

Abstract
Fibroblast growth factor 23 (FGF23) is responsible for phosphate wasting and the phenotypic changes observed in human diseases such as X-Linked Hypophosphatemia (XLH). Targeted over-expression of nuclear high molecular weight fibroblast growth factor 2 isoforms (HMW isoforms) in osteoblasts resulted in a transgenic mouse with phenotypic changes similar to XLH, including increased FGF23, hypophosphatemia and rickets/osteomalacia. The goal of this study was to assess whether HMW isoforms also reduced mineralized bone formation via phosphate-independent effects in bone marrow stromal cells (BMSCs) by modulating FGF23/FGFR/ERKsignaling. To determine if decreased bone formation in BMSC cultures from HMW transgenic mice could be rescued by blocking this pathway, an FGF23 neutralizing antibody, the FGF receptor tyrosine kinase inhibitor SU5402 and the MAP kinase inhibitor PD98059 were used. FGF23 levels in the conditioned medium of HMW BMSC cultures were dramatically increased compared to BMSC from control (Vector) mice. Mineralized nodule formation was significantly decreased in HMW BMSC cultures compared with control cultures. The decreased nodule formation in HMW cultures was partially rescued by the FGF23 neutralizing antibody, SU5402 and PD98059. mRNA levels for the osteoblast-related genes, osteocalcin, Runx2, and osterix, and the osteocyte-related gene Dmp1 were significantly decreased in HMW cultures compared with control cultures, and the decreases were partially rescued by SU5402 or PD98059 treatment. Matrix-gla-protein (Mgp) mRNA was significantly higher in HMW cultures compared with control cultures, reduced by SU5402, but further increased byPD98059. Our results suggest that phosphate-independent effects of HMW isoforms in vitro may be directly mediated in part via FGF23 and that HMW isoforms signal via FGF23/FGFR/MAPK to inhibit bone formation in vitro.
Keywords: HMWFGF2, FGF23, PHOSPHATE, MAPK, FGF RECEPTOR INHIBITOR
Introduction
Fibroblast growth factor 2 (FGF2) is widely expressed and is a mitogen for many cell types, including osteoblasts and chondrocytes (1). The Fgf2 gene encodes multiple protein isoforms of FGF2 that are ubiquitously expressed in cells including osteoblasts. In humans, there are 4 high molecular weight nuclear isoforms (HMW) and a low molecular weight 18kDa FGF2 protein (LMW) that is exported from cells. HMW isoforms are directly translocated into the nucleus to mediate biological effects (2). HMW isoforms remain intracellular and are usually not released from cells; but they have nuclear localization sequences and function in an intracrine manner (3). Previous studies showed that global over-expression of all isoforms of human FGF2 protein resulted in dwarfism in mice (4) and decreased bone mineral density (BMD) and bone mass (5). To determine whether there are differential biological effects of the LMW versus HMW isoforms in bone, we developed transgenic mice in which either the LMW or HMW isoforms are expressed in osteoblast lineage cells. We previously made novel observations that targeted over-expression of the LMW isoform in osteoblasts resulted in increased BMD and bone mass in mice via modulation of Wnt signaling (6). In contrast, targeted over-expression of nuclear HMW isoforms in osteoblasts resulted in dwarfism, decreased BMD and bone mass and hypophosphatemia in mice via modulation of FGF23 and Klotho (7). Interestingly, the decreased bone mass and hypophosphatemia of HMW transgenic mice is similar to results reported in mice that over-express FGF23 (8) and in the Hyp mouse, a mouse model of human X-linked hypophosphatemic rickets/osteomalacia (XLH) (9).
As noted above, HMW isoforms are ordinarily not released from the cells but function in an intracrine manner (3). The HMW isoforms mediate their effects on bone and kidney by modulating the expression of the phosphatonin FGF23 (7), which is secreted and acts locally or systemically in a paracrine or endocrine manner (10). Intriguingly, in the Hyp mouse, a model of XLH, Fgf2 mRNA and protein were increased in bones/osteoblasts, and FGF23 and HMW protein were co-localized in osteocytes (7). These novel findings provided support for the idea that HMW isoforms play a role in FGF23 regulation. Indeed, our published studies demonstrated that transfection of Fgf2-null osteoblasts with an expression vector for the HMW isoforms resulted in increased Fgf23 mRNA expression (7), whereas transfection with a LMW expression construct did not (7).
The present study investigates the role of HMW isoforms in bone formation in vitro and the involvement of FGF23 in the action of HMW isoforms on bone nodule formation in vitro in a phosphate replete mouse bone marrow stromal cell (BMSC) culture model. This study demonstrates that HMW isoforms inhibit bone formation independently of their systemic effects on phosphate homeostasis. Furthermore, the results reported here revealed novel mechanisms of bone formation regulated by HMW isoforms through activation of FGF23/FGFR1/MAPK signaling.
Materials and Methods
Vector and HMW transgenic mice
Development of the Vector and HMW transgenic mice was previously described in detail (7). Briefly, Col3.6-HMWFgf2 isoforms-IRES-GFPsaph (Green Fluorescent Protein-Sapphire) was built by replacing a chloramphenicol acetyltransferase fragment in previously made Col3.6-CAT-IRES-GFPsaph with HMW isoforms of human Fgf2 cDNA. This expression vector concurrently overexpresses HMW and GFPsaph from a single bicistronic mRNA. Col3.6-IRES/GFPsaph (Vector) construct was also prepared as a control. The construct inserts were released from Col3.6-IRES/GFP (Vector) or Col3.6-HMWFgf2 isoforms-IRES-GFPsaph by digestion with AseI and AflII(7). Microinjections into the pronuclei of fertilized oocytes were performed at the Gene Targeting and Transgenic Facility at the University of Connecticut Health Center. Founder mice of the F2 (FVBN) strain were bred with wild type mice to establish individual transgenic lines. The University of Connecticut Health Center, Institutional Animal Care and Use Committee approved all animal procedures.
Mouse BMSC cultures
Mouse BMSCs were isolated from Vector and HMW mice as previously described (11).The studies were conducted using 2-month-old male homozygote mice. Tibiae and femurs were dissected free of adhering tissue. The bone ends were removed and the marrow cavity flushed with alpha minimal essential medium (αMEM, Invitrogen, Grand Island, NY). To perform an in vitro analysis of osteogenesis, BMSCs from both genotypes were plated at 2 × 106/well in 6-well dishes and cultured in basal medium [alpha-MEM+10% heat inactivated fetal calf serum (FBS)+ penicillin (100 U/ml) and streptomycin (50 µg/ml)] for 3 days, then switched to osteogenic media [basal medium + phospho-ascorbate (50 µg/ml) + β-glycerophosphate (8 mM)] for an additional 19 days. Media were changed every other day. Cells cultured in osteogenic media were stained for alkaline phosphatase (ALP) using a commercial kit (Sigma, St. Louis, MO), scanned and counter-stained for total cells by crystal violet and for mineral by von Kossa (12). Some dishes were stained for calcium using alizarin red S (Sigma Chemical Co, St Louis, MO)(13). Quantitative analysis of the calcium content was performed after solubilizing alizarin red S staining. To determine whether an FGF23 neutralizing antibody could rescue defective bone nodule formation in BMSC cultures from HMW transgenic mice, BMSCs from both genotypes were plated at 2 × 106 cells/well in 6 well dishes. Cells were treated with control IgG (Control) or a rat anti-rat FGF23 neutralizing antibody (clone 58.5, 100 nM) (a gift from Amgen Inc., Thousand Oaks, CA) throughout the 21 days of culture. The control antibody was rat-anti-NGFPb-3F8-raIgG2a. The FGF23 and control antibodies were dissolved in A5su buffer (9% sucrose in sodium acetate buffer, pH 5.0). The IC50 of the antibody is about 2nM for inhibiting FGF23 induction of Elk1 signaling and complete inhibition was obtained at 100nM(communication with Amgen). Based on this report, we used 100nM of the FGF23 antibody for the in vitro studies. Conditioned media were collected for an FGF23 ELISA (kindly measured by Immutopics, Inc., San Clemente, CA). Cultures were harvested at 21 days and stained for calcium by alizarin red S(13).
