grown for 4–20 hours. Cells were then infected with RH Δhxgprt, RH Δrop16, GT1, Pru Δhxgprt, Pru Δgra15, ME49, CEP Δhxgprt, or VEG parasites at varying MOIs for 1–4 hours, and subsequently stimulated with human IFNγ for 12–24 hours. Cells were lysed with 20 µl Cell Culture Lysis Reagent (Promega) containing 1× protease inhibitors (Roche), and plates were frozen at −80°C. Luciferase activity in plates was detected using a Varioskan Flash Reader (Roche) after addition of 100 µl Luciferase assay substrate (Promega), according to the manufacturer’s instructions. Data were normalized to the uninfected, unstimulated sample and averaged over at least two experiments per condition.
Microarray
1.5×106 RAW264.7 cells were plated in 6-well plates and grown for 24 hours. The cells were then left uninfected or infected with RH 1–1, Pru Δhxgprt A7, or CEP hxgprt - C22 parasites at an MOI ∼5 for 18 hours and subsequently stimulated with murine IFNγ for six hours. The RH infection was done at one time and Pru and CEP infections were done together at a later time. Uninfected controls were included for both sets of infections. RNA was isolated and microarray analysis, including analysis with the DiRE server, was performed as described previously [19], with Mouse 430A 2.0 Affymetrix gene chips. Microarray data has been uploaded to NCBI Gene Expression Omnibus and are accessible through GEO Series accession number GSE34913.
Plaque Assay
For Western blot, luciferase reporter, and microarray assays, a plaque assay was done to determine the viability of each strain and the actual MOI. One hundred parasites per well were added to confluent HFFs in a 24-well plate and were incubated undisturbed for 5–7 days at 37°C, and the number of plaques was counted. Samples with similar MOIs were then picked for analysis.
Results
A Type II Strain Activates IRF1 via GRA15 and NF-κB
Infection of HFFs with a type II Pru strain of Toxoplasma was previously shown to induce the expression of 46 genes that were also defined as IFNγ regulated [13], raising the possibility that type II strains are not as good at inhibiting IFNγ induced gene expression as other clonal lineages. To compare the ability of type I, II, and III strains to inhibit the IFNγ response we pre-infected HFFs with RH(I), Pru(II), or CEP(III) strains, or left cells uninfected, and subsequently stimulated the cells with IFNγ or left them unstimulated. We then visualized and quantified the amount of IRF1 in the nucleus by immunofluorescence. IRF1 is a primary response gene induced directly by STAT1 upon IFNγ stimulation. After three hours of infection, with IFNγ stimulation for the last two hours, cells pre-infected with either RH(I), Pru(II), or CEP(III) all had significantly lower levels of IRF1 in their nuclei than uninfected cells (Fig. 1A, B), as was previously seen for type I, II, and III strains [8], [9], [13]. However, after 24 hours of infection, with IFNγ stimulation for the last six hours, while RH(I), Pru(II), and CEP(III) infection all significantly inhibited IRF1 expression compared to uninfected cells, cells pre-infected with a Pru(II) strain had significantly higher IRF1 in their nuclei than cells pre-infected with a RH(I) strain (Fig. 1A, B). Cells pre-infected with a Pru(II) strain also had higher levels of IRF1 than cells pre-infected with a CEP(III) strain but this difference was not statistically significant. These data suggest that a Pru(II) strain does not inhibit IRF1 expression as well as RH(I) or CEP(III) strains.
Figure 1. Type II GRA15 affects the expression of IRF1.

HFFs were infected with RH(I), RH GRA15II, Pru(II), PruΔgra15, or CEP(III) parasites and/or stimulated with 100 U/ml IFNγ, subsequently fixed and permeabilized and stained with an antibody against IRF1 (red) and with Hoechst dye (blue, nucleus). A, B. HFFs were infected for three or 24 hours with GFP-expressing parasites (green), or left uninfected, and stimulated with IFNγ for the last two or six hours, or left unstimulated. Nuclear localization of IRF1 was quantified in at least 12 randomly selected cells per condition and normalized to uninfected, unstimulated cells (A) and a representative cell for each condition is shown (B). Scale bar represents 10 µm. This experiment was performed three times with similar results. Data and standard deviation from one representative experiment are shown. Asterisk (*) indicates p<0.05 compared to uninfected cells, dollar sign ($) indicates p<0.05 compared to type II infected cells. C. HFFs were infected with an RH(I) or RH GRA15II strain, left uninfected, or left uninfected and stimulated with IFNγ for 24 hours. Nuclear localization of IRF1 was quantified in at least 30 randomly selected cells and normalized to uninfected, unstimulated cells. Data and standard deviation from one experiment are shown. Asterisk (*) indicates p<0.05 compared to uninfected cells or as represented by brackets. D. U3A STAT1-deficient cells were infected with Pru(II) or PruΔgra15 parasites for 20 hours, left uninfected, or stimulated with IFNγ for 1 hour. Nuclear localization of IRF1 was quantified in at least 8 randomly selected cells per condition, and normalized to uninfected, unstimulated cells. This experiment was performed twice with similar results, data from one representative experiment are shown. Asterisk (*) indicates p<0.05 compared to uninfected cells, dollar sign ($) indicates p<0.05 compared to type II infected cells.
We next determined IRF1 levels after infection in the absence of IFNγ. In unstimulated cells infected with Pru(II) for 24 hours, we find ∼2.5 fold higher nuclear IRF1 levels than in uninfected cells or cells infected with either RH(I) or CEP(III) (Fig. 1A, B). These data suggest that the different IRF1 protein levels observed in Pru(II) and RH(I) infected cells after IFNγ treatment may not be due to differences in the ability of these strains to inhibit IFNγ induced gene expression but instead due to the induction of IRF1 by Pru(II) infection, independently of IFNγ.
