

SUMMARY
Innate immune sensors are required for induction of pathogen-specific immune responses. Retroviruses are notorious for their ability to evade immune defenses and establish long-term persistence in susceptible hosts. However, some infected animals are able to develop efficient virus-specific immune responses, and thus can be employed for identification of critical innate virus-sensing mechanisms. With mice from two inbred strains that control retroviruses via adaptive immune mechanisms, we found that of all steps in viral replication, the ability to enter the host cell was sufficient to induce antivirus humoral immune responses. Virus sensing occurred in endosomes via a MyD88-Toll-like receptor 7-dependent mechanism and stimulated virus-neutralizing immunity independently of type I interferons. Thus, efficient adaptive immunity to retroviruses is induced in vivo by innate sensing of the early stages of retroviral infection.
INTRODUCTION
The demand for producing highly efficient vaccines against human immunodeficiency virus (HIV) is great. However, the approaches for making vaccines currently available may not be relevant to retroviral infections, because none of the trials conducted to date have succeeded. A basic understanding of how the immune system detects and responds to retroviruses must be gained first in order to apply this knowledge to the production of antiretrovirus vaccines.
A required step in the development of a pathogen-specific protective immune response (Medzhitov and Janeway, 1997) is the recognition of pathogen-associated molecular patterns (PAMPs) by pattern recognition receptors (PRRs). PAMPs represent highly conserved microbial molecular structures that are not found in the host cells or in the compartment of infected cells in which the pathogen replicates. Bacterial pathogens are detected by Toll-like receptors (TLRs), which recognize bacterial lipids, peptidoglycans, or proteins that are foreign to eukaryotic cells (Medzhitov, 2007). Unlike bacterial cell surfaces, viral exteriors lack specific structures that can distinguish them from the surfaces of eukaryotic cells. Consequently, viral recognition occurs through cytosolic or endocytic PRRs that detect virally produced replication intermediates (e.g., various forms of nucleic acids) (Kawai and Akira, 2010) or through inflammasomes, which detect the activities of some virally encoded proteins (Ichinohe et al., 2010). Although many viral sensors have been identified in vitro, very few of these have been proven to play an essential role in virus sensing in vivo (Kawai and Akira, 2010). In some instances, viral sensors identified in vitro were shown to be dispensable for generation of effective antiviral immune responses in vivo (Ammann et al., 2009; Bhoj et al., 2008; Edelmann et al., 2004). Therefore, sensing documented in vitro cannot be accepted as meaningful without the scrutiny of in vivo experiments, making animal models essential for evaluating the requirements and consequences of virus sensing.
Various retroviral replication intermediates can potentially be recognized by the innate immune system. The question is: which sensors are important for a successful adaptive immune response? To address this, it is critically important to select the most appropriate in vivo model system. Most mouse strains are susceptible to various retroviruses, making them ineffective for identification of retrovirus-sensing mechanisms. In contrast, mice that are genetically resistant to retroviruses are able to initiate and maintain robust antiviral responses. Because adaptive immune responses require an innate immune trigger, genetic inactivation of virus-detection mechanisms should result in complete loss of resistance in these animals. If the sensing mechanisms were to be identified in these mice, they would probably be shared with other vertebrate species, because retroviruses use the same replication strategy in all vertebrates.
Therefore, we consider retrovirus-resistant mice to be a natural choice for addressing the three outstanding issues concerning retrovirus-specific immunity: is viral replication required for induction of a virus-specific immune response? What are the necessary and sufficient steps in the viral replication cycle that trigger this response? And what is the nature of the PRR(s) that senses retroviral pathogens?
Retroviruses from two distinct genera were used to search for retrovirus-sensing mechanisms: mouse mammary tumor virus (MMTV; a betaretrovirus) and murine leukemia virus (MuLV; a gammaretrovirus). MMTV is transmitted as an exogenous virus passed either through the milk of lactating females or as an endogenous stably integrated provirus (Coffin, 1990). Lymphoid cells are the first targets of infection; they then spread the virus to the mammary glands, leading to tumor development. MuLV is also transmitted via both exogenous and endogenous routes (Rosenberg and Jolicoeur, 1997). Exogenous MuLV is passed through the blood and milk and primarily infects cells of lymphoid origin. Susceptible mice develop severe splenomegaly and succumb to leukemia.
Mice from retrovirus-susceptible strains detect retroviral pathogens, as indicated by the fact that they initiate an antiretroviral response. However, this response is not long lasting and is unsuccessful in controlling virus replication (Chesebro et al., 1990; Purdy et al., 2003), which is probably due to the numerous mechanisms of immune evasion employed by retroviruses (Dittmer et al., 2004; Evans and Desrosiers, 2001; Jude et al., 2003; Malim and Emerman, 2008). In contrast, pathogen detection in resistant mice translates into a robust, long-lasting, and virus-neutralizing immune response (Chesebro et al., 1990; Purdy et al., 2003).
Two retrovirus-resistant mouse models were chosen for studying mechanisms of retrovirus detection. Mice from these strains become infected but mount powerful antivirus immune responses that either eliminate the virus or efficiently control viral replication and transmission. C57BL/6J (B6) mice infected with MuLV eliminate the virus through a dominant mechanism that includes humoral and cell-mediated responses (Miyazawa et al., 2008). Antiretroviral production of antibody (Ab) in B6 mice is mediated by the single dominant gene, recovery from Friend virus 3 (rfv3), mapped to chromosome 15 (Hasenkrug et al., 1995). Two closely linked genes have been recently implicated as candidates for rfv3: deoxycytidine deaminase Apobec3 (Santiago et al., 2008, 2011) and B cell-activating factor receptor (Tnfrsf13c)(Tsuji-Kawahara et al., 2010).
Although B6 mice are resistant to MuLV, they are susceptible to MMTV (Buggiano et al., 1999; Wrona and Dudley, 1996). In contrast, mice from the I/LnJ strain are the only known inbred mice capable of controlling both MMTV and MuLV infections via Ab (Case et al., 2005; Purdy et al., 2003) and cell-mediated responses (Case et al., 2008; Kane et al., 2011). The mechanism of resistance inherited by I/LnJ mice is recessive and is mapped to a single locus on chromosome 17 (Case et al., 2008; Golovkina, 2000). The gene within this locus responsible for retroviral resistance is yet to be identified.
It is necessary to emphasize that a knowledge of the specific mechanisms underlying antiviral immune responses in resistant mice is not required for identification of retrovirus innate immune sensor(s), because these mechanisms operate downstream of virus detection events. Consequently, abrogation of innate immune sensing is expected to reverse the resistance phenotype and provide a straightforward readout of retroviral detection. With these retrovirus-resistance mice, we found that endosomal Toll-like receptor 7 (TLR7) is an innate immune receptor that detects retroviruses.
RESULTS
Antiretroviral Responses against MMTV
As previous studies have shown, retrovirus-resistant I/LnJ mice do not react against their endogenous MMTVs (Case et al., 2008; Purdy et al., 2003). The unresponsiveness to endogenous viruses may be explained by immunological tolerance. Alternatively, it could be due to the lack of specific stimuli associated with the replicating retroviral pathogen, because the vast majority of endogenous viruses are defective or silent (Boeke and Stoye, 1997).