To determine whether SU5402 (EMD Chemicals, Inc. Gibbstown, NJ), a FGFR1-specific tyrosine kinase inhibitor, or PD98059 (Sigma, St. Louis, MO), a specific p42/44 mitogen-activated protein (MAP) kinase cascade inhibitor, could rescue defective bone nodule formation in HMW BMSC cultures, BMSCs from both genotypes were plated at 2 × 106 cells/well in 6 well dishes. Cells were treated with vehicle dimethyl sulfoxide (DMSO)(Sigma, St. Louis, MO), SU5402 (25uM) or PD98059 (25uM) from day 14 of culture. SU5402 and PD98059 were added with every media change. On day 21 of culture, xylenol orange (20uM) was added to the culture to stain for mineralization in living cells for 2 hours and then the dishes were scanned for the expression of GFP and xylenol orange using an Olympus IX50 inverted system microscope equipped with an IX-FLA inverted reflected light fluorescence (Olympus America, Inc., Melville, NY) (6). Fluorescent images were taken with equal exposure times applied to cultures derived from Vector and HMW mice. Then, the cultures were harvested, stained for alizarin red S, and scanned by HP Precision Scan Pro, followed by solubilization of alizarin red S and spectroscopic quantification (14).
Regulation of cell growth in BMSCs
To assess the effects of HMW isoforms on metabolic activity, BMSCs were plated at 0.3x106 cells/well in 96-well dishes in basal medium. After overnight culture, non-adherent cells were washed away with PBS and adherent cells were cultured for 1,2,3, or 4 days. For the last 1 h of culture, 20 ml per well of the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) Reagent (Promega Corporation, Madison, WI) was added and metabolic activity was measured by the MTT assay. Absorbance is directly proportional to the metabolic activity of the cells.
To assess the effects of overexpression of HMW isoforms on cell proliferation, cell number was counted. BMSCs were plated at 0.03x106 cells /well in 96-well dishes in basal medium. Non-adherent cells were washed away after overnight culture. Adherent cells were counted on day 1,2,3, and 4.
RNA isolation and mRNA expression
Total RNA was extracted from cells of sister plates using Trizol reagent (Invitrogen Life Technologies, Carlsbad, CA) according to the manufacturer's instructions. For real-time, quantitative RT-PCR (qRT-PCR) analysis, RNA was reverse-transcribed by the Super-Script™ First-Strand Synthesis System (Invitrogen Life Technologies, Carlsbad, CA). Quantitative PCR was carried out using the QuantiTect™ SYBR Green PCR kit (Qiagen) on a MyiQ™ instrument (BIO-RAD Laboratories Inc. Hercules, CA). The primer sequences for the genes of interest are shown in Table 1. β-actin was used as an internal reference for each sample. Using a formula described previously (15), mRNA was normalized to the β-actin mRNA level and expressed as the fold-change relative to the appropriate Vector group.
Table 1.
Primers used in qRT-PCR.
| Gene | Forward | Reverse |
|---|---|---|
| β actin | 5′-atggctggggtgttgaaggt-3′ | 5′-atctggcaccacaccttctacaa-3′ |
| Runx2/Cbfa1 | 5′-acaaacaaccacagaaccacaagt-3′ | 5′-gtctcggtggctggtagtga-3′ |
| osterix | 5′-actggctaggtggtggtcag-3′ | 5′-ggtagggagctgggttaagg-3′ |
| OCN | 5′-gagggcaataaggtagtgaacaga-3′ | 5′-aagccatactggtttgatagctcg-3′ |
| Fgf2 | 5’-gtgtgtgctaaccgttacct-3’ | 5’-gctcttagcagacattggaag-3’ |
| Fgf23 | 5’-acttgtcgcagaagcatc-3’ | 5’-gtgggcgaacagtgtagaa-3’ |
| Fgfr1c | 5’-gactgctggagttaatacca-3’ | 5’-ctggtctctcttccagggct-3’ |
| Dmp1 | 5’-caactggcttttctgtggcaa-3’ | 5’-tgggtgcgctgatgtttgct-3’ |
| Mgp | 5’-gtggcgagctaaagcccaa-3’ | 5’-cgtagcgctcacacagcttg-3’ |
Western blot analysis
Nuclear and cytosolic protein were extracted from cultured BMSCs using a Nuclear Extraction Kit (Panomics, Inc., Fremont, CA) following the instructions provided by the manufacturer. Protein concentration was assayed with BCA protein assay reagent (Pierce, Rockford, IL). After SDS-polyacrylamide gel electrophoresis on 4–15% gels, proteins were transferred to Immun-Blot PVD membranes (Bio-Rad Laboratories, Hercules, CA). Membranes were blocked overnight in TBS-T containing 5% nonfat dry milk (BIO-RAD). Membranes were then incubated with an anti-FGF Receptor 1 antibody (Cell Signaling Technology, Danvers, MA), anti-mouse FGF-23 antibody (R&D Systems, Minneapolis, MN), anti phospho-p44/42 MAPK antibody (Cell Signaling Technology, Inc., Danvers, MA) or anti-mouse FGF2 antibody (BD Biosciences, Billerica, MA) for 1 h, washed 1 h with TBS-T, and then incubated with a donkey anti-rabbit secondary antibody (Amersham Biosciences Piscataway, NJ) for FGFR1 and phosphate-p44/42, rabbit anti-rat secondary (Stressgen, Ann Arbor, MI) for FGF23 or sheep anti mouse secondary antibody for FGF2 (GE Healthcare, Giles, Buckinghamshire HP8 4SP, United Kingdom) in TBS-T/1% nonfat milk for 1 h. After incubation with antibodies, membranes were washed 1 h with TBS-T. After further washing with TBS–T, immunoreactive bands were visualized using AmershamTM ECL Plus Western Blotting Detection System (GE Healthcare Buckinghamshire, UK) and Hyperfilm-ECL film (Amersham Biosciences, Europe, GMBH) in accordance with the manufacturer’s instructions. To normalize the bands, filters were stripped and re-probed with actin antibody (Santa Cruz Biotechnology) or rabbit p44/42 antibody (Cell Signaling). Mouse anti-α-tubulin antibody (Sigma–Aldrich) was used to show the purity of the nuclear fraction. Band densities were quantified densitometrically by NIH Image. Since histone and MEK antibodies, commonly used for nuclear fraction normalization, are regulated by FGF2, they were not used for nuclear fraction normalization in this study. Additional Western blots were performed with antibodies to tubulin (cytoplasmic only) to demonstrate that the nuclear extracts did not contain cytoplasmic proteins. In Sup Fig.1, cytosolic extracts were run and additional Western blots were performed with antibodies to tubulin (cytoplasmic only) and histone (nuclear only) to demonstrate that the cytosolic fractions did not contain nuclear proteins.