Although IRF1 is primarily induced by interferons it also has three NF-κB binding sites in its promoter [4] and these are important for the synergistic induction of genes by IFNγ and TNFα [26]. Type II parasites activate NF-κB-mediated transcription via the dense granule protein GRA15 [19] and we hypothesized that GRA15-mediated NF-κB activation could drive the expression of IRF1. To test this hypothesis, we also infected HFFs with a PruΔgra15 strain (Fig. 1A, B). After 24 hours of infection, this strain induced significantly less IRF1 protein than a Pru strain (p<0.001) and PruΔgra15 infected cells have similar IRF1 levels as cells infected with RH(I) and CEP(III) strains which possess inactive copies of GRA15 [19]. An RH(I) strain ectopically expressing a type II copy of GRA15 also induced IRF1 expression in HFF host cells (Fig. 1C).
To determine whether this GRA15-mediated activation of IRF1 is dependent on STAT1, we infected U3A STAT1-deficient cells [30], [31] with either Pru(II) or PruΔgra15 parasites, or stimulated the cells with IFNγ. While IFNγ treatment, which relies on STAT1 signaling, does not activate IRF1 expression in these cells, infection with Pru(II) parasites does, and this activation is again dependent on the presence of GRA15 (Fig. 1D), demonstrating that the GRA15-mediated induction of IRF1 is through a different transcription factor. GRA15 is known to activate the NF-κB p65 transcription factor [19], and since it is also known that NF-κB can activate the expression of IRF1 [4], [26], we hypothesized that GRA15 was inducing IRF1 through the activation of NF-κB p65. Indeed, in a previous microarray analysis [19], while Irf1 transcript was induced by infection of wild-type MEFs with GRA15-expressing Pru(II) parasites, infection with this strain did not induce Irf1 transcript in p65−/− MEFs, strongly suggesting that induction of IRF1 expression by GRA15 is through the NF-κB p65 transcription factor.
IRF1 is itself a transcription factor and to test whether GRA15 might be responsible for more than just the expression of IRF1, but also the expression of other IFNγ regulated genes that were found to be induced by Pru infection [13], we re-analyzed the microarray data from which this observation was made. We found 775 oligonucleotide probes that were at least two-fold induced in HFFs by IFNγ treatment and by Pru infection. These 775 probes correspond to 374 genes also present in a microarray analysis of HFFs infected with GRA15-deficient and GRA15-overexpressing Toxoplasma strains [19]. Of these 374 genes, 43 were previously found to be at least two-fold GRA15-regulated in at least one parasite genetic background [19], a significant enrichment (p = 0.03, hypergeometric test), indicating that GRA15 does induce the expression of a subset of IFNγ responsive genes (Data S1). Therefore, while type I, II, and III Toxoplasma strains can all inhibit the IFNγ induced expression of IRF1, type II strains also induce IRF1 expression, independently of STAT1, most likely through GRA15-mediated activation of NF-κB p65. This IRF1 induction also leads to the expression of a small subset of other IFNγ responsive genes.
Toxoplasma Infection Affects STAT1 Phosphorylation
After IFNγ treatment, STAT1 is tyrosine phosphorylated in the cytoplasm which allows it to traffic to the nucleus. Most recently, it was shown that infection of cells with type II Pru [13] or NTE parasites [9], [14] does not inhibit IFNγ-induced STAT1 trafficking into the nucleus. Previously however, the nuclear translocation of STAT1 was reported to be inhibited by type II (NTE) Toxoplasma infection [8]; the tyrosine phosphorylation of STAT1 was reported to be inhibited by type I (BK) infection [12], and type I (RH), type II (Pru), and type III (CL14) Toxoplasma strains were suggested to cause dephosphorylation of STAT1 in the nucleus [13].
To determine if there are strain differences in the effect of infection on IFNγ-induced STAT1 phosphorylation and localization, we infected HFFs for one hour with either RH(I), Pru(II), or CEP(III) parasites, subsequently stimulated the cells for two hours with IFNγ, and quantified STAT1 tyrosine phosphorylation and nuclear translocation by immunofluorescence (Fig. 2A, B). Quantification of the immunofluorescence signal revealed that levels of IFNγ induced nuclear phospho-STAT1Tyr were actually higher in infected cells compared to uninfected cells (Fig. 2A). We therefore find that none of the tested Toxoplasma strains inhibit the tyrosine phosphorylation or nuclear accumulation of phospho-STAT1Tyr after IFNγ treatment, which agrees with the majority of previous results.
Figure 2. ROP16 activates STAT1 tyrosine phosphorylation and nuclear translocation.

A, B. HFFs were infected with a GFP (green) expressing RH(I), RHΔrop16, Pru(II), Pru ROP16I, or CEP(III) strain, or left uninfected, for 3 hours, and 100 U/ml IFNγ was added for the last two hours of infection, or cells were left unstimulated. Cells were fixed, permeabilized, and stained with anti-phospho-STAT1Tyr (red) and Hoechst dye (nucleus, blue). Nuclear localization of phospho-STAT1Tyr was quantified in at least 30 randomly selected cells infected with at least three parasites (A) and a representative cell for each condition is shown (B). Scale bar represents 10 µm. This experiment was performed for each condition at least two times with similar results. Data and standard deviation from one representative experiment are shown. Asterisk (*) indicates p<0.05 compared to uninfected cells or as indicated by brackets.
Since we observed higher levels of phospho-STAT1Tyr in infected cells as compared to uninfected cells after IFNγ stimulation, we wondered whether infection with type I, II, or III parasites induces nuclear phospho-STAT1Tyr in the absence of IFNγ. We infected HFFs for three hours with either RH(I), Pru(II), or CEP(III) parasites, and quantified STAT1 tyrosine phosphorylation and nuclear translocation by immunofluorescence. Indeed, we observed nuclear phospho-STAT1Tyr in unstimulated cells infected with three or more RH(I) or CEP(III) parasites, and to a lower level in cells infected with three or more Pru(II) parasites (Fig. 2B). We quantified this signal in cells infected with three or more parasites and find that infection results in a significant increase in nuclear phospho-STAT1Tyr levels in RH(I) and CEP(III) infected cells (Fig. 2A). Pru(II) infection also significantly induces phospho-STAT1Tyr, although not as highly as RH(I) or CEP(III) parasites (Fig. 2A).