Endogenous Mtv7 and Mtv17 inherited by I/LnJ mice (Mac-Dearmid et al., 2006) are unable to produce infectious virions because of premature stop codons in their env genes (the Mouse Genome Project [http://www.sanger.ac.uk/Projects/M_musculus/] and data not shown). To determine whether a replication-competent endogenous virus is required for induction of an antivirus immune response in I/LnJ mice, we introduced a new MMTV provirus capable of producing infectious virions, termed hybrid provirus (HP) (Shackleford and Varmus, 1988), as a transgene into the I/LnJ genome (I/LnJ-HP mice) (see Figure S1A available online). The HP transgene encodes a T cell receptor (TCR) Vβ14-specific superantigen (SAg) and thus causes negative selection of Vβ14+ T cells (Golovkina et al., 1994). As expected, SAg-cognate T cells were deleted in I/LnJ-HP mice (0.53% ± 0.4% [n = 10] of Vβ14+ T cells among CD4+ T cells in transgenic versus 8.79% ± 1.0% [n = 8] in nontransgenic mice). I/LnJ-HP mice produced infectious virions, as shown by the fact that viral RNA was present in the milk of I/LnJ-HP mice (Figure S1B) and newly integrated HP proviruses were present in the splenic DNA (Figure S1C).
To test whether I/LnJ mice are capable of responding to infectious virus produced from the endogenous HP transgene, sera from I/LnJ-HP multiparous females (MMTV expression is controlled by lactogenic hormones) (Golovkina et al., 1995; Yamamoto, 1985) were analyzed for the presence of Abs reactive with MMTV virion proteins. Whereas I/LnJ-HP mice produced virus-specific Abs, nontransgenic I/LnJ mice did not (Figures 1A and 1B). Because the production of virus-specific Abs was similar in I/LnJ-HP mice fostered by either HP transgenic or nontransgenic females, the response did not require HP transmission through milk, but rather was induced by infection with the virus encoded by the transgene. The neutralizing Ab response was specific to virus-resistant I/LnJ-HP mice, as shown by the fact that virus-susceptible HP transgenic (C3H/HeN-HP) mice did not respond to the virus (Figure 1A).
Figure 1. Endogenous Replication-Competent MMTV Elicits Antivirus Immune Responses in Retrovirus-Resistant Mice.
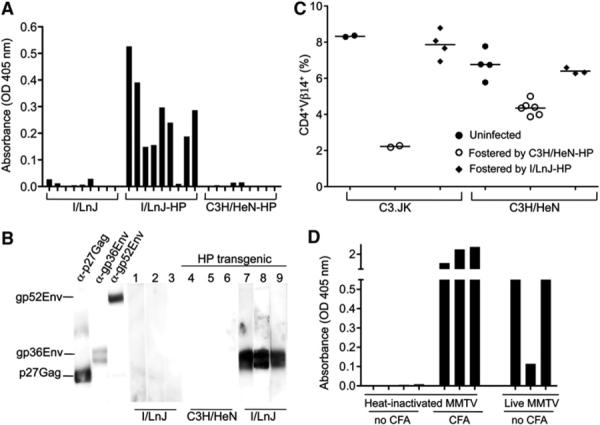
(A) Serum samples from I/LnJ, I/LnJ-HP, and control C3H/HeN-HP multiparous females were tested for reactivity against MMTV virion proteins by ELISA. Because the virus-specific humoral immune response in I/LnJ mice is IFN-γ mediated (Purdy et al., 2003), anti-mouse IgG2a-specific Abs coupled to AP were used at the second step. OD 405: optical density at 405 nm. Nine mice per each group were tested.
(B) PVDF strips with separated virion proteins were incubated with mouse monoclonal Abs against capsid Gag (αp27Gag), transmembrane Env (αgp36Env), surface Env (αgp52Env), or indicated mouse sera. Anti-mouse polyisotypic Abs (for monoclonal Abs) or anti-mouse IgG2a-specific Abs coupled to horseradish peroxidase (HRP) were used at the second step. Numbers correspond to individual mice.
(C) Frequencies of peripheral Vβ14+ T cells among CD4+ T cells were determined in 10-week-old virus-susceptible mice fostered by either I/LnJ-HP or control C3H/HeN-HP females by FACS. Each symbol represents one mouse. None of more than 50 individual C3H/HeN-HP transgenic females tested produced uninfected animals.
(D) Sera from I/LnJ mice injected with either heat-inactivated or biologically active virus were tested for reactivity against MMTV virion proteins by ELISA. In parallel, a group of I/LnJ mice was immunized with heated MMTV virions in CFA. Anti-mouse IgG2a-specific Abs coupled to AP were used at the second step. Three to four mice were used per group in two independent experiments.
In I/LnJ mice infected with exogenous MMTVs, virus transmission to the offspring is abolished, because virions secreted into the milk are coated with Abs (Purdy et al., 2003). Abs produced by I/LnJ-HP females were also virus neutralizing, and mice fostered by these females remained uninfected (Figure 1C). Thus, I/LnJ-HP mice were capable of mounting a neutralizing response against an infectious virus produced by a replication-competent endogenous provirus.
Activation of the innate system produces three different signals that are required for the initiation of adaptive immune responses: active antigen processing and presentation, costimulation, and cytokine production. These signals can be upregulated by external adjuvants, often used for vaccine production. The data presented thus far suggested that replicating virus could substitute for the external adjuvant. To test whether denatured virus retains adjuvant properties, I/LnJ mice were injected with heat-inactivated virions in either the presence or absence of Complete Freund's Adjuvant (CFA). Immunization of I/LnJ mice with heat-inactivated virus in CFA resulted in production of virus-specific Abs, similar to the response to infection with biologically active virus (Figure 1D). However, I/LnJ mice injected with heat-inactivated virus in the absence of CFA were unresponsive (Figure 1D). Thus, although infectious virus is able to upregulate adaptive immunity in the absence of an external adjuvant, denatured virions are incapable of inducing this response.
Antiretroviral Responses against MuLV
In addition to endogenous MMTVs, the mouse genome contains multiple copies of endogenous MuLVs, which are classified as ecotropic, xenotropic, or polytropic mink cell focus forming (MCF) viruses based on their receptor usage (Risser et al., 1983). Although many endogenous MuLVs are defective, some are capable of producing infectious particles (Cloyd et al., 1980; Hook et al., 2002; Stoye et al., 1991). I/LnJ mice inherit endogenous MuLVs (Figure S2A), and ecotropic viruses were upregulated in aged I/LnJ mice, as evidenced by the presence of infectious virions in their spleens (Table 1). To determine whether these replication-competent endogenous MuLVs were proficient in inducing an Ab response akin to that triggered by endogenous replication-competent MMTVs, we investigated anti-MuLV responses in aged I/LnJ mice. All MuLV-shedding I/LnJ mice produced virus-specific Abs (Table 1; Figure 2A). Importantly, the ability to produce Abs was not dependent on the virus titer, as shown by the fact that both high and low virus producers were able to mount a virus-specific response (Table 1). The Abs were directed against major virion proteins (Figure 2B). Similar to responses against exogenous retroviruses (Purdy et al., 2003), humoral responses against endogenous viruses were dependent on IFN-γ (Table 1; Figure 2).
Table 1.
Production of Infectious Ecotropic MuLV by BALB/cJ and I/LnJ Lymphocytes
| Mice | Age | ODa | PFU/107Cellsb |
|---|---|---|---|
| BALB/cJ | >150 days | 0.0 | 44 |
| 0.0 | 0 | ||
| 0.0 | 145 | ||
| 0.0 | 44 | ||
| 0.0 | 141 | ||
| 0.0 | 0 | ||
| 0.0 | 49 | ||
| I/LnJ | <90 days | 0 (n=10) | 7 ± 4 (n = 3) |
| I/LnJ | >150 days | 1.3 | 204 |
| 0.7 | 22 | ||
| 0.3 | 31 | ||
| 0.3 | 572 | ||
| 0.2 | 78 | ||
| 0.1 | 508 | ||
| 0.1 | 84 | ||
| I/LnJ lfng−/− | >120 days | 0 (n = 6) | 467 ± 64 (n = 6) |
Abbreviations: PFUs, plaque forming units; n, number of mice used; ND, nondetermined.