Flow cytometry
Fluorescence activated cell sorting (FACS) was performed to evaluate the cell surface FGFR1 on BMSCs. BMSCs from both genotypes cultured for 1 week were collected with 0.2%EDTA in PBS. Fc receptors were blocked with Fcγ RIIb/CD16-2 (2.4G2) antibody (Santa Cruz Biotechnology) for 10 minutes. Then BMSCs were stained with rabbit polyclonal FGFR1 antibody (Santa Cruz Biotechnology) for 15 minutes. After washing, the cells were incubated with goat anti-rabbit Texas Res-X (Invitrogen). Flow cytometric analysis was performed after two washes using PBS. Live events (50,000) were acquired for analysis.
Statistical analysis
Data are presented as means±SE. T-test, one-way or two-way ANOVA followed by least significant difference (LSD) for Post Hoc Multiple Comparisons were used. SPSS software was used for statistical analysis, and the results were considered significantly different at p<0.05.
Results
Analysis of BMSC cultures from Vector and HMW mice
Since we reported that overexpression of HMW increases FGF23/FGFR/KLOTHO signaling in kidneys of HMW mice to down-regulate the sodium phosphate transporter 2a (NPT2a), causing phosphate wasting, osteomalacia, and decreased BMD in vivo (7), we wanted to determine whether HMW regulation of bone formation was totally phosphate dependent. Therefore, BMSCs were cultured in phosphate replete media under osteogenic conditions for 21days. The time course of formation of crystal violet stained colonies, ALP-positive colonies and mineralized nodules is shown in Fig. 1A, and quantification of crystal violet stained colony area, ALP positive colony area, mineralized colony area is shown in Fig. 1B~1F. At one week of culture, ALP positive colony area was significantly increased by 79% in HMW BMSC cultures compared with Vector (Fig. 1C). Dishes that were initially stained for ALP after 1 week of culture were counterstained with crystal violet to stain all colonies. The stained dishes and graphic representations of total colonies are shown in Fig. 1A&1B. There was a 71% significant increase in total colony area of crystal violet staining in HMW cultures compared with Vector. In order to determine if the increased ALP staining was due to increased colony number, the ALP positive area was normalized to crystal violet area (Fig. 1D). After normalization, no difference was observed between Vector and HMW cultures. However, when the cells were cultured for 2 and 3 weeks, mineralized bone nodule formation was decreased by 39% (Fig. 1E) and 33% (Fig. 1F), respectively, in HMW BMSC cultures compared with Vector.
Fig. 1.
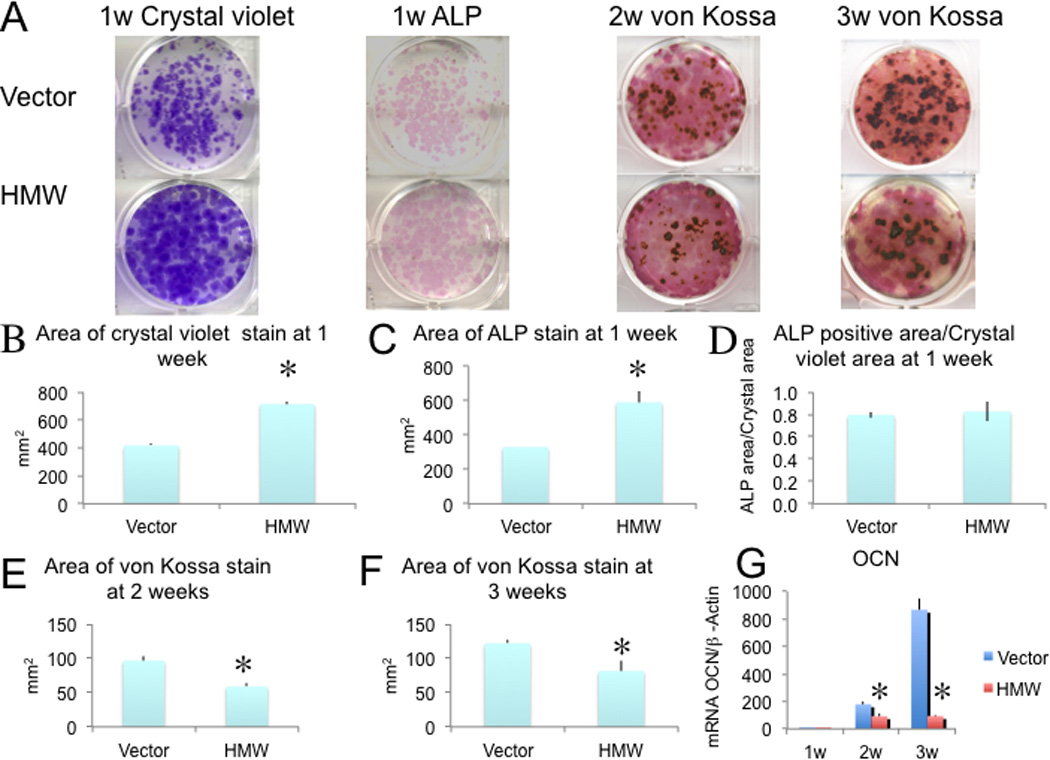
Analysis of bone nodule formation and OCN gene expression in BMSCs from Vector and HMW mice. BMSCs from both genotypes were cultured under osteogenic condition for 3 weeks. (A) Increased crystal violet stained colonies and ALP positive colonies at 1 week of culture of HMW compared with Vector. Decreased mineralized nodules (von Kossa) were observed in HMW BMSC cultures at 2 and 3 weeks. (B-F). Colony area was measured by NIH Image. (G) qRT-PCR analysis of OCN mRNA. Beta-actin was used as an internal control. The mRNA expression levels were expressed as relative (fold changes) to the expression of the Vector 1 week group.*Significantly different from Vector. p < 0.05. Values are Mean ± S.E for three independent experiments. T-test was used in B-F; Two-way ANOVA followed by LSD was used in G.
We also assessed the mRNA expression for osteocalcin (OCN), a late osteoblast marker gene. As shown in Fig. 1G, there was a progressive increase in OCN mRNA expression over 3 weeks of cultures in both Vector and HMW cultures. However, OCN mRNA expression was significantly decreased by 48% and 89%,respectively, at 2 and 3 weeks in HMW BMSC cultures compared with Vector.
Taken together, these data indicate that overexpression of HMW isoforms in BMSCs increased cell proliferation but decreased their terminal differentiation and mineralization. The data also suggest that HMW isoforms decrease bone nodule mineralization in vitro independent of systemic effects on phosphate homeostasis.
In vitro analysis of Fgf2, Fgf23 and Fgfr1c gene expression in BMSC cultures from Vector and HMW mice
Since we observed increased Fgf23 mRNA and, FGF23 protein in bone and increased FGF23 in serum from HMW mice in vivo(7), we determined whether there was modulation of Fgf23 gene expression in HMW versus Vector BMSCs that were cultured for up to 3 weeks. As shown in Fig. 2A, Fgf2 mRNA expression was increased over time in HMW cultures and the expression level was significantly higher in HMW cultures compared with Vector at each time point, as expected. Fgf23 mRNA expression (Fig. 2B) was also significantly higher in HMW cultures compared with Vector at all time points.