We next sought to determine what parasite factor induces phospho-STAT1Tyr after host cell infection. It is known that the secreted rhoptry kinase ROP16 from type I and III strains can directly tyrosine phosphorylate STAT3 and STAT6 [20], [21]. The first 700 amino acid residues of STATs 1–6 share up to 40% identity [32], raising the possibility that ROP16 also induces the tyrosine phosphorylation of STAT1. To determine if ROP16 is required for the tyrosine phosphorylation of STAT1 by RH(I) parasites in non-IFNγ-stimulated conditions, we also infected HFFs with RHΔrop16 parasites and again visualized phospho-STAT1Tyr nuclear accumulation by immunofluorescence. As compared to cells infected with RH(I) parasites, cells infected with RHΔrop16 parasites had significantly less phospho-STAT1Tyr in their nuclei, with levels almost as low as in uninfected cells (Fig. 2A, B). We next infected HFFs with a Pru strain that overexpresses the type I copy of ROP16. The ectopic expression of ROP16I in a type II background led to an increase in phospho-STAT1Tyr after infection (Fig. 2A, B). However, deletion of ROP16 from a type I parasite background or overexpression of ROP16I in a type II parasite background did not affect the level of phospho-STAT1Tyr in infected cells after IFNγ treatment, indicating that the increase in phospho-STAT1Tyr in infected cells after IFNγ stimulation occurs independently of ROP16 (Fig. 2A, B). Together, these results demonstrate that in the absence of IFNγ, ROP16 can induce the tyrosine phosphorylation of STAT1.
ROP16 Activated STAT1 is not Transcriptionally Active
Our results indicate that ROP16 can directly activate STAT1 and it is therefore possible that strains with an active ROP16 (I and III) might be less efficient in inhibiting IFNγ mediated STAT1 activation. On the other hand, ROP16 also activates STAT3 and STAT6, both of which can induce SOCS gene expression, which might inhibit IFNγ-STAT1 signaling [33], [34]. Indeed, we previously showed that Socs1, a potent inhibitor of the IFNγ-STAT1 signaling pathway, is one of the host genes most highly induced by ROP16 expression [25]. To determine if ROP16 might play a role in the modulation of the IFNγ response, we infected HFFs with an RH(I) or RHΔrop16 strain for three hours, or left cells uninfected, and subsequently stimulated with IFNγ for one hour, or left cells unstimulated, and analyzed IRF1 protein levels by Western blot. While only RH(I) infection induced the tyrosine phosphorylation of STAT1 in the absence of IFNγ (Fig. 3A), RH(I) and RHΔrop16 parasites both completely inhibited IFNγ induced IRF1 expression (Fig. 3A), indicating that ROP16 induced phospho-STAT1Tyr is not transcriptionally active and that ROP16 is not required for the ability of Toxoplasma infection to block IFNγ induced STAT1 mediated gene expression. These results were confirmed in an immunofluorescence assay. After two hours of infection with either RH or RHΔrop16 parasites, HFFs were subsequently stimulated with IFNγ for two hours, cells were fixed and permeabilized, and IRF1 expression and STAT1 tyrosine phosphorylation were visualized. As seen previously (Fig. 2A, B), after IFNγ treatment, cells infected with either RH or RHΔrop16 had higher nuclear phospho-STAT1Tyr than uninfected cells (Fig. 3B). But, as in the Western blot results (Fig. 3A), both strains clearly inhibited IFNγ induced IRF1 expression (Fig. 3B).
Figure 3. ROP16-activated STAT1 is not transcriptionally active. A.
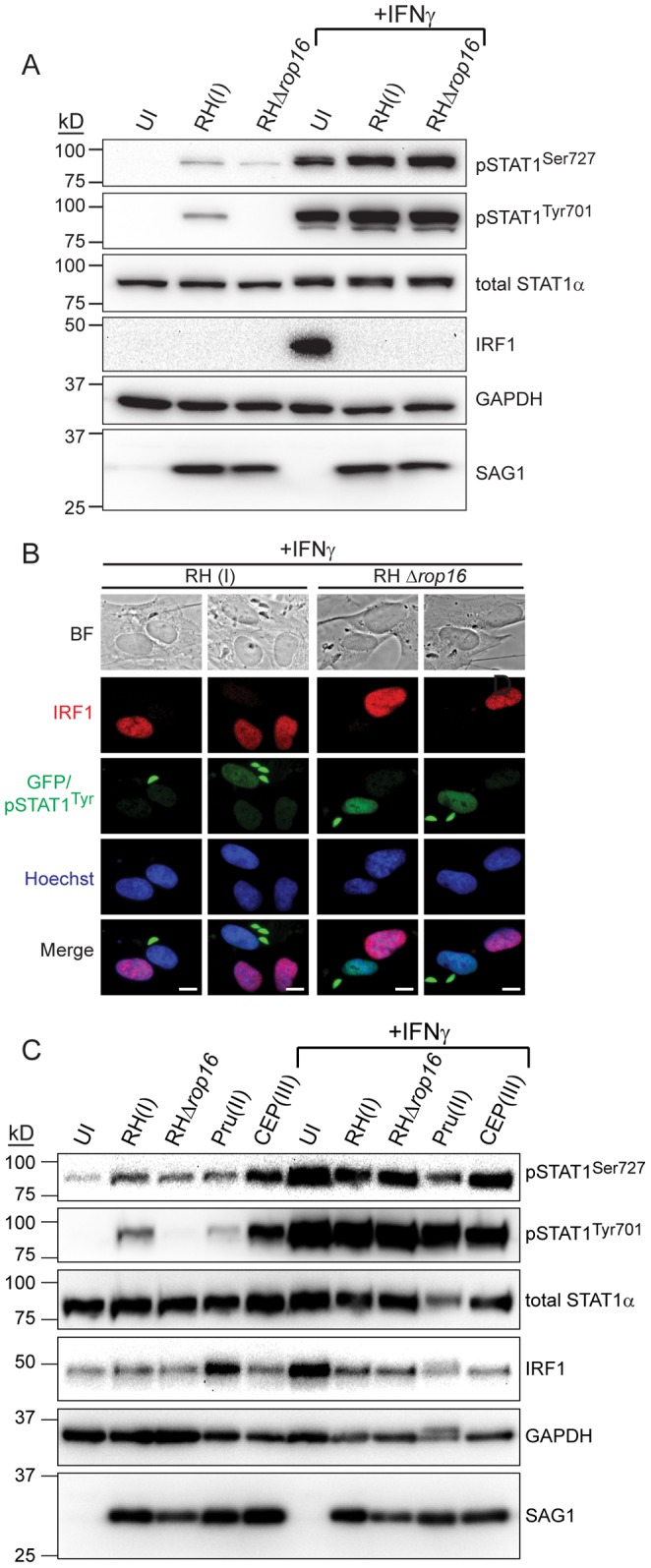
HFFs were infected with RH(I) or RHΔrop16 parasites at an MOI ∼7, or left uninfected, for four hours. Cells were stimulated, or not, with 100 U/ml human IFNγ for the last hour of infection and cell lysates were collected, run on an SDS-PAGE gel, and Western blotted for phospho-STAT1Ser727, phospho-STAT1Tyr701, total STAT1α, IRF1, GAPDH (host cell loading control) and SAG1 (parasite loading control). This experiment has been performed three times with similar results. B. HFFs were infected with RH(I) or RHΔrop16 parasites for four hours, stimulated with 100 U/ml IFNγ for the last two hours of infection, fixed, and stained with anti-IRF1 (red), anti-phospho-STAT1Tyr (green), and Hoechst dye (nucleus, blue). Parasites also express GFP (green). Scale bar represents 10 µm. This experiment was performed three times with similar results. C. HFFs were infected with RH(I), RHΔrop16, Pru(II), or CEP(III) parasites at an MOI ∼1, or left uninfected, for four hours. Cells were stimulated, or not, with 100 U/ml human IFNγ for the last hour of infection and cell lysates were collected, run on an SDS-PAGE gel, and Western blotted for phospho-STAT1Ser727, phospho-STAT1Tyr701, total STAT1α, IRF1, GAPDH (host cell loading control) and SAG1 (parasite loading control). This experiment has been performed two times with similar results.