Optical density (OD) at 405 nm obtained with mouse sera minus OD obtained with secondary Ab alone in MuLV-specific ELISA. Animals are ranked from high to low Ab producers.
Spleen cells from BALB/cJ, I/LnJ, and I/LnJ Ifng-deficient (Ifng−/−) mice were subjected to an infectious center assay.
Figure 2. Infectious Virus Produced by Endogenous MuLVs Stimulates an Antiviral Response.

(A) Serum samples from BALB/cJ, I/LnJ, and I/LnJ Ifng−/− mice were tested for reactivity against MuLV virion proteins by ELISA. Six mice per each group were tested.
(B) Serum samples were tested for reactivity against MuLV virion proteins by immunoblot. gp70Env, surface glycoprotein, product of the env gene; p30Gag, capsid protein, product of the gag gene; p25Gag, immature capsid protein, product of the gag gene. Numbers correspond to individual mice.
Anti-mouse IgG2a-specific Abs coupled to HRP were used at the second step for all but I/LnJ Ifng−/− mice for which anti-mouse polyisotypic Abs coupled to HRP were utilized.
We then asked whether virus derived from infectious endogenous provirus is capable of inducing an immune response when introduced as an exogenous virus. Accordingly, we injected young I/LnJ mice with virions harvested from SC-1 cells cocultured with virus-shedding splenocytes derived from aged I/LnJ mice (Figure S2B). All I/LnJ mice that received the virus mounted antiviral humoral immune responses (Figure S2C). Thus, virus-resistant mice are not tolerant to endogenous retroviruses, but require some step(s) of the viral replication cycle in order to activate an antivirus immune response.
Endosomal Uptake of Nonreplicating Virus Stimulates an Immune Response in Virus-Resistant Mice
Both MuLV and MMTV enter via receptor-mediated endocytosis (Katen et al., 2001; Wang et al., 2008). To investigate whether viral entry is sufficient for activation of virus-specific immune response, we used UV irradiation, a treatment that blocks replication of RNA viruses but does not affect their entry (Bender et al., 1995; Ogura, 1976). Likewise, both live and UV-irradiated MMTV and MuLV entered endosomes (Figure 3A; Figure S3A). To determine whether viral entry is sufficient for induction of antiretroviral immunity, we injected I/LnJ mice with live or UV-irradiated MMTV or MuLV virions. As expected, UV irradiation abolished virus infectivity, because injected mice did not show deletion of SAg-cognate T cells (Figure 3B) and did not contain newly integrated exogenous proviruses in their spleens (data not shown). UV irradiation also rendered MuLV noninfectious, as shown by the fact that susceptible BALB/cJ mice injected with UV-irradiated MuLV virions did not become infected (Figure 3C). Within 6 weeks of injection, virus-specific Abs became evident in mice injected with either live or UV-irradiated virus (Figures 3B and 3C). The Ab response was seen only in virus-resistant I/LnJ mice, whereas virus-susceptible C3H/HeN and BALB/cJ mice did not respond to UV-irradiated virus, indicating that Ab production by I/LnJ mice was not a result of immunization (Figures S3B and S3C). Collectively, these results demonstrate that the ability to enter the host cell is sufficient to induce an antivirus humoral immune response, because equivalent starting titers of UV-irradiated and live virus were comparably immunogenic.
Figure 3. Retroviral Entry Is Necessary and Sufficient to Stimulate an Antivirus Immune Response.

(A) Live or UV-irradiated MMTV or MuLV were incubated with NMuMG or XC cells, respectively. Cells were stained with Abs against the late endosomal marker Rab7 (green) and respective viral Gag proteins (red). Top: Maximum intensity 3D reconstruction of z-stacks. Middle: Enlarged single 0.2 μM thick optical slices of boxed areas. Asterisks indicate colocalization of viral Gag and Rab7; corresponding images with orthogonal views (XZ projection) are shown below. Bottom: Uninfected cells and cells infected with live virus were stained with Abs against Gag (red). In all panels, nuclei were stained with DAPI (blue). Three independent experiments were performed.
(B and C) I/LnJ mice were injected with live or UV-irradiated MMTV (B) or MuLV (C). Deletion of SAg-cognate CD4+Vβ14+ T cells (shown as percent of all CD4+ T cells) was used as a read-out for MMTV infectivity. ELISA was utilized to screen sera for Abs reactive against virion proteins.
(B) In experiment 1, mice were analyzed 2 weeks after injection; in experiment 2, mice were analyzed at 2–6 weeks after injection (6 week time-point is shown).
(C) Ab reactivity against MuLV virion proteins was tested 2 weeks after injection (left). Goat anti-mouse Abs coupled to alkaline phosphatase (AP) were used at the second step. None: uninfected I/LnJ mice. Bars correspond to individual mice. To confirm lack of replication capability of UV-irradiated virus, BALB/cJ mice were injected with UV-irradiated virus (or live virus as control) and their spleen cells were subjected to XC-PFU test 6 weeks postinjection (right). n, number of mice tested in a single experiment.
A MyD88-Dependent TLR7-Mediated Mechanism Controls MMTV-Specific Ab Responses
Because nonreplicating entry-competent retrovirus was capable of inducing an immune response, we reasoned that an endosomal PRR(s) would be sensing the pathogen. Three sensors are localized within the endosomal compartment: TLR3, TLR9, and TLR7, which detect dsRNA, DNA, and ssRNA, respectively. Whereas TLR3 signals via a MyD88-independent mechanism, both TLR7 and TLR9 require MyD88 for signal transduction (Kawai and Akira, 2010). We first tested whether a TLR3-mediated innate immune mechanism underlies retrovirus-specific Ab responses in virus-resistant mice. TLR3 signals via an IRF-3-dependent mechanism (Kawai and Akira, 2010), and this signaling mechanism is ablated in both Tlr3−/− and Irf3−/− mice (Alexopoulou et al., 2001; Sato et al., 2000). Accordingly, Tlr3−/− I/LnJ mice and Irf3−/− B6 mice infected with MMTV or MuLV, respectively, were screened for retrovirus-specific Abs. Animals from both groups responded to retroviruses by producing virus-specific Abs (Figures 4A and 4B), indicating that TLR3 is dispensable for the retroviral detection that controls antivirus humoral immune responses. These data suggested that a MyD88-dependent receptor(s) must be the retrovirus sensor in the endosome.
Figure 4. Antivirus Humoral Responses Are Not TLR3 Dependent.
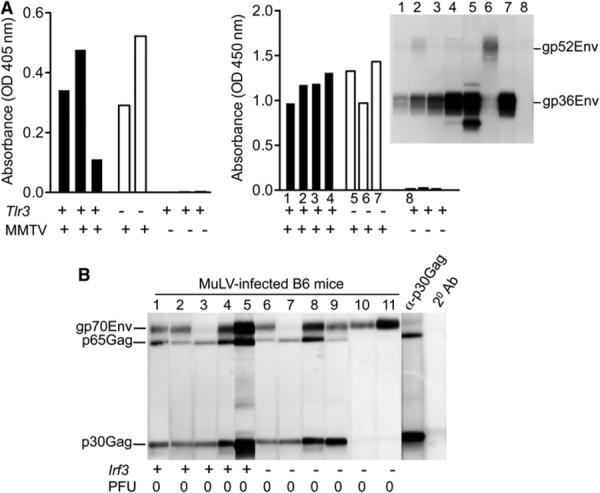
(A) Tlr3−/− and Tlr3+/+ I/LnJ mice were fostered on MMTV-infected C3H/HeN females. Sera from fostered mice were tested for reactivity against MMTV virion proteins by ELISA and/or immunoblot at 10 weeks of age. Mice in experiment 1 (left) were from N7 intercross, whereas mice in experiment 2 (right) were from N10 intercross. In the second experiment (right), the same serum samples were subjected to both ELISA and immunoblot (inset, right). Anti-mouse IgG2a-specific Abs coupled to either AP or HRP were used at the second step. Numbers correspond to individual mice whose sera were used for immunoblot.