Fig. 2.
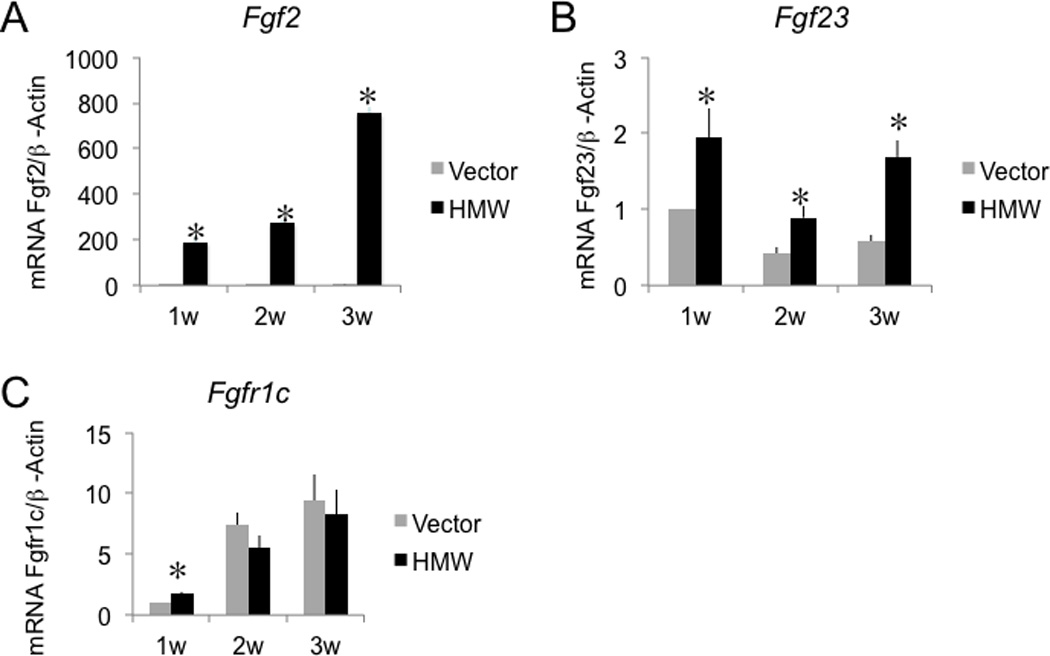
Fgf2, Fgf23, Fgfr1c mRNA expression in BMSC cultures from Vector and HMW mice. (A), Fgf2 mRNA, (B), Fgf23 mRNA, and (C), Fgfr1c mRNA expression in BMSC cultures. RNA was extracted from BMSCs cultured in osteogenic media for 1–3 weeks, and Fgf2, Fgf23, Fgfr1c mRNA was measured using qRT-PCR. Values are Mean ± S.E for three (A and C) or four (B) independent experiments. *: p < 0.05 compared with Vector. Beta-actin was used as an internal control. The mRNA expression level was expressed as relative value (fold changes) to the expression in Vector cultures at 1 week. Two-way ANOVA followed by LSD was used.
It was previously reported that the FGFR1 signaling response to an excess of FGF23 in osteoblasts negatively regulated mineralized nodule formation in rat calvarial cells (16). It was also reported that co-transfection of the HMW isoforms increased the nuclear content of FGFR1 in human astrocytes (17,18). Since FGF2 is known to have a higher affinity for FGFR1c than for the other FGFR isoforms (19), we determined whether Fgfr1c gene expression was modulated in HMW versus Vector BMSCs that were cultured for up to 3 weeks (Fig. 2C). There was a progressive increase in Fgfr1c mRNA expression over 3 weeks of culture that was similar in both Vector and HMW. However, at 1 week of culture Fgfr1c mRNA was significantly increased by 71% in BMSCs from HMW mice relative to Vector mice. These data suggest that overexpression of HMW isoforms up-regulate FGF23 and FGFR1 mRNA expression in vitro.
FGF23 expression in cells and conditioned media of Vector and HMW BMSC cultures
FGF23 is secreted from bone, circulates in blood and then functions at target organs such as kidney (20)(21).We previously reported that FGF23 was significantly increased in sera from HMW mice (7). We therefore examined whether FGF23 protein was over-expressed in cultured BMSCs from HMW mice and secreted in conditioned media. BMSCs from both genotypes were cultured for up to 3 weeks under osteogenic conditions. Conditioned media were collected at the end of culture and nuclear protein was extracted from the cells. As shown in Fig. 3A and 3B, overexpression of HMW isoforms increased FGF23 and FGF2 protein in nuclear fractions of BMSCs from HMW mice, as measured by Western blot, and increased FGF23 levels in conditioned media, as measured by ELISA. In contrast, the FGF23 level in culture media from Vector was almost undetectable.
Fig. 3.
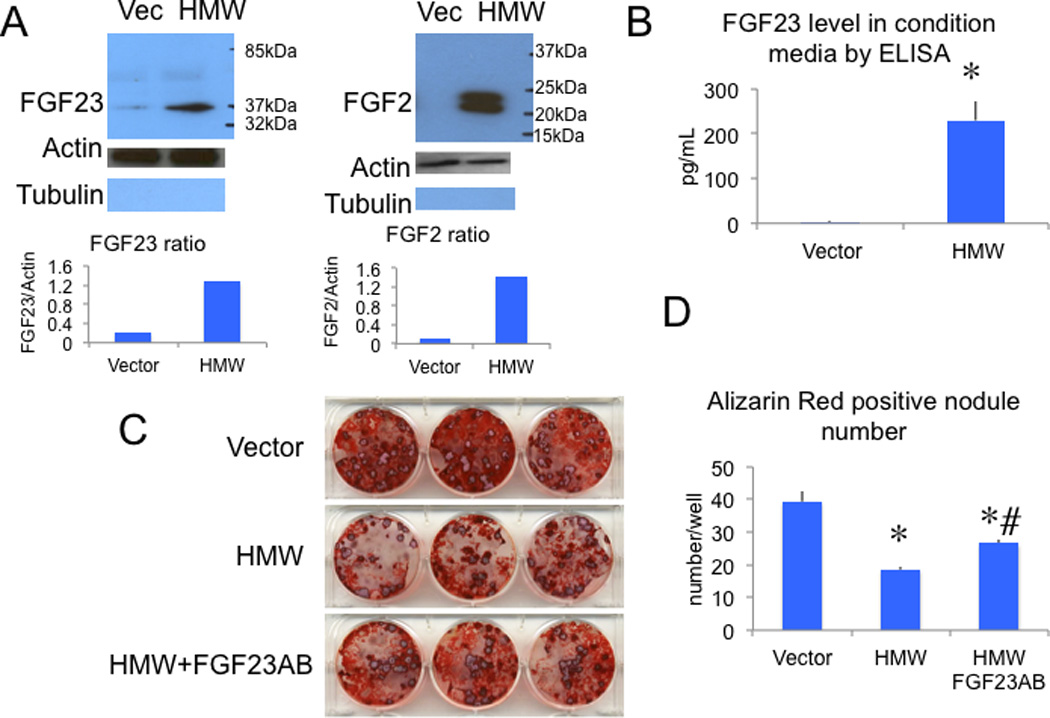
FGF23 protein expression in cultured BMSCs and conditioned media from Vector and HMW mice and the effect of neutralizing FGF23 antibody on mineralized bone nodule formation. (A), Increased nuclear FGF23 protein expression was accompanied by overexpression of HMW protein detected by Western blot at 3 weeks of culture. Nuclear protein was extracted using a Nuclear Extraction Kit from Panomics (Panomics, Inc., Fremont, CA) according to the manufacturer’s instructions. Graphs show the ratio of FGF23/Actin and FGF2/Actin. Blots were re-probed with α-tubulin to show the purity of nuclear fraction. (B), FGF23 was increased in culture media in BMSCs from HMW but not Vector mice as measured by ELISA. T-test was used. (C) Alizarin red S staining shows suppression of bone nodule formation in BMSC cultures expressing HMW compared with Vector when the cells were treated with a control antibody. Treatment with an FGF23 neutralizing antibody increased alizarin red S staining in HMW cultures. (D) Quantitative analysis of alizarin red S stain after solubilization of alizarin red S. One-way ANOVA followed by LSD was used. *: Compared with Vector P < 0.05; # compared with HMW P < 0.05. Values are Mean ± S.E for three independent experiments.