In addition to STAT1 tyrosine phosphorylation at residue 701, which is required for dimerization and nuclear translocation, STAT1 also must be serine phosphorylated at residue 727 for full transcriptional activity [35]. We wondered whether ROP16 or type I, II, or III strains of Toxoplasma affect this serine phosphorylation. It was previously shown that infection with a Pru(II) strain of Toxoplasma does not interfere with IFNγ induced serine phosphorylation of STAT1 in HFFs [13], but this has not been shown for any type I or III strains. We infected HFFs with an RH(I), RHΔrop16, Pru(II), or CEP(III) strain, or left cells uninfected, for three hours, subsequently stimulated cells with IFNγ for one hour, or left cells unstimulated, and analyzed lysates by Western blot. We first blotted for IRF1 as a control to confirm that infection with any of these strains inhibited the IFNγ induced accumulation of IRF1 (Fig. 3C), as we have shown by immunofluorescence (Fig. 1A, B, Fig. 3B). IRF1 was not inhibited as strongly in this infection as compared to the previous Western blot (Fig. 3A) due to a lower MOI. Additionally, Pru(II) infection alone led to the induction of IRF1 protein, in concordance with previous immunofluorescence experiments (Fig. 1A,B). We next analyzed STAT1 phosphorylation in these lysates. Consistent with our immunofluorescence data (Fig. 2A,B), infection with RH(I) or CEP(III) led to a high level of phospho-STAT1Tyr as compared to uninfected cells while a Pru(II) strain also induced phospho-STAT1Tyr but to a lesser extent (Fig. 3C). Deletion of ROP16 from RH almost completely abolished this tyrosine phosphorylation (Fig. 3C). In addition, none of these strains inhibited the IFNγ induced accumulation of phospho-STAT1Tyr. Looking next at STAT1 serine phosphorylation, we found that infection with any of the Toxoplasma strains that we tested induced the serine phosphorylation of STAT1 slightly, but none of these strains strongly inhibited IFNγ induced serine phosphorylation of STAT1 (Fig. 3C). These results indicate that ROP16 does not play a role in serine phosphorylation of STAT1 and that type I, II, and III strains do not differentially modulate STAT1 serine phosphorylation. Thus, Toxoplasma infection alone can induce low levels of STAT1 serine phosphorylation independently of ROP16 and ROP16 mediates the tyrosine phosphorylation and subsequent nuclear translocation of STAT1, but this nuclear phospho-STAT1Tyr701/Ser727 does not seem to be transcriptionally active.
Type I, II, and III Strains All Inhibit STAT1 Transcriptional Activity
While our results demonstrate that type I, II, and III parasites can all inhibit the IFNγ induced expression of IRF1, we have also shown that type I, II, and III parasites can differentially modulate specific aspects of the IFNγ/STAT1 signaling pathway. The type II GRA15 protein induces IRF1 expression independently of STAT1 (Fig. 1D), and the rhoptry kinase ROP16 induces STAT1 tyrosine phosphorylation (Fig. 2, 3). Additionally, the expression of many IFNγ-regulated genes can be induced by transcription factors other than STAT1; for example the activation of IRF1 by GRA15 via NF-κB (Fig. 1A, B) and the induction of Socs1 by ROP16 via STAT3 or 6 [25]. It is therefore difficult to interpret the modulation of the STAT1 transcriptional response by different Toxoplasma strains using the expression of particular genes as a read out. We instead decided to use a stable STAT1 reporter cell line to determine the ability of the Toxoplasma clonal lineages to interfere with STAT1’s activity in the nucleus. One previous report used two different luciferase reporters to demonstrate that infection with Toxoplasma inhibits STAT1 transcriptional activity [9]. However, one of these reporters was a stable reporter but comprised the entire CIITA pIV promoter, containing binding sites for IRF1, AP-1, and NF-GMa transcription factors and an E-box site as well as a GAS site, making it difficult to determine whether STAT1 activity itself was being affected by Toxoplasma infection or if one of the other transcription factors was being affected. The second reporter measured STAT1 activity more clearly as it contained only a minimal GAS site, however the reporter vector was transiently transfected into cells. Given recent results that indicate that chromatin remodeling factors such as Brahma-related gene (BRG)-1 are differentially recruited to GAS sites after Toxoplasma infection to inhibit STAT1-mediated transcription [14], and that Toxoplasma infection can modulate chromatin modifications resulting in changes in gene expression [36], a transient plasmid reporter that is not integrated into the genome and does not have a normal chromatin structure also may not be an accurate measure of STAT1 transcriptional activity [37], [38]. Additionally, potential strain differences in the inhibition of these reporters were not investigated.