(B) Sera from MuLV-infected WT B6 and Irf3−/− B6 mice were tested for reactivity against MuLV virion proteins by immunoblot. Anti-mouse IgG2c-specific Abs coupled to HRP were used at the second step. α-p30Gag, monoclonal Ab against capsid Gag; p65Gag, immature capsid protein, product of the gag gene; 2° Ab, a strip incubated with secondary Ab alone. Numbers correspond to individual mice used in a single experiment. PFU, virus titers in spleens (106 cells) 10 weeks after infection.
Previously, Browne and Littman (2009) established that MyD88 is required for neutralizing Ab responses against MuLV in B6 mice. To extend these findings to another retroviral system, we used MMTV-resistant I/LnJ mice. Unlike MMTV-infected wild-type (WT) I/LnJ mice, infected Myd88−/− I/LnJ mice failed to produce virus-specific Abs (Figure 5A) nor did virus-exposed Myd88−/− splenocytes secrete proinflammatory cytokines (Figure S4A). Together these data indicate that MyD88 is required for MMTV-mediated production of proinflammatory cytokines and virus-specific Abs.
Figure 5. A TLR7-Dependent Mechanism Detects Retroviruses.
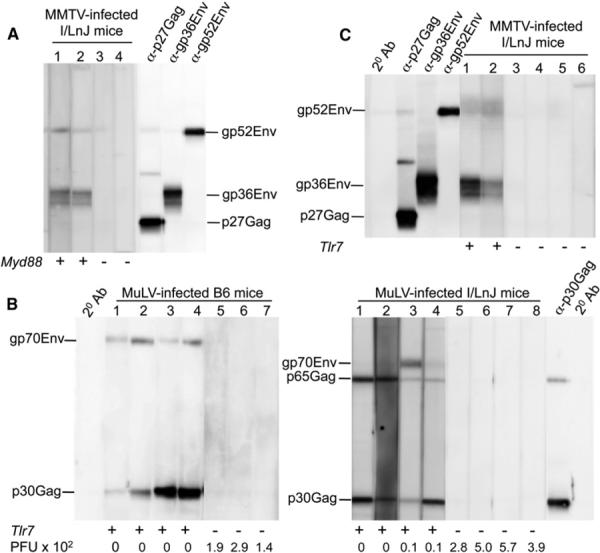
(A) WT I/LnJ and Myd88−/− I/LnJ mice were i.p. injected with MMTV. Sera were analyzed for reactivity against MMTV virion proteins by immunoblot 10 weeks after infection.
(B) Sera from RL-MuLV-infected B6 (left) and I/LnJ (right) mice with indicated genotypes were tested for reactivity against MuLV virion proteins by immunoblot 10 weeks after infection. PFU × 102: virus titers in mouse spleens (105 cells) 10 weeks after infection.
(C) WT I/LnJ and Tlr7−/− I/LnJ mice were i.p. injected with MMTV. Sera were analyzed for reactivity against MMTV virion proteins by immunoblot 10 weeks after infection.
Goat anti-mouse Abs coupled to HRP were used at the second step. Numbers correspond to individual mice within a single experiment.
To identify which of the two MyD88-dependent endosomal PRRs detects retroviruses, we infected retrovirus-resistant mice with deficiency in either TLR9 or TLR7. Both MuLV-infected Tlr9+/+ and Tlr9−/− B6 mice produced virus-specific Abs and cleared the virus (Figure S4B), indicating that TLR9 is not required for Ab-mediated control of retroviral infections. TLR7 is an endosomal MyD88-dependent PRR, which detects ssRNA (Diebold et al., 2004), and is therefore a likely candidate for a retroviral sensor. To determine whether TLR7-MyD88-mediated signaling is indeed the basis for detection of retroviral pathogens, we infected Tlr7−/− B6 and I/LnJ mice with MuLV and monitored them for antiviral immune responses. Although WT B6 and I/LnJ mice were able to efficiently control infection, Tlr7−/− B6 and I/LnJ mice retained infectious virus (Figure 5B). Restriction of viral replication in WT mice correlated with the production of virus-specific Abs (Figure 5B).
To determine whether recognition of MMTV infection is also TLR7 dependent, we infected Tlr7−/− I/LnJ and B6vic1I/LnJ mice (congenic B6 mice that carry the vic1 region from I/LnJ mice responsible for virus control) (Figure S5A) with MMTV and monitored them for virus-specific Ab production. Whereas WT I/LnJ and B6vic1I/LnJ mice mounted anti-MMTV humoral responses (Figure 5C; Figure S5B) and class-switched to an IgG2a- or IgG2c-specific response (not shown and Figure S5B), Tlr7−/− mice of both backgrounds did not produce virus-specific Abs of any isotype (Figure 5C; Figure S5B). These data indicate that retroviruses are detected via a TLR7-dependent mechanism and that this detection leads to an antivirus humoral immune response.
Type I IFNs Are Dispensable for Antiretroviral Humoral Responses
Stimulation of TLR7 results in upregulation of both type I IFNs (via interferon regulatory factor 7 [IRF7]) and inflammatory cytokines (via NF-κB) (Kawai and Akira, 2010). Type I IFNs have been implicated as crucial factors for antiviral immunity in the early stages of MuLV infection (Gerlach et al., 2006). To address whether type I IFNs contribute to protective antiretrovirus immunity, we infected IFN-αβ receptor 1-deficient (Ifnar1−/−) B6 mice with MuLV and compared their antivirus response to the response generated by infected WT B6 mice. Ifnar1−/− B6 mice produced antiretroviral Abs and cleared viral infection similarly to WT B6 mice (Figure 6). These results indicate that type I IFNs are not essential for the production of antiretroviral neutralizing Abs.
Figure 6. Virus Clearance Is Type I IFN Independent.
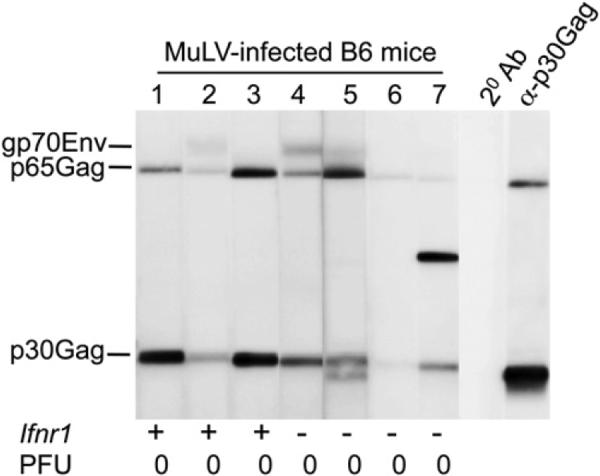
Sera from MuLV-infected B6 mice with indicated genotypes were tested for reactivity against MuLV virion proteins by immunoblot 10 weeks postinfection. Anti-mouse IgG2c-specific Abs coupled to HRP were used at the second step. PFU, virus titers in spleens (106 cells) 10 weeks after infection. Numbers correspond to individual mice within a single experiment.