Ability of FGF23 neutralization to rescue the decreased mineralization in BMSCs from HMW mice
Recently published data showed that overexpression of FGF23 suppressed osteoblast differentiation and matrix mineralization in vitro (16).Since we observed increased FGF23 in conditioned media from BMSC scultured from HMW mice, we assessed whether the decreased mineralization of HMW BMSC cultures was due to the direct action of FGF23. We repeated the BMSC culture studies in the presence and absence of a neutralizing FGF23 antibody. As shown in Fig. 3C and 3D, mineralized bone nodule formation was decreased in HMW cultures compared with Vector (19±3 vs. 39±1/well). However in the presence of the FGF23 neutralizing antibody, decreased bone nodule formation in HMW BMSC cultures was partially rescued (27±1/well).
Time course expression of FGFR1 and phospho-ERK in nuclear fractions of BMSCs from Vector and HMW mice
FGF23 induced phosphorylation of ERK is detected in a limited number of tissues including kidney and parathyroid and pituitary gland but not in bone (22)(23)(20)(24)(25)(26). Klotho, an anti-aging protein not detected in bone (27), is required to form aFGF23-FGFR-Klotho complex for induction of phospho-ERK signaling in kidney (24,25). More recent studies showed (16) that FGF23 has a direct role in bone formation. To examine the effect of overexpression of HMW isoforms on FGF23/FGFR/ERK signaling, we assessed FGFR1 expression in the nuclear fractions of BMSCs cultured from both genotypes (Fig. 4 A-B) and phospho-ERK expression in both cytosolic (Fig. 4 C-D) and nuclear fractions (Sup Fig. 6 A-B). FACS analysis was also performed on BMSCs cultured for 1 week to determine if plasma membrane bound FGFR1 was elevated in the HMW cultures (Fig. 4E Sup Fig. 5).We observed that nuclear FGFR1 protein was increased in HMW BMSC cultures at 1 week compared with Vector. Increased phospho-ERK protein was observed in cytosolic fractions from HMW BMSCs compared with Vector BMSCs at 2 and 3 weeks of culture. However, increased phospho-ERK protein nuclear accumulation was observed in HMW BMSCs compared with Vector BMSCs as early as 1 week of culture. Plasma membrane bound FGFR1 was elevated in HMW cultures at 1 week. These data suggest the possibility that secreted FGF23 is acting in a paracrine manner to increase Fgfr1 levels and that overexpression of HMW isoforms activate FGF23/FGFR/ERK signaling pathway.
Fig. 4.
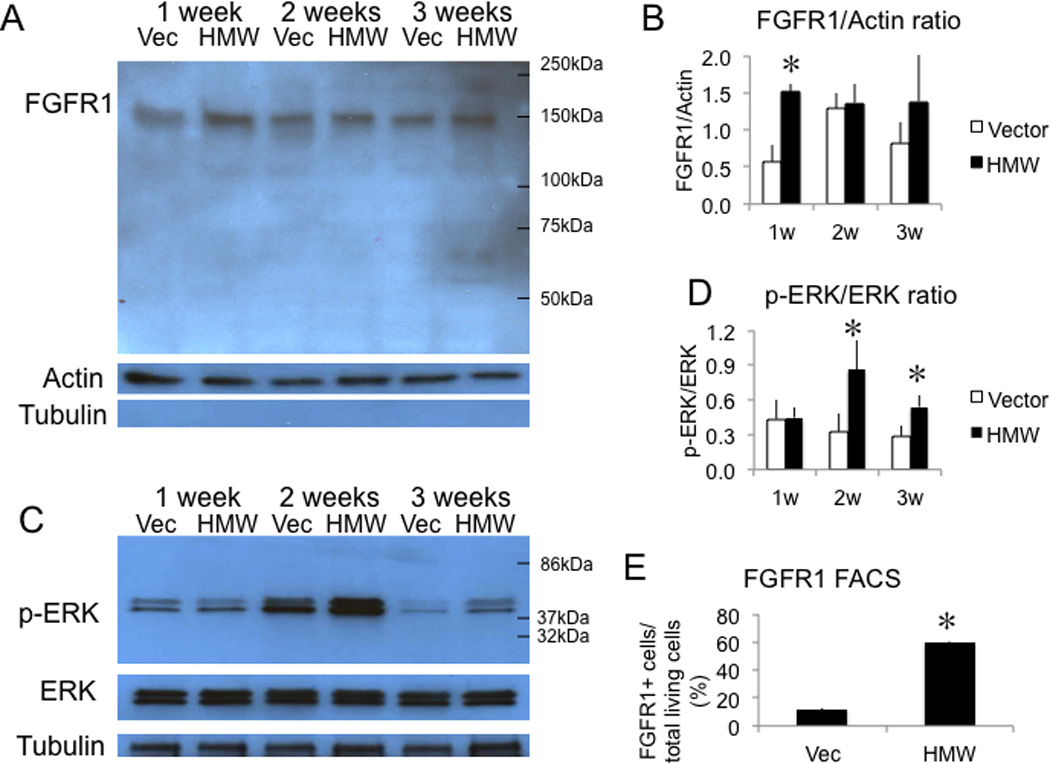
FGFR1 and phospho-ERK protein expression in nuclear fractions of BMSCs and cell surface FGFR1 expression on BMSCs from Vector and HMW mice cultured in osteogenic media. (A) Western blots showed increased FGFR1 protein expression in HMW BMSC cultures at 1 week compared with Vector. α-tubulin was used to show no cytosolic contamination. (B) Statistical analysis of Western blots of FGFR1 from three independent experiments showing the ratio of FGFR1/Actin. Two-way ANOVA followed by LSD was used.(C) Increased phospho-ERK protein expression was observed in cytosolic fractions from HMW BMSC cultures compared with Vector at 2 and 3 weeks.(D) Statistical analysis of Western blots of pERK from three independent experiments showing the ratio of p-ERK/ERK. Values are Mean ± S.E for three independent experiments. Two-way ANOVA followed by LSD was used. (E) FACS analysis showed increased cell surface FGFR1 expression in BMSCs from HMW compared with Vector at 1 week of culture. T-test was used. *: Compared with Vector, P < 0.05. Values are Mean ± S.E for three determinations/group.