We therefore developed a stable GAS reporter cell line in the easily transduced HEK293 cell line to measure STAT1 transcriptional activity directly. Treatment of this GAS reporter cell line with IFNγ, but not IFNβ, TNFα, or IL4, results in the robust induction of luciferase activity (Fig. 4). We infected this cell line with RH(I), RHΔrop16, Pru(II), PruΔgra15, or CEP(III) parasites, stimulated the cells with IFNγ, and measured the induction of luciferase activity. Infection with any of these strains significantly inhibited IFNγ induced luciferase activity, and the extent of this inhibition did not vary significantly between the strains (Fig. 5A). These reporter experiments also confirmed that while ROP16 can activate the tyrosine phosphorylation and nuclear translocation of STAT1 (Fig. 2), this STAT1 is not transcriptionally active; infection of this cell line with any of these strains did not result in the induction of luciferase (Fig. 5A). To verify that this ability to inhibit STAT1 transcriptional activity is common to the type I, II, and III clonal lineages and not just RH(I), Pru(II), and CEP(III) strains, we also infected this cell line with other representative strains from these lineages, GT1(I), ME49(II), or VEG(III), as well as RH(I) (Fig. 5B). Again, all of these strains were able to inhibit IFNγ induced luciferase activity. These results indicate that type I, II, and III strains can all inhibit IFNγ induced STAT1 transcriptional activity to a similar extent.
Figure 4. Characterization of HEK293 GAS reporter cell line.
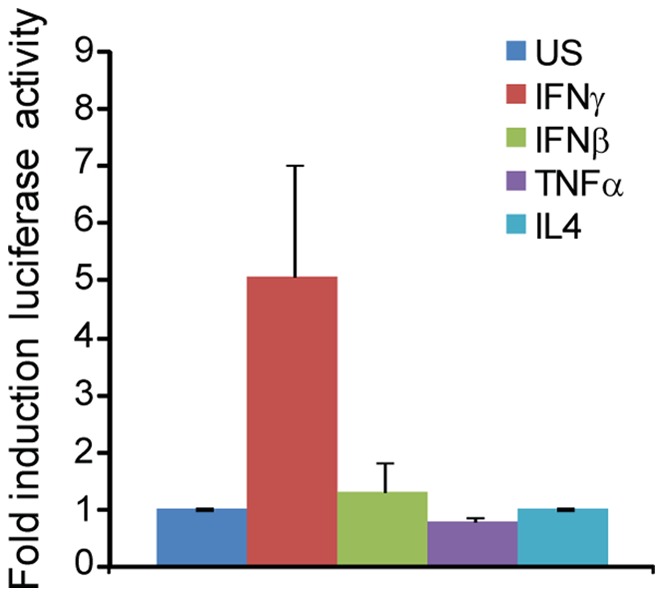
A HEK293 GAS luciferase reporter cell line was left unstimulated or stimulated with 100 U/ml IFNγ, 100 U/ml IFNβ, 20 ng/ml TNFα, or 50 ng/ml IL4. Cells were lysed 6–20 hours later and luciferase activity was measured. Average luciferase induction normalized to unstimulated cells from three experiments is shown and error bars represent standard error.
Figure 5. All three clonal lineages of Toxoplasma inhibit STAT1-mediated gene expression. A, B.

A HEK293 GAS luciferase reporter cell line was infected with RH(I), RHΔrop16, GT1(I), Pru(II), PruΔgra15, ME49(II), CEP(III), or VEG(III) parasites, or left uninfected, and subsequently stimulated, or not, with 100 U/ml IFNγ. Cells were then lysed and luciferase activity was measured. Results are from 2–8 experiments per condition, with a pre-infection time of 1–5 hours followed by a stimulation of 12–24 hours, and represent the average induction over uninfected, unstimulated samples. Error bars represent SEM. Asterisk (*) indicates p<0.05 compared to uninfected cells in the same condition.
Type I, II, and III Strains All Inhibit Global IFNγ Induced Transcription
Although all Toxoplasma strains that we have tested inhibit a stable GAS reporter cell line, we have seen that Toxoplasma strains can differentially affect particular aspects of the IFNγ signaling pathway through GRA15 and ROP16, and it is therefore unclear whether the ability to inhibit STAT1 activity corresponds to the ability of type I, II, and III strains to similarly inhibit global IFNγ induced gene expression. We therefore analyzed the effect of infection with an RH(I), Pru(II), or CEP(III) strain on IFNγ induced transcription using microarray analysis. As more genes have been found to be induced by IFNγ in macrophages than fibroblasts [13], [14], we pre-infected a murine macrophage cell line (RAW264.7) with the above strains for 24 hours, adding IFNγ for the last six hours. We isolated RNA from these cells as well as uninfected control cells, with and without IFNγ stimulation, and analyzed gene expression with Affymetrix microarrays. In this macrophage cell line, 514 genes were more than 2-fold upregulated by IFNγ treatment, while the expression of 481 genes was more than 2-fold repressed (Fig. 6). In the pre-infected samples, 431 of the 514 induced genes were at least 2-fold inhibited by at least one strain, with 314 genes being inhibited by all strains (Fig. 6). Interestingly, the expression of genes that are important for control of Toxoplasma infection, Nos2 [39], Iigp1/Irga6 [40], [41], Iigp2/Irgm2 [42], and Tgtp/Irgb6 [42] were at least 2-fold inhibited by all three strains. Of the 481 IFNγ repressed genes, the repression of 312 of them was more than 2-fold inhibited by at least one strain while 147 genes were inhibited by all three strains (Fig. 6). It seems then that while Toxoplasma strains may differentially inhibit small subsets of IFNγ responsive genes, all three of the clonal lineages significantly inhibit global IFNγ induced gene expression.