DISCUSSION
Most retroviral infections result in indefinite viral persistence with an undetectable or inefficient antivirus immune response. Host failure to clear retroviral infections is probably due to the unique ability of retroviruses to evade or extinguish host immunity (Johnson and Desrosiers, 2002; Jude et al., 2003; Schneider-Schaulies and Dittmer, 2006), rather than the inability to detect infection. However, this lack of virus-neutralizing responses makes the identification of retroviral sensors in susceptible individuals an unachievable goal. To overcome this obstacle, we utilized mice from retrovirus-resistant strains, in which innate immune detection is translated into efficient virus-neutralizing immune responses. We found that replicating exogenous retrovirus did not require an external adjuvant to stimulate an antivirus immune response, and endogenous retroviruses (MMTV and MuLV) were capable of inducing the response as long as infectious virions were produced. Importantly, complete replication was dispensable for triggering of the sensor, as shown by the fact that UV-treated virions were still capable of upregulating the response. Thus, viral entry was documented as the critical step essential for induction of antiretroviral immunity.
Our findings pointed at the endosome as the most likely site for innate recognition of the retroviruses. Subsequent experiments revealed that TLR7 is in fact the retrovirus-sensing receptor, as indicated by the fact that MuLV- or MMTV-infected Tlr7−/− retrovirus-resistant mice did not produce virus-specific Abs and failed to clear infection. TLR7 is expressed in plasmacytoid dendritic cells (pDCs), CD11b+ DCs, and B cells (Iwasaki and Medzhitov, 2004). This receptor senses ssRNA derived from RNA viruses such as orthmyxoviruses (influenza A virus) (Diebold et al., 2004), paramyxoviruses (Sendai virus, SeV) (Lee et al., 2007), rhabdoviruses (vesicular stomatitis virus, VSV) (Lund et al., 2004), and arteriviruses (lactate dehydrogenase-elevating virus, LDV) (Ammann et al., 2009), and also from retroviruses (HIV) (Beignon et al., 2005; Hardy et al., 2007). Signaling through TLR7 triggered by these viruses induces secretion of type I IFNs and of proinflammatory cytokines as determined by in vitro and in vivo studies. However, one must exercise caution when addressing the role of PRR signaling in antiviral responses in vivo. For example, strong TLR7-dependent upregulation of type I IFNs by LDV appears to be completely dispensable for controlling virus replication in vivo (Ammann et al., 2009). The end-point of antiviral responses in retrovirus-resistant mice used in our study was viral clearance/control, so our results provide definitive evidence that TLR7-dependent recognition is required for an efficient immune response against retroviruses in vivo.
Our results further demonstrate that type I IFNs are not required for protective antiviral immunity and ultimate virus clearance. Although our experiments do not address the role for type I IFNs in the early stages of infection, they do suggest that production of type I IFNs is not an adequate read-out for the induction of antiviral adaptive immune responses, because they are dispensable for the control of retroviral infection in vivo.
Identification of viral PRR ligands in in vitro experiments has created an oversimplified view of innate sensing of viral pathogens, because these studies assume that viral nucleic acids are abundant in the compartments in which the sensors are located. The major problem with this assumption is that viral replication is tightly regulated and viral nucleic acids are protected from exposure to cellular factors by various means. Both viral RNA and unintegrated DNA reside within a protein core, termed preintegration complex (PIC), and are isolated from cellular factors and/or sensors at all times during the virus' journey from the cell membrane to the nucleus. Only when the PIC reaches the nucleus does it become accessible by other proteins (Bowerman et al., 1989). How then is the viral RNA being sensed by endosomal TLR7? Because there is no evidence documenting degradation of viral particles in endosomes prior to viral RNA release, we favor other potential explanations. One possibility is related to defective viral particles that enter cells but fail to complete the life cycle. Indeed, it has been estimated that only 10% of retroviral particles in a viral stock are able to productively infect cells (Andreadis et al., 2000). Thus, it is plausible that some defective virions enter cells and fuse with endosomes but fail to release their RNA into cytosol. Alternatively, cells that express TLR7 may somehow uniquely support the release of viral RNA into endosomes. The precise mechanism of retroviral RNA exposure to TLR7 is yet to be identified.
Immune control of retroviruses in virus-resistant B6 and I/LnJ mice requires both neutralizing Ab- and cell-mediated responses (Miyazawa et al., 2008). Although MuLV-infected B6.Tlr7−/− mice failed to raise a sustained antivirus Ab response, they did not develop splenomegaly by 12 weeks after infection (a phenotype controlled partly by cell-mediated responses), suggesting that cell-mediated responses were unaffected by TLR7 deficiency and that immune responses to retroviruses are probably regulated by other receptor(s) in addition to TLR7. Further supporting this notion, the cytotoxic T lymphocyte (CTL) response in MuLV-infected Myd88−/− B6 mice was also unchanged (Browne and Littman, 2009). Similarly, the CTL response against another ssRNA virus, influenza, is regulated by an additional unidentified factor(s) distinct from TLR7 (Heer et al., 2007; Koyama et al., 2007). Thus, it is safe to say that humoral and cellular responses against retroviruses are stimulated through distinct groups of innate sensors. In vivo experiments will be required to reveal other sensing mechanisms that lead to protective immunity.
As previously discussed, the genetic mechanisms underlying successful antiviral responses in B6 and I/LnJ mice are distinct. All mice appear capable of detecting retroviruses because both retrovirus-susceptible and retrovirus-resistant animals can initiate antivirus immune responses (Miyazawa et al., 2008; Purdy et al., 2003). Therefore, mutations within the genes that function to translate virus detection into beneficial adaptive immune responses or affect the virus's ability to evade immune responses are most likely to underlie resistance phenotypes in retrovirus-resistant mice.
Most HIV-infected people are able to detect infection, as shown by the fact that they mount CTL and Ab responses, but these responses generally do not result in protection against the virus (Pantaleo and Fauci, 1995). This further supports the notion that appropriate virus detection does not always lead to a neutralizing immune response; additional factors are required to translate this detection into protective responses, as occurs in the case of HIV “elite controllers.” These elite controllers represent approximately 1% of all HIV-1 infections (Deeks and Walker, 2007) and exhibit mechanisms similar to retrovirus-resistant mice: they control viral replication through productive and sustained antiretroviral immune responses. An understanding of the genetic mechanisms underlying retroviral resistance will help to design antiretroviral vaccines that utilize inactivated viruses combined with targeting of the immune pathways employed successfully in resistant organisms.
EXPERIMENTAL PROCEDURES
Mice
Mice used in this study were bred and maintained at the animal facility of the University of Chicago. B6, I/LnJ, BALB/cJ, C3.JK-H2j H2-T18b/Sn (C3.JK, C3H/He mice congenic by the MHC H-2j haplotype to I/LnJ mice), and B6;129S1-Tlr3tm1Flv/J mice (Alexopoulou et al., 2001) were purchased from The Jackson Laboratory. B6.Myd88−/− mice (Adachi et al., 1998) were obtained from R. Medzhitov, Yale University. B6.Tlr7−/− mice (Hemmi et al., 2002) were obtained from D. Roopenian, The Jackson Laboratory. B6.Ifnar1−/− mice (Müller et al., 1994) were obtained from D. Portnoy, University of California at Berkley. B6.Irf3−/− mice (Sato et al., 2000) were obtained from K. Fitzgerald, University of Massachusetts. C3H/HeN mice were originally purchased from NCI and have been maintained in our colony for more than 12 years. I/LnJ hybrid provirus (HP) transgenic mice were generated by crossing C3H/HeN HP transgenic mice (Golovkina et al., 1994) to I/LnJ mice for 10 generations. I/LnJ Ifng−/− mice have been previously described (Purdy et al., 2003). B6;129S1-Tlr3tm1Flv/J mice were crossed to I/LnJ mice for seven generations, and heterozygous mice were intercrossed to produce I/LnJ Tlr3−/− mice. I/LnJ mice were crossed to B6.Myd88−/− mice for eight generations, and heterozygous N8 mice were intercrossed to generate I/LnJ Myd88−/− mice. B6.Tlr7−/− were crossed to I/LnJ mice for four generations. Heterozygous animals were intercrossed and Tlr7−/− mice homozygous for the I/LnJ alleles of vic1 (Case et al., 2008) and rfv3 (Super et al., 1999) were selected for experimentation. All imported animals were rederived by standard embryo transfer at Charles River before introduction to our colony. The studies described in this paper have been reviewed and approved by the Animal Care and Use Committee at the University of Chicago.