Effect of SU5402 and PD98059 to rescue decreased mineralization in BMSCs from HMW mice
Our data show that overexpression of HMW isoforms induced an excess secretion of FGF23. This excess secretion of FGF23 acted directly on BMSCs to increase FGFR1 and phospho-ERK protein expression. We next examined whether blockade of FGFR signaling could rescue the decreased bone nodule formation by overexpression of HMW isoforms. We used SU5402, an inhibitor of FGFR1 kinase activity. As shown in Sup Fig.2, there were more GFP positive colonies in HMW cultures compared with Vector cultures. However, the xylenol orange scans showed decreased mineralized nodules. These results suggest that, consistent with the findings by the ALP and von Kossa staining (Fig. 1), overexpression of HMW isoforms promotes osteoblast proliferation but inhibits their terminal differentiation and mineralization. SU5402 slightly increased the number of mineralized bone nodules in Vector BMSC cultures but significantly increased the number in HMW BMSC cultures. These findings were confirmed by alizarin red staining and quantitative analysis of solubilized alizarin red staining (Fig. 5A & B). Overexpression of HMW isoforms resulted in decreased alizarin red nodules and concentration compared with Vector BMSC cultures (1.06±0.03 vs. 1.46±0.03mM). Treatment with SU5402 (1.45±0.02 mM) increased mineralization in BMSC cultures from HMW mice.
Fig. 5.
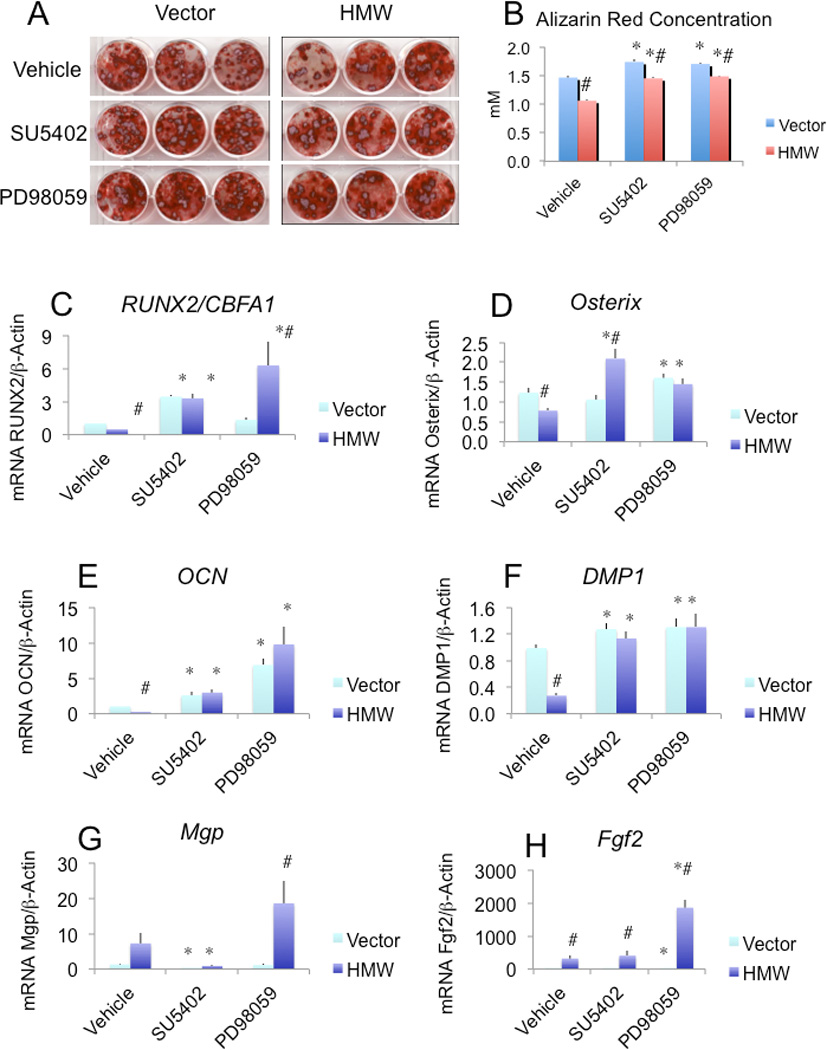
Effect of SU5402 and PD98059 on impaired mineralization induced by HMW overexpression. On day 14, cultures were treated with SU5402 or PD98059 and then refreshed with the effectors at every subsequent media change. (A) Alizarin red S staining of BMSC cultures on day 21. (B) Quantitative analysis of alizarin red S staining. (C-H) Runx2/Cbfa1, osterix, OCN, Dmp1, Mgp, and Fgf2 mRNA expression. mRNA was extracted from parallel dishes and qRT-PCR analysis of interested genes was performed. *:compared with vehicle-treated BMSCs from each genotype p<0.05. #: compared with corresponding Vector group p<0.05.Values are Mean ± S.E for three independent experiments. Two-way ANOVA followed by LSD was used in B-H.
A major intracellular signaling pathway of FGF23 in kidney cells is via activation of the MAPK pathway (23). To examine the functional relevance of the activation of MAPKs in BMSCs, we next examined the effects of PD98059, a MAPK inhibitor, on the reduced bone nodule formation in HMW BMSC cultures. As shown in Fig. 5A and B and Sup Fig. 2, PD98059 partially rescued the decreased bone nodule formation in HMW BMSC cultures. Alizarin red staining and quantization confirmed that PD98059 blocked the inhibition of bone nodule formation by FGF23. This indicates the involvement of the MAPK pathway in the inhibition of the nodule formation by FGF23 in cultured BMSCs. Western blots confirmed that PD98059 blocked ERK phosphorylation (data not show).
We also examined the expression of osteoblast marker genes in the presence and absence of SU5402 and PD98059. As shown in Fig. 5C, at 3 weeks of culture, Runx2 mRNA was decreased by 52% in BMSCs from HMW mice compared with BMSCs from Vector mice. After treatment with SU5402 or PD98059, Runx2 mRNA expression increased by 7-fold and 13-fold, respectively, in HMW cultures. Since the transcription factor osterix can modulate osteoblast differentiation (28) and is regulated by Runx2 (29), we also measured osterix mRNA levels. As shown in Fig. 5D, osterix mRNA was significantly decreased by 36% in HMW cultures compared with Vector cultures. This decrease in osterix mRNA expression was blocked by SU5402 and PD98059 treatment. As shown in Fig. 5E, the mRNA for OCN, a target gene for Runx2 (30), was significantly decreased by 77% in BMSCs from HMW mice and was rescued by SU5402 or PD98059 treatment. The mRNA for dentin matrix protein 1 (Dmp1), an osteocyte marker, was also significantly decreased by72% in cultures from HMW mice compared with Vector mice (Fig. 5F). Decreased Dmp1 expression was rescued by SU5402 or PD98059 treatment. Since the mineralization defect in HMW overexpressing BMSC cultures was not completely rescued by blocking FGFR/MAPK signaling, we speculated that HMW might also modulate key genes involved in bone matrix mineralization. We found that the expression of matrix Gla-protein (Mgp), an inhibitor of mineralization (31), was significantly increased by 451% in HMW BMSC cultures and blocked bySU5402, but was further increased by PD98059 (Fig. 5G).Fig. 5H showed that the colonies responding to the Fgfr and Erk inhibitors overexpressed FGF2.
These data indicate that HMW isoforms suppress mineralization in cultured BMSCs at least in part in a FGF23/FGFR/MAPK dependent manner.