Figure 6. All three clonal lineages of Toxoplasma inhibit global IFNγ-induced gene expression.

RAW264.7 macrophages were infected with RH(I), Pru(II), or CEP(III) parasites, or left uninfected (UI) for 24 hours with 100 U/ml IFNγ added for the last 6 hours of infection, and host gene expression was analyzed by microarray analysis. Greater than 2-fold IFNγ induced (left) and repressed (right) genes were determined from the uninfected samples. Boxplots are shown of the log2 expression of these genes in all samples. Data are from two arrays for the uninfected conditions and one array for each infected sample.
Discussion
The expression level of many genes is regulated by multiple transcription factors allowing more precise control and responsiveness to varying stimuli. While we find that strains representing three Toxoplasma clonal lineages, types I, II, and III, can all inhibit IFNγ induced STAT1 transcriptional activity, these strains also differentially modulate certain IFNγ responsive genes through the activity of at least two known polymorphic effectors, GRA15 and ROP16. In studying the ability of Toxoplasma to inhibit the IFNγ response, the choice of readout for IFNγ induced gene expression is therefore very important, as some IFNγ responsive genes are also activated by Toxoplasma through GRA15, ROP16, and likely other secreted proteins.
GRA15II-mediated activation of NF-κB can induce expression of IRF1, and the levels of IRF1 in Pru(II) infected cells stimulated with IFNγ are virtually identical to those of Pru(II) infected cells that were not stimulated (Fig. 1A). This indicates that Pru(II) parasites can inhibit IFNγ induced expression of IRF1, even though they induce IRF1 through GRA15-mediated activation of NF-κB (Fig. 1A, D). Similarly, ROP16I/III induces Socs1 expression by 10-fold in murine BMDM [25], likely through STAT3 or STAT6. But, our microarray data from the murine macrophage RAW264.7 cell line shows that pre-infection with RH(I) parasites can still inhibit IFNγ induced Socs1 transcript by two-fold. Thus, although Toxoplasma is able to inhibit the STAT1-mediated induction of genes such as IRF1 and Socs1, it does not inhibit the expression of these genes activated by other transcription factors. This indicates that whatever mechanism Toxoplasma employs to inhibit the IFNγ-induced transcriptional response must specifically target STAT1-mediated transcriptional activation of genes.
While neither GRA15 nor ROP16 affects the ability of Toxoplasma strains to inhibit the STAT1-mediated global induction of IFNγ responsive gene expression, it is unclear how large of an effect GRA15 and ROP16 have on subsets of IFNγ responsive genes as our experiments were done in a different cell line than previous transcriptional analyses on GRA15 and ROP16. However, IRF1 is an important secondary transcription factor in the response to IFNγ. Additionally, NF-κB is likely to co-regulate other IFNγ responsive genes besides IRF1. A significant number of genes induced by both IFNγ and Pru(II) infection are GRA15-regulated (Data S1). While one microarray analysis in HFFs found that IFNγ responsive genes that were also induced by Pru(II) infection alone were associated with TNFα signaling and included many NF-κB target genes [13], another microarray analysis in murine BMDM did not find an enrichment in NF-κB target genes among genes induced by both IFNγ and another type II strain, NTE [14]. However, it is unknown whether the NTE(II) strain has an active copy of GRA15 and activates NF-κB.
The strongest effect of ROP16 on IFNγ signaling seems be on the phosphorylation status of STAT1 (Fig. 2). Since ROP16 directly tyrosine phosphorylates STAT3 and STAT6 [20], [21], it is likely that tyrosine 701 on STAT1 is also a direct target. It seems that either the affinity or catalytic efficiency of ROP16 for STAT1 is lower than for at least STAT6 because clear phospho-STAT1Tyr activation was only observed in cells infected with at least three parasites, whereas only one parasite needs to inject its rhoptry contents into a host cell to activate STAT6 [43].
It is still unclear why we observe a higher level of IFNγ induced phospho-STAT1Tyr after pre-infection with any strain of Toxoplasma (Fig. 2A). This phenotype is not dependent on ROP16 as it also occurs in RHΔrop16 infected cells. The transcripts of the main components of this pathway, IFNγ receptor 1 and 2, JAK1 and 2, and STAT1, are not upregulated by type I Toxoplasma infection in HFFs, according to previous microarray data [19]. Additionally, SOCS proteins that can downregulate JAK and STAT1 phosphorylation are actually induced by Toxoplasma infection [12], and the expression of the protein tyrosine phosphatases (PTPs) that are known to dephosphorylate JAK1, JAK2, or STAT1 [44] are not downregulated by infection alone [19].
Our data suggest that the type I, II, and III strains use a similar mechanism to inhibit STAT1 transcriptional activity in the nucleus. This inhibition is independent of how STAT1 is activated; Toxoplasma can also inhibit the activity of ROP16 induced phospho-STAT1Tyr, and this interference is specific for STAT1, as ROP16-activated STAT3 and STAT6 are transcriptionally active [18], [25]. A recent study, which focused mainly on the IFNγ induced expression of CIITA and MHC class II genes, concluded that Toxoplasma inhibits IFNγ induced gene expression through impaired BRG-1 chromatin remodeling [14]. Although that may be the mechanism for CIITA, the IFNγ induced expression of IRF1 does not require BRG-1 remodeling [45]. It is therefore important for future studies to determine the mechanism by which Toxoplasma inhibits the STAT1-mediated induction of primary response genes such as IRF1.
Supporting Information
GRA15-regulated IFNγ responsive genes. 374 genes induced by IFNγ treatment and by Pru infection in a published microarray analysis and also present in a microarray analysis of HFFs infected with GRA15-deficient and GRA15-overexpressing Toxoplasma strains are listed. Whether these genes were also found to be at least two-fold GRA15-regulated in at least one parasite genetic background is also indicated.