MMTV Infection and Immunization
Two exogenous MMTV variants were used in these studies. An MMTV(LA) viral variant consisting of three different viruses (BALBLA, BALB2, and BALB14, with Vβ6- and 8-, Vβ2-, and Vβ14-specific superantigens [Sags], respectively) (Golovkina et al., 1997; Piazzon et al., 1994) was propagated in BALB/cJ and C3H/HeN mice. Deletion of CD4+Vβ6+, CD4+Vβ14+, and CD4+Vβ2+ SAgcognate T cells was used as a readout for MMTV(LA) infection (Purdy et al., 2003). I/LnJ mice inherit Mtv7 and thus do not have Vβ6+ T cells (MacDearmid et al., 2006; Purdy et al., 2003). Consequently, MMTV(LA)-infected I/LnJ mice were screened for virus infection by following deletion of CD4+Vβ14+ and CD4+Vβ2+ T cells among CD4+ T cells. HP is an exogenous MMTV produced by the HP transgene (Figure S1A; Golovkina et al., 1994; Hook et al., 2000), which encodes for the Vβ14-specific SAg (Choi et al., 1991). As a result, mice infected with exogenous HP show deletion of CD4+Vβ14+ T cells (Hook et al., 2000).
Mice were infected by either fostering on viremic females or by i.p. injection of purified virions. For injection, MMTV virions were purified from solidified milk removed from the stomachs of 2- to 4-day-old MMTV-infected pups, diluted in 10 volumes of phosphate-buffered saline, and centrifuged at 2000 × g for 15 min. The cream and pellet were discarded, and the skim milk was injected at an equivalent of ~50 μl of milk per mouse.
In some instances, mice were injected i.p. with MMTV virions heated at 85° C for 10 min, or immunized with 85° C heated MMTV virions in CFA (DIFCO) as previously described (Purdy et al., 2003).
MuLV Isolation and Infection
Rauscher-like-MuLV (RL-MuLV) virus mixture has been described previously (Hook et al., 2002) and was propagated in SC-1 cells (ATCC) (Case et al., 2008). Viral titers in supernatants were determined by an infectious center assay (Rowe et al., 1970) and mice were injected i.p. with 2 × 104 PFU of virus per mouse.
To test for ecotropic MuLVs, 105 to 107 splenocytes isolated from I/LnJ and BALB/cJ mice were irradiated at 2000 Rad and subjected to the infectious center assay. To isolate exogenous virus produced by the endogenous ecotropic provirus, irradiated splenocytes from an aged I/LnJ mouse were cocultured with SC-1 cells. Supernatants were collected from SC-1 cells after four passages. Titers of infectious virus were determined by infectious center assay. Virus was injected at 2 × 104 PFU per mouse.
UV Irradiation
Within each experiment, a single viral prep was divided equally into live and UV-irradiated fractions and the same amount was injected into mice. For UV irradiation, purified MMTV or MuLV virions were exposed to 72 Ergs/cm2 UV irradiation for 45 min on ice 25 cm from the source. Live and UV-irradiated virions were injected i.p. at an equivalent of 50 μl milk (MMTV) and 2 × 104 PFU (MuLV) per mouse.
ELISA
To detect MMTV and MuLV Abs in mouse sera, an enzyme-linked immunosorbent assay (ELISA) was performed as previously described (Case et al., 2008; Purdy et al., 2003). All sera were used at 5 × 10−3 dilution. Mouse IgG2a-, IgG2c-specific, or polyisotypic immunoglobulin Abs coupled to alkaline phosphatase (AP) or horseradish peroxidase (HRP) were used at the second step (Southern Biotech; Jackson ImmunoResearch). Backgrounds obtained from incubation with secondary Abs alone were subtracted.
Immunoblot Analysis
MMTV virions were isolated from the milk as previously described (Purdy et al., 2003). MuLV virions were isolated from supernatants of infected SC-1 cells via centrifugation at 95,000 × g. MMTV and MuLV virion proteins were electrophoresed on 15% polyacrylamide gels under reducing conditions, transferred to polyvinylidene fluoride (PVDF) membranes (Bio-Rad), and incubated with sera or monoclonal Abs. All sera were used at 5 × 10−3 dilution. Anti-Env and anti-Gag MMTV monoclonal Abs (Purdy et al., 2003) were used as positive controls for the MMTV-specific immunoblot. Anti-p30CA, a rat monoclonal Ab against p30CAGag (ATCC CRL-1912, generous gift of W. Mothes, Yale University) was used as control for MuLV-specific immunoblots. For monoclonal Abs, anti-mouse or anti-rat polyisotypic Abs coupled to HRP were used at the second step. For sera, either mouse IgG2a-, IgG2c-specific, or polyisotypic immunoglobulin Abs coupled to HRP were used at the second step (Southern Biotech; Jackson ImmunoResearch; Bio-Rad). Blots were developed with immunoblot detection reagents (GE Healthcare Life Sciences).
Immunofluorescence
NMuMG (ATCC #CRL-1636) or XC (ATCC #CCL-165) cells seeded on CultureWell 8-well Coverslips (Sigma-Aldrich) were incubated with live or UV-treated MMTV (purified from ~10 μl milk) or MuLV (1 × 105 PFU), respectively, for 90 min at 4°C, then shifted to 37°C for 15 min (as previously described by Wang et al., 2008). Cells were then fixed with 4% paraformaldehyde in PBS for 15 min and permeabilized with 0.2% Triton X-100. After blocking with 30% sheep-serum, cells were incubated with primary and secondary Abs and mounted on DAPI-containing mounting medium (Invitrogen).
For detailed methods, please refer to Supplemental Experimental Procedures.
Supplementary Material
ACKNOWLEDGMENTS
We are thankful to the members of the T.V.G. laboratory, J. Bubeck-Warden-burg, and A. Chervonsky for helpful discussion. We are also thankful to A. Kozlova and The University of Chicago Light Microscopy director V. Bindokas for technical support. This work was supported by PHS grant CA134667 to T.V.G. and by a grant (P30 CA014599) to the University of Chicago.
Footnotes
SUPPLEMENTAL INFORMATION Supplemental Information includes Supplemental Experimental Procedures and five figures and can be found with this article online at doi:10.1016/j.immuni.2011.05.011.