Discussion
Targeted overexpression of HMW in pre-osteoblasts and osteoblasts had a positive effect on osteoprogenitor cell proliferation but significantly suppressed not only terminal osteoblast differentiation but also matrix mineralization in mouse BMSC cultures. BMSCs overexpressing HMW isoforms have increased FGF23 mRNA and protein in osteoblasts and osteocytes(7). Overexpression of HMW isoforms also increased Fgfr1c mRNA and nuclear accumulation of FGFR1 protein and increased phospho-ERK in osteoblasts. An FGF23 neutralizing antibody, SU5402, an inhibitor of the tyrosine kinase activity of FGFR1, and PD98059, a MAPK inhibitor, partially rescued HMW BMSC cultures from the inhibition of matrix mineralization induced by an excess of FGF23. Mgp, a key gene involved in mineralization, was up-regulated in BMSCs from HMW mice, and was further increased after PD98059 treatment. These data suggest that overexpression of HMW isoforms negatively regulates bone formation, independent of systemic effects on phosphate homeostasis, through the FGF23/FGFR/ERK signaling pathway in cultured BMSCs.
ALP positive area was normalized to crystal violet area. After normalization, no difference was observed between Vector and HMW. This indicates that increased ALP staining in HMW cultures is due to an increase in colony number. MTT assay and cell counting showed that HMW promotes metabolic activity and cell proliferation at an early stage. However HMW isoforms significantly reduced matrix mineralization at 2 and 3 weeks of culture. Therefore, the HMW transgene dissociates proliferation and mineralization. Matrix mineralization in our BMSC culture model can be analyzed separately from proliferation. Therefore, by observing decreased bone nodule formation at 3 weeks after ALP nodules had fully formed but before their mineralization, we found a direct role for HMW isoforms in matrix mineralization independent of their effects on proliferation. Although, treatment with recombinant human FGF2, which is a LMW isoform of FGF2, inhibited matrix mineralization in fetal rat calvarial cell cultures (16), we observed the opposite effect of overexpression of LMW isoform on mineralization in our BMSC culture model (6). These data suggest that the defective mineralization by overexpression of HMW is isoform-specific.
We previously reported, in osteoblastic like ROS17/2.8 cell (32), that in the presence of 1 or 10% serum, DNA synthesis was increased in cells expressing HMW isoforms compared with Vector. A neutralizing FGF2 antibody did not block the mitogenic effect of cells harboring the HMW isoform. In the current study, there was increased proliferation in the HMW BMSCs cultured for 1 week. One could argue that decreased bone nodule formation in HMW BMSC cultures at 2 and 3 weeks was due to the fact that the cells were still in the proliferation stage so they could not differentiate. To exclude this possibility, we performed longer cultures for up to 4 weeks. The decreased mineralization of nodules in HMW BMSC cultures never catches up to the mineralization of nodules in Vector BMSC cultures. This indicates that decreased nodule formation in BMSC cultures from HMW mice is due to inhibition of terminal differentiation and mineralization.
We also believe that HMW isoforms regulate mineralization through FGF23. This regulation is independent of the pathway by which HMW isoforms up-regulate proliferation. First, there is no evidence that FGF23 alone stimulates osteoprogenitor cell proliferation (16)(26). Second, even though FGF2 isoforms including HMW isoforms increase BMSC proliferation (6), only HMW isoforms decrease mineralized bone nodule formation. Thus we cannot attribute the effects of HMW overexpression on osteoblast marker expression, decreased nodule formation, and mineralization in HMW BMSCs to an effect on proliferation. Of course, a systemic and comprehensive analysis of FGFs and FGF2 isoforms in osteoblast remains of interest.
Establishing the direct effects of HMW isoforms on mineralization has not been reported. HMW mice in which type I collagen regulatory sequences drive HMW isoforms in osteoblasts have similar phenotypic abnormalities as the Hyp mouse, which is a mouse model of human XHL (7). These phenotypes include dwarfism, decreased BMD, osteomalacia, and decreased serum phosphate. Serum FGF23 was increased in HMW mice. Overexpression of HMW increased FGF23/FGFR/KLOTHO signaling in kidney to down-regulate the sodium/phosphate transporter (NPT2a), causing phosphate wasting (7). Our current in vitro data clearly show a direct effect of HMW isoforms overexpression on bone formation without confounding changes in systemic parameters.
Currently the regulation of FGF23 by HMW isoforms is still undefined. HMW isoforms mediate their effects on bone and kidney by modulating the expression of FGF23, which is secreted to act locally or systemically in a paracrine or endocrine manner (7). HMW isoforms are ordinarily not released from the cells, but function in an intracrine manner (3). Intriguingly, in the Hyp mouse, a model of XLH, we found that Fgf2 mRNA and protein were increased in bones/osteoblasts, and that FGF23 and HMW protein were co-localized in osteocytes (7). Consistent with this, we found increased FGF23 and HMW protein expression in the nuclear lysates of cultured HMW BMSCs. This novel finding provides support for the idea that HMW isoforms do play a role in FGF23 regulation. Indeed, our published studies demonstrate that transfection of Fgf2-null osteoblasts with expression vector for the HMW isoforms resulted in increased FGF23 expression, whereas transfection with the LMW construct did not (7). Our results are in contrast to another study showing that commercially available FGF2, which is similar to the LMW isoform, can stimulate Fgf23 promoter activity in ROS17/2.8 cells (33). It should be noted that we also observed normal phosphate homeostasis in transgenic mice over-expressing LMW isoform targeted to osteoblasts (6). In support of our published studies that the LMW isoform does not regulate FGF23 (7), a recent report by Kyono et al found that exogenous FGF2 treatment did not increase FGF23 in osteocytic MLO-Y4 cells (34). Furthermore, we did not observe an increase in serum FGF23 in LMW transgenic mice (unpublished observation).
Phenotypic changes observed in HMW mice included hypophosphatemia and increased serum FGF23 as well as increased Fgf23 mRNA and protein in bone. Most importantly, we showed that FGF2 was over-expressed in Hyp mice, a model of human XLH (7). Our published data strongly suggest that HMW isoforms regulate FGF23 at the transcriptional level. We have published that HMW isoforms up-regulate Fgf23 mRNA expression in Fgf2 null osteoblasts. Further, in preliminary unpublished studies of a 3550kb fragment of the FGF23 promoter, we observed that HMW isoforms also stimulate p3550Fgf23-luc activity in ROS17/2.8 cells and Fgf2-null calvarial osteoblasts whereas the LMW isoform did not. VDR, CREB and E4F have putative transcription factor (4) binding sites in the Fgf23 promoter region found to be up-regulated in ROS17/2.8 cells over-expressing HMW isoforms. Since nuclear FGFR1 and FGF2 were reported to activate tyrosine hydroxylase promoter through the CRE (35) in bovine adrenal medullar cells, it is possible that HMWisoforms similarly modulate Fgf23 promoter activity in osteoblasts through this integrative nuclear FGFR1 signaling pathway. This is worthy of further investigation.
It is well known that Klotho, an anti-aging protein, is needed to form a complex with FGF23/FGFR for induction of FGF23-specific pathways (24)(25). Klotho is not expressed in bone. Recently Wang et al reported that overexpression of FGF23 negatively regulates bone formation independent of its systemic effects on Pi homeostasis through FGFR without Klotho (16). Consistent with this finding, we also clearly show that FGF23/FGFR signaling is involved in HMW-dependent bone formation defects in the BMSC cultures. Thus, our findings provide new insight into the role of HMW isoforms in skeletal development. In other words, HMW isoforms act on bone formation directly and independently of their systemic effect on phosphate homeostasis. The mineralization defect observed in HMW BMSC cultures is unique for HMW isoforms since we did not observe similar findings in LMW BMSC cultures (6).