(XLSX)
Acknowledgments
We thank the members of the Saeij laboratory for useful comments and discussion, Lindsay Julien for technical assistance, and the MIT BioMicro Center and Manlin Luo for technical assistance with the microarray experiments.
Funding Statement
J. Saeij was supported by National Institutes of Health R01-AI080621 and by a Scientist Development Grant from the American Heart Association (0835099N). E. Rosowski was supported by a pre-doctoral grant in the Biological Sciences (5-T32- GM007287-33) and the Cleo and Paul Schimmel Fund. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Scharton-Kersten TM, Wynn TA, Denkers EY, Bala S, Grunvald E, et al. (1996) In the Absence of Endogenous IFN-y, Mice Develop Unimpaired IL-12 Responses to Toxoplasma gondii While Failing to Control Acute Infection. The Journal of Immunology 157: 4045. [PubMed] [Google Scholar]
- 2. Yap GS, Sher A (1999) Effector cells of both nonhemopoietic and hemopoietic origin are required for interferon (IFN)-gamma- and tumor necrosis factor (TNF)-alpha-dependent host resistance to the intracellular pathogen, Toxoplasma gondii. Journal of Experimental Medicine 189: 1083–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lieberman L, Banica M, Reiner SL, Hunter C (2004) STAT1 plays a critical role in the regulation of antimicrobial effector mechanisms, but not in the development of Th1-type responses during toxoplasmosis. Journal of immunology 172: 457–463. [DOI] [PubMed] [Google Scholar]
- 4. Saha B, Jyothi Prasanna S, Chandrasekar B, Nandi D (2010) Gene modulation and immunoregulatory roles of interferon gamma. Cytokine 50: 1–14. [DOI] [PubMed] [Google Scholar]
- 5. Yang T, Aosai F, Norose K, Ueda M, Yano A (1996) Differential regulation of HLA-DR expression and antigen presentation in Toxoplasma gondii-infected melanoma cells by interleukin 6 and interferon gamma. Microbiology and immunology 40: 443–449. [DOI] [PubMed] [Google Scholar]
- 6. Lüder CGK, Lang T, Beuerle B, Gross U (1998) Down-regulation of MHC class II molecules and inability to up-regulate class I molecules in murine macrophages after infection with Toxoplasma gondii. Clinical and experimental immunology 112: 308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lüder CGK, Lang C, Giraldo-Velasquez M, Algner M, Gerdes J, et al. (2003) Toxoplasma gondii inhibits MHC class II expression in neural antigen-presenting cells by down-regulating the class II transactivator CIITA. Journal of neuroimmunology 134: 12–24. [DOI] [PubMed] [Google Scholar]
- 8. Lüder CGK, Walter W, Beuerle B, Maeurer MJ, Gross U (2001) Toxoplasma gondii down-regulates MHC class II gene expression and antigen presentation by murine macrophages via interference with nuclear translocation of STAT1alpha. European journal of immunology 31: 1475–1484. [DOI] [PubMed] [Google Scholar]
- 9. Lang C, Algner M, Beinert N, Gross U, Lüder CGK (2006) Diverse mechanisms employed by Toxoplasma gondii to inhibit IFN-gamma-induced major histocompatibility complex class II gene expression. Microbes and infection 8: 1994–2005. [DOI] [PubMed] [Google Scholar]
- 10. Seabra SH, de Souza W, DaMatta RA (2002) Toxoplasma gondii partially inhibits nitric oxide production of activated murine macrophages. Experimental parasitology 100: 62–70. [DOI] [PubMed] [Google Scholar]
- 11. Rozenfeld C, Martinez R, Seabra S, Sant’Anna C, Gonçalves JGR, et al. (2005) Toxoplasma gondii Prevents Neuron Degeneration by Interferon-γ-Activated Microglia in a Mechanism Involving Inhibition of Inducible Nitric Oxide Synthase and Transforming Growth Factor-β1 Production by Infected Microglia. The American Journal of Pathology 167: 1021–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zimmermann S, Murray PJ, Heeg K, Dalpke AH (2006) Induction of suppressor of cytokine signaling-1 by Toxoplasma gondii contributes to immune evasion in macrophages by blocking IFN-gamma signaling. Journal of immunology 176: 1840–1847. [DOI] [PubMed] [Google Scholar]
- 13. Kim SK, Fouts AE, Boothroyd JC (2007) Toxoplasma gondii dysregulates IFN-gamma-inducible gene expression in human fibroblasts: insights from a genome-wide transcriptional profiling. The Journal of Immunology 178: 5154. [DOI] [PubMed] [Google Scholar]
- 14. Lang C, Hildebrandt A, Brand F, Opitz L, Dihazi H, et al. (2012) Impaired Chromatin Remodelling at STAT1-Regulated Promoters Leads to Global Unresponsiveness of Toxoplasma gondii-Infected Macrophages to IFN-γ. PLoS Pathogens 8: e1002483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Montoya JG, Liesenfeld O (2004) Toxoplasmosis. Lancet 363: 1965–1976. [DOI] [PubMed] [Google Scholar]
- 16. Howe DK, Sibley LD (1995) Toxoplasma gondii comprises three clonal lineages: correlation of parasite genotype with human disease. Journal of Infectious Diseases 172: 1561. [DOI] [PubMed] [Google Scholar]
- 17. Melo MB, Jensen KDC, Saeij JPJ (2011) Toxoplasma gondii effectors are master regulators of the inflammatory response. Trends in parasitology 27: 487–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saeij JPJ, Coller S, Boyle JP, Jerome ME, White MW, et al. (2007) Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature 445: 324–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rosowski EE, Lu D, Julien L, Rodda L, Gaiser RA, et al. (2011) Strain-specific activation of the NF-kappaB pathway by GRA15, a novel Toxoplasma gondii dense granule protein. The Journal of experimental medicine 208: 195–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yamamoto M, Standley DM, Takashima S, Saiga H, Okuyama M, et al. (2009) A single polymorphic amino acid on Toxoplasma gondii kinase ROP16 determines the direct and strain-specific activation of Stat3. The Journal of experimental medicine 206: 2747–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ong Y-C, Reese ML, Boothroyd JC (2010) Toxoplasma rhoptry protein 16 (ROP16) subverts host function by direct tyrosine phosphorylation of STAT6. The Journal of biological chemistry 285: 28731–28740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Costa-Pereira AP, Tininini S, Strobl B, Alonzi T, Schlaak JF, et al. (2002) Mutational switch of an IL-6 response to an interferon-gamma-like response. Proceedings of the National Academy of Sciences 99: 8043–8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baus D, Nonnenmacher F, Jankowski S, Döring C, Bräutigam C, et al. (2009) STAT6 and STAT1 are essential antagonistic regulators of cell survival in classical Hodgkin lymphoma cell line. Leukemia 23: 1885–1893. [DOI] [PubMed] [Google Scholar]