REFERENCES
- Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Ammann CG, Messer RJ, Peterson KE, Hasenkrug KJ. Lactate dehydrogenase-elevating virus induces systemic lymphocyte activation via TLR7-dependent IFNalpha responses by plasmacytoid dendritic cells. PLoS ONE. 2009;4:e6105. doi: 10.1371/journal.pone.0006105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreadis S, Lavery T, Davis HE, Le Doux JM, Yarmush ML, Morgan JR. Toward a more accurate quantitation of the activity of recombinant retroviruses: alternatives to titer and multiplicity of infection. J. Virol. 2000;74:3431–3439. doi: 10.1128/jvi.74.7.3431-3431.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beignon AS, McKenna K, Skoberne M, Manches O, DaSilva I, Kavanagh DG, Larsson M, Gorelick RJ, Lifson JD, Bhardwaj N. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J. Clin. Invest. 2005;115:3265–3275. doi: 10.1172/JCI26032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A, Bui LK, Feldman MA, Larsson M, Bhardwaj N. Inactivated influenza virus, when presented on dendritic cells, elicits human CD8+ cytolytic T cell responses. J. Exp. Med. 1995;182:1663–1671. doi: 10.1084/jem.182.6.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhoj VG, Sun Q, Bhoj EJ, Somers C, Chen X, Torres JP, Mejias A, Gomez AM, Jafri H, Ramilo O, Chen ZJ. MAVS and MyD88 are essential for innate immunity but not cytotoxic T lymphocyte response against respiratory syncytial virus. Proc. Natl. Acad. Sci. USA. 2008;105:14046–14051. doi: 10.1073/pnas.0804717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeke JD, Stoye JP. Retrotransposons, endogenous retroviruses, and the evolution. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1997. pp. 343–436. [PubMed] [Google Scholar]
- Bowerman B, Brown PO, Bishop JM, Varmus HE. A nucleoprotein complex mediates the integration of retroviral DNA. Genes Dev. 1989;3:469–478. doi: 10.1101/gad.3.4.469. [DOI] [PubMed] [Google Scholar]
- Browne EP, Littman DR. Myd88 is required for an antibody response to retroviral infection. PLoS Pathog. 2009;5:e1000298. doi: 10.1371/journal.ppat.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buggiano V, Goldman A, Nepomnaschy I, Bekinschtein P, Berguer P, Lombardi G, Deroche A, Francisco MV, Piazzon I. Characterization of two infectious mouse mammary tumour viruses: Superantigenicity and tumorigenicity. Scand. J. Immunol. 1999;49:269–277. doi: 10.1046/j.1365-3083.1999.00502.x. [DOI] [PubMed] [Google Scholar]
- Case LK, Purdy A, Golovkina TV. Molecular and cellular basis of the retrovirus resistance in I/LnJ mice. J. Immunol. 2005;175:7543–7549. doi: 10.4049/jimmunol.175.11.7543. [DOI] [PubMed] [Google Scholar]
- Case LK, Petell L, Yurkovetskiy L, Purdy A, Savage KJ, Golovkina TV. Replication of beta- and gammaretroviruses is restricted in I/LnJ mice via the same genetic mechanism. J. Virol. 2008;82:1438–1447. doi: 10.1128/JVI.01991-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesebro B, Miyazawa M, Britt WJ. Host genetic control of spontaneous and induced immunity to Friend murine retrovirus infection. Annu. Rev. Immunol. 1990;8:477–499. doi: 10.1146/annurev.iy.08.040190.002401. [DOI] [PubMed] [Google Scholar]
- Choi Y, Kappler JW, Marrack P. A superantigen encoded in the open reading frame of the 3′ long terminal repeat of mouse mammary tumour virus. Nature. 1991;350:203–207. doi: 10.1038/350203a0. [DOI] [PubMed] [Google Scholar]
- Cloyd MW, Hartley JW, Rowe WP. Lymphomagenicity of recombinant mink cell focus-inducing murine leukemia viruses. J. Exp. Med. 1980;151:542–552. doi: 10.1084/jem.151.3.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffin JM. Retroviridae and their replication. In: Fields BN, Knipe DM, editors. Virology. New York: Raven Press: 1990. pp. 1437–1500. [Google Scholar]
- Deeks SG, Walker BD. Human immunodeficiency virus controllers: Mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity. 2007;27:406–416. doi: 10.1016/j.immuni.2007.08.010. [DOI] [PubMed] [Google Scholar]
- Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- Dittmer U, He H, Messer RJ, Schimmer S, Olbrich AR, Ohlen C, Greenberg PD, Stromnes IM, Iwashiro M, Sakaguchi S, et al. Functional impairment of CD8(+) T cells by regulatory T cells during persistent retroviral infection. Immunity. 2004;20:293–303. doi: 10.1016/s1074-7613(04)00054-8. [DOI] [PubMed] [Google Scholar]
- Edelmann KH, Richardson-Burns S, Alexopoulou L, Tyler KL, Flavell RA, Oldstone MB. Does Toll-like receptor 3 play a biological role in virus infections? Virology. 2004;322:231–238. doi: 10.1016/j.virol.2004.01.033. [DOI] [PubMed] [Google Scholar]
- Evans DT, Desrosiers RC. Immune evasion strategies of the primate lentiviruses. Immunol. Rev. 2001;183:141–158. doi: 10.1034/j.1600-065x.2001.1830112.x. [DOI] [PubMed] [Google Scholar]
- Gerlach N, Schimmer S, Weiss S, Kalinke U, Dittmer U. Effects of type I interferons on Friend retrovirus infection. J. Virol. 2006;80:3438–3444. doi: 10.1128/JVI.80.7.3438-3444.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golovkina TV. A novel mechanism of resistance to mouse mammary tumor virus infection. J. Virol. 2000;74:2752–2759. doi: 10.1128/jvi.74.6.2752-2759.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golovkina TV, Chervonsky A, Prescott JA, Janeway CA, Jr., Ross SR. The mouse mammary tumor virus envelope gene product is required for superantigen presentation to T cells. J. Exp. Med. 1994;179:439–446. doi: 10.1084/jem.179.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golovkina TV, Dudley JP, Jaffe AB, Ross SR. Mouse mammary tumor viruses with functional superantigen genes are selected during in vivo infection. Proc. Natl. Acad. Sci. USA. 1995;92:4828–4832. doi: 10.1073/pnas.92.11.4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golovkina TV, Piazzon I, Nepomnaschy I, Buggiano V, de Olano Vela M, Ross SR. Generation of a tumorigenic milk-borne mouse mammary tumor virus by recombination between endogenous and exogenous viruses. J. Virol. 1997;71:3895–3903. doi: 10.1128/jvi.71.5.3895-3903.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy AW, Graham DR, Shearer GM, Herbeuval JP. HIV turns plasmacytoid dendritic cells (pDC) into TRAIL-expressing killer pDC and down-regulates HIV coreceptors by Toll-like receptor 7-induced IFN-alpha. Proc. Natl. Acad. Sci. USA. 2007;104:17453–17458. doi: 10.1073/pnas.0707244104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasenkrug KJ, Valenzuela A, Letts VA, Nishio J, Chesebro B, Frankel WN. Chromosome mapping of Rfv3, a host resistance gene to Friend murine retrovirus. J. Virol. 1995;69:2617–2620. doi: 10.1128/jvi.69.4.2617-2620.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heer AK, Shamshiev A, Donda A, Uematsu S, Akira S, Kopf M, Marsland BJ. TLR signaling fine-tunes anti-influenza B cell responses without regulating effector T cell responses. J. Immunol. 2007;178:2182–2191. doi: 10.4049/jimmunol.178.4.2182. [DOI] [PubMed] [Google Scholar]
- Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, Horiuchi T, Tomizawa H, Takeda K, Akira S. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat. Immunol. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- Hook LM, Agafonova Y, Ross SR, Turner SJ, Golovkina TV. Genetics of mouse mammary tumor virus-induced mammary tumors: Linkage of tumor induction to the gag gene. J. Virol. 2000;74:8876–8883. doi: 10.1128/jvi.74.19.8876-8883.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook LM, Jude BA, Ter-Grigorov VS, Hartley JW, Morse HC, 3rd, Trainin Z, Toder V, Chervonsky AV, Golovkina TV. Characterization of a novel murine retrovirus mixture that facilitates hematopoiesis. J. Virol. 2002;76:12112–12122. doi: 10.1128/JVI.76.23.12112-12122.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinohe T, Pang IK, Iwasaki A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 2010;11:404–410. doi: 10.1038/ni.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- Johnson WE, Desrosiers RC. Viral persistence: HIV's strategies of immune system evasion. Annu. Rev. Med. 2002;53:499–518. doi: 10.1146/annurev.med.53.082901.104053. [DOI] [PubMed] [Google Scholar]
- Jude BA, Pobezinskaya Y, Bishop J, Parke S, Medzhitov RM, Chervonsky AV, Golovkina TV. Subversion of the innate immune system by a retrovirus. Nat. Immunol. 2003;4:573–578. doi: 10.1038/ni926. [DOI] [PubMed] [Google Scholar]
- Kane M, Case LK, Golovkina TV. Vital role for CD8+ cells in controlling retroviral infections. J. Virol. 2011;85:3415–3423. doi: 10.1128/JVI.01768-10. Published online January 19, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katen LJ, Januszeski MM, Anderson WF, Hasenkrug KJ, Evans LH. Infectious entry by amphotropic as well as ecotropic murine leukemia viruses occurs through an endocytic pathway. J. Virol. 2001;75:5018–5026. doi: 10.1128/JVI.75.11.5018-5026.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- Koyama S, Ishii KJ, Kumar H, Tanimoto T, Coban C, Uematsu S, Kawai T, Akira S. Differential role of TLR- and RLR-signaling in the immune responses to influenza A virus infection and vaccination. J. Immunol. 2007;179:4711–4720. doi: 10.4049/jimmunol.179.7.4711. [DOI] [PubMed] [Google Scholar]
- Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315:1398–1401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA. 2004;101:5598–5603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDearmid CC, Case LK, Starling CL, Golovkina TV. Gradual elimination of retroviruses in YBR/Ei mice. J. Virol. 2006;80:2206–2215. doi: 10.1128/JVI.80.5.2206-2215.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malim MH, Emerman M. HIV-1 accessory proteins—Ensuring viral survival in a hostile environment. Cell Host Microbe. 2008;3:388–398. doi: 10.1016/j.chom.2008.04.008. [DOI] [PubMed] [Google Scholar]
- Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Janeway CA., Jr. Innate immunity: The virtues of a nonclonal system of recognition. Cell. 1997;91:295–298. doi: 10.1016/s0092-8674(00)80412-2. [DOI] [PubMed] [Google Scholar]
- Miyazawa M, Tsuji-Kawahara S, Kanari Y. Host genetic factors that control immune responses to retrovirus infections. Vaccine. 2008;26:2981–2996. doi: 10.1016/j.vaccine.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Müller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Ogura H. XC cell fusion by murine leukemia viruses: Fusion from without. Med. Microbiol. Immunol. (Berl.) 1976;162:175–181. doi: 10.1007/BF02120995. [DOI] [PubMed] [Google Scholar]
- Pantaleo G, Fauci AS. New concepts in the immunopathogenesis of HIV infection. Annu. Rev. Immunol. 1995;13:487–512. doi: 10.1146/annurev.iy.13.040195.002415. [DOI] [PubMed] [Google Scholar]
- Piazzon I, Goldman A, Torello S, Nepomnaschy I, Deroche A, Dran G. Transmission of an Mls-1a-like superantigen to BALB/c mice by foster-nursing on F1 Mls-1bxa mothers. Sex-influenced onset of clonal deletion. J. Immunol. 1994;153:1553–1562. [PubMed] [Google Scholar]
- Purdy A, Case L, Duvall M, Overstrom-Coleman M, Monnier N, Chervonsky A, Golovkina T. Unique resistance of I/LnJ mice to a retrovirus is due to sustained interferon gamma-dependent production of virus-neutralizing antibodies. J. Exp. Med. 2003;197:233–243. doi: 10.1084/jem.20021499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risser R, Horowitz JM, McCubrey J. Endogenous mouse leukemia viruses. Annu. Rev. Genet. 1983;17:85–121. doi: 10.1146/annurev.ge.17.120183.000505. [DOI] [PubMed] [Google Scholar]
- Rosenberg N, Jolicoeur P. Retroviral pathogenesis. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1997. pp. 475–586. [PubMed] [Google Scholar]
- Rowe WP, Pugh WE, Hartley JW. Plaque assay techniques for murine leukemia viruses. Virology. 1970;42:1136–1139. doi: 10.1016/0042-6822(70)90362-4. [DOI] [PubMed] [Google Scholar]
- Santiago ML, Montano M, Benitez R, Messer RJ, Yonemoto W, Chesebro B, Hasenkrug KJ, Greene WC. Apobec3 encodes Rfv3, a gene influencing neutralizing antibody control of retrovirus infection. Science. 2008;321:1343–1346. doi: 10.1126/science.1161121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago ML, Smith DS, Barrett BS, Montano M, Benitez RL, Pelanda R, Hasenkrug KJ, Greene WC. Persistent Friend virus replication and disease in Apobec3-deficient mice expressing functional B-cell-activating factor receptor. J. Virol. 2011;85:189–199. doi: 10.1128/JVI.01838-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- Schneider-Schaulies S, Dittmer U. Silencing T cells or T-cell silencing: Concepts in virus-induced immunosuppression. J. Gen. Virol. 2006;87:1423–1438. doi: 10.1099/vir.0.81713-0. [DOI] [PubMed] [Google Scholar]
- Shackleford GM, Varmus HE. Construction of a clonable, infectious, and tumorigenic mouse mammary tumor virus provirus and a derivative genetic vector. Proc. Natl. Acad. Sci. USA. 1988;85:9655–9659. doi: 10.1073/pnas.85.24.9655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoye JP, Moroni C, Coffin JM. Virological events leading to spontaneous AKR thymomas. J. Virol. 1991;65:1273–1285. doi: 10.1128/jvi.65.3.1273-1285.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Super HJ, Hasenkrug KJ, Simmons S, Brooks DM, Konzek R, Sarge KD, Morimoto RI, Jenkins NA, Gilbert DJ, Copeland NG, et al. Fine mapping of the friend retrovirus resistance gene, Rfv3, on mouse chromo-some 15. J. Virol. 1999;73:7848–7852. doi: 10.1128/jvi.73.9.7848-7852.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji-Kawahara S, Chikaishi T, Takeda E, Kato M, Kinoshita S, Kajiwara E, Takamura S, Miyazawa M. Persistence of viremia and production of neutralizing antibodies differentially regulated by polymorphic APOBEC3 and BAFF-R loci in friend virus-infected mice. J. Virol. 2010;84:6082–6095. doi: 10.1128/JVI.02516-09. Published online April 7, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E, Obeng-Adjei N, Ying Q, Meertens L, Dragic T, Davey RA, Ross SR. Mouse mammary tumor virus uses mouse but not human transferrin receptor 1 to reach a low pH compartment and infect cells. Virology. 2008;381:230–240. doi: 10.1016/j.virol.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrona T, Dudley JP. Major histocompatibility complex class II I-E-independent transmission of C3H mouse mammary tumor virus. J. Virol. 1996;70:1246–1249. doi: 10.1128/jvi.70.2.1246-1249.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto KR. Steroid receptor regulated transcription of specific genes and gene networks. Annu. Rev. Genet. 1985;19:209–252. doi: 10.1146/annurev.ge.19.120185.001233. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.