- 24. Ohmori Y, Hamilton TA (1997) IL-4-induced STAT6 suppresses IFN-gamma-stimulated STAT1-dependent transcription in mouse macrophages. The Journal of Immunology 159: 5474. [PubMed] [Google Scholar]
- 25. Jensen KDC, Wang Y, Wojno EDT, Shastri AJ, Hu K, et al. (2011) Toxoplasma polymorphic effectors determine macrophage polarization and intestinal inflammation. Cell host & microbe 9: 472–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Robinson CM, Hale PT, Carlin JM (2006) NF-kappa B activation contributes to indoleamine dioxygenase transcriptional synergy induced by IFN-gamma and tumor necrosis factor-alpha. Cytokine 35: 53–61. [DOI] [PubMed] [Google Scholar]
- 27. Saeij JPJ, Boyle JP, Coller S, Taylor S, Sibley LD, et al. (2006) Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science 314: 1780–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim S-K, Karasov A, Boothroyd JC (2007) Bradyzoite-specific surface antigen SRS9 plays a role in maintaining Toxoplasma gondii persistence in the brain and in host control of parasite replication in the intestine. Infection and immunity 75: 1626–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boyle JP, Saeij JPJ, Boothroyd JC (2007) Toxoplasma gondii: Inconsistent dissemination patterns following oral infection in mice. Experimental Parasitology 116: 302–305. [DOI] [PubMed] [Google Scholar]
- 30. McKendry R, John J, Flavell D, Müller M, Kerr IM, et al. (1991) High-frequency mutagenesis of human cells and characterization of a mutant unresponsive to both alpha and gamma interferons. Proceedings of the National Academy of Sciences 88: 11455–11459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Müller M, Laxton C, Briscoe J, Schindler C, Improta T, et al. (1993) Complementation of a mutant cell line: central role of the 91 kDa polypeptide of ISGF3 in the interferon-alpha and -gamma signal transduction pathways. The EMBO journal 12: 4221–4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schindler C, Darnell JE (1995) Transcriptional responses to polypeptide ligands: the JAK-STAT pathway. Annual review of biochemistry 64: 621–651. [DOI] [PubMed] [Google Scholar]
- 33. Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, et al. (1997) Structure and function of a new STAT-induced STAT inhibitor. Nature 387: 924–929. [DOI] [PubMed] [Google Scholar]
- 34. Hebenstreit D, Luft P, Schmiedlechner A, Regl G, Frischauf A-M, et al. (2003) IL-4 and IL-13 induce SOCS-1 gene expression in A549 cells by three functional STAT6-binding motifs located upstream of the transcription initiation site. Journal of immunology 171: 5901–5907. [DOI] [PubMed] [Google Scholar]
- 35. Platanias LC (2005) Mechanisms of type-I- and type-II-interferon-mediated signalling. Nature Reviews Immunology 5: 375–386. [DOI] [PubMed] [Google Scholar]
- 36. Leng J, Butcher B, Egan CE, Abi Abdallah DS, Denkers EY (2009) Toxoplasma gondii prevents chromatin remodeling initiated by TLR-triggered macrophage activation. The Journal of Immunology 182: 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smith CL, Hager GL (1997) Transcriptional regulation of mammalian genes in vivo. A tale of two templates. Journal of Biological Chemistry 272: 27493–27496. [DOI] [PubMed] [Google Scholar]
- 38. Hebbar PB, Archer TK (2008) Altered histone H1 stoichiometry and an absence of nucleosome positioning on transfected DNA. The Journal of biological chemistry 283: 4595–4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Khan I, Schwartzman JD, Matsuura T, Kasper LH (1997) A dichotomous role for nitric oxide during acute Toxoplasma gondii infection in mice. Proceedings of the National Academy of Sciences of the United States of America 94: 13955–13960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Martens S, Parvanova I, Zerrahn J, Griffiths G, Schell G, et al. (2005) Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLoS pathogens 1: e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liesenfeld O, Parvanova I, Zerrahn J, Han S-J, Heinrich F, et al. (2011) The IFN-γ-Inducible GTPase, Irga6, Protects Mice against Toxoplasma gondii but Not against Plasmodium berghei and Some Other Intracellular Pathogens. PloS one 6: e20568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hunn JP, Koenen-Waisman S, Papic N, Schroeder N, Pawlowski N, et al. (2008) Regulatory interactions between IRG resistance GTPases in the cellular response to Toxoplasma gondii. The EMBO journal 27: 2495–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Koshy A, Dietrich HK, Christian D, Melehani JH, Shastri AJ, et al. (2012) Toxoplasma Co-opts Host Cells It Does Not Invade. PLoS Pathogens 8: e1002825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shuai K, Liu B (2003) Regulation of JAK-STAT signalling in the immune system. Nature reviews Immunology 3: 900–911. [DOI] [PubMed] [Google Scholar]
- 45. Wang Y, Gao B, Xu W, Xiong S (2011) BRG1 is indispensable for IFN-γ-induced TRIM22 expression, which is dependent on the recruitment of IRF-1. Biochemical and biophysical research communications 410: 549–554. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
GRA15-regulated IFNγ responsive genes. 374 genes induced by IFNγ treatment and by Pru infection in a published microarray analysis and also present in a microarray analysis of HFFs infected with GRA15-deficient and GRA15-overexpressing Toxoplasma strains are listed. Whether these genes were also found to be at least two-fold GRA15-regulated in at least one parasite genetic background is also indicated.
(XLSX)