Our published in vivo data showed a decrease in OCN mRNA levels in tibiae of HMW versus Vector mice (7). OCN is a late osteoblast differentiation gene. It is known to influence mineralization through binding to hydroxyapatite and functions in cell signaling and recruitment of osteoblasts (36). Dmp1 is an osteocyte marker. The effect of overexpression of HMW isoforms on osteoblast and osteocyte marker expression reported here are consistent with our in vivo findings that reflect the defective terminal differentiation of ostoblasts. It is interesting that Runx2 and osterix, which are upstream of OCN, were significantly affected in HMWBMSC cultures and both rescued by blocking FGFR signaling and MAPK pathway. Our results showed that overexpression of HMW isoforms increased FGFR mRNA and protein expression. The selective FGFR1 tyrosine kinase inhibitor, SU5402, and the MAPK cascade inhibitor PD98059, partially rescued mineralization defects in HMW isoform cultures, suggesting that biological relevant HMW isoforms signaling in osteoblast may be mediated, at least in part, by FGFR1 and MAPK. In any case, a search of receptors involved in osteoblast should be useful to further address these issues. Our data do not preclude the possibility that other FGFRs may participate in HMW isoforms-specific signaling in BMSCs. BMSCs from HMW mice showed an intrinsic mineralization defect, indicating that the bone defects observed in HMW mice (7) are not due entirely to either the elevated FGF23or hypophosphatemia; thus other phosphatonins may be involved.
In summary, this study shows that an excess of HMW isoforms negatively regulate BMSC mineralization in vitro, which is independent of their systemic effects on phosphate homeostasis. This regulation is at least in part through FGF23/FGFR1/MAKP signaling pathway. Further studies will be necessary to understand the local effects of HMW isoforms on bone formation and the pathology of phosphate wasting diseases.
Supplementary Material
Fig. 6.
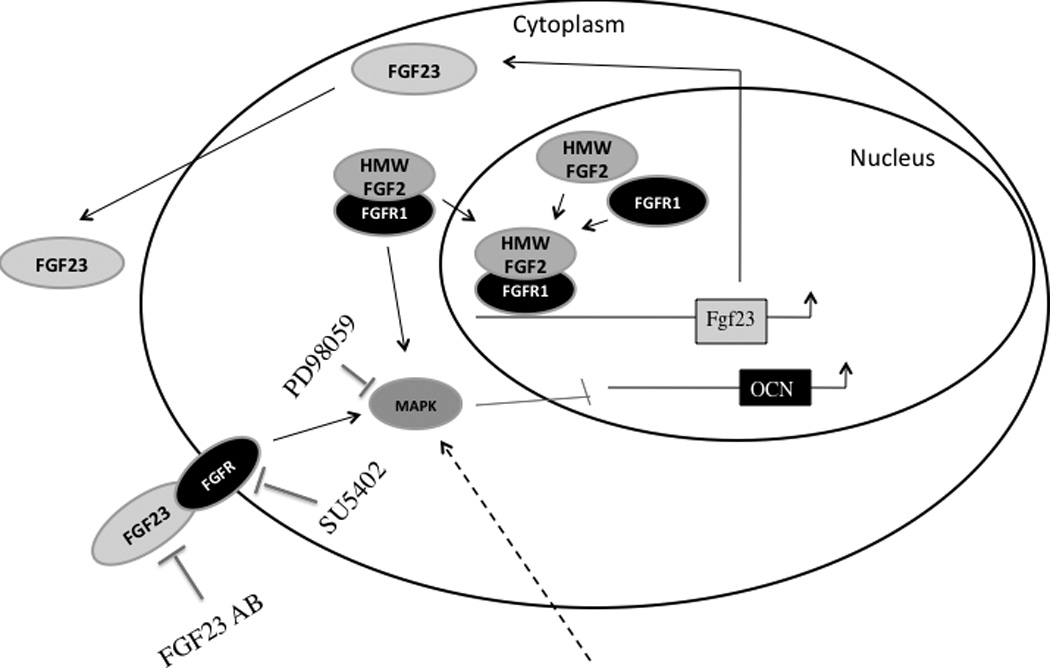
A schematic model depicting how HMW isoforms regulate bone formation. HMW isoforms, which have nuclear targeting sequences, trans locate to the nucleus to up regulate FGF23 expression. FGF23 is secreted into the media and activates the tyrosine kinase domain of FGFR on the cell surface, triggering downstream ERK signaling pathway.
Acknowledgments
We thank Amgen Inc. for supplying the neutralizing FGF23 antibody; and Immutopics, Inc. for performing the FGF23 ELISA measurements. We thank Dr. Joseph A. Lorenzo for his valuable critique of the manuscript. We also wish to thank Professor Joseph Burleson for his help with the statistical analysis. This project was supported in part by NIH grant AG021189 to Marja M. Hurley.
Footnotes
Disclosures
All the authors state that they have no conflicts of interest.
Authors’ roles: LX performed, analyzed, and interpreted the data and wrote the manuscript. AE, under the direction of LX, carried out the Western blot and qPCR analysis. MMH conceived of the study, reviewed the data and approved the final version of the manuscript.
References
- 1.Globus RK, Plouet J, Gospodarowicz D. Cultured bovine bone cells synthesize basic fibroblast growth factor and store it in their extracellular matrix. Endocrinology. 1989;124(3):1539–1547. doi: 10.1210/endo-124-3-1539. [DOI] [PubMed] [Google Scholar]
- 2.Florkiewicz RZ, Sommer A. Human basic fibroblast growth factor gene encodes four polypeptides: three initiate translation from non-AUG codons. Proc Natl Acad Sci U S A. 1989;86(11):3978–3981. doi: 10.1073/pnas.86.11.3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arese M, Chen Y, Florkiewicz RZ, Gualandris A, Shen B, Rifkin DB. Nuclear activities of basic fibroblast growth factor: potentiation of low-serum growth mediated by natural or chimeric nuclear localization signals. Mol Biol Cell. 1999;10(5):1429–1444. doi: 10.1091/mbc.10.5.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coffin JD, Florkiewicz RZ, Neumann J, Mort-Hopkins T, Dorn GW, 2nd, Lightfoot P, German R, Howles PN, Kier A, O’Toole BA, et al. Abnormal bone growth and selective translational regulation in basic fibroblast growth factor (FGF-2) transgenic mice. Mol Biol Cell. 1995;6(12):1861–1873. doi: 10.1091/mbc.6.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sobue T, Naganawa T, Xiao L, Okada Y, Tanaka Y, Ito M, Okimoto N, Nakamura T, Coffin JD, Hurley MM. Over-expression of fibroblast growth factor-2 causes defective bone mineralization and osteopenia in transgenic mice. J Cell Biochem. 2005;95(1):83–94. doi: 10.1002/jcb.20389. [DOI] [PubMed] [Google Scholar]
- 6.Xiao L, Liu P, Li X, Doetschman T, Coffin JD, Drissi H, Hurley MM. Exported 18- kDa isoform of fibroblast growth factor-2 is a critical determinant of bone mass in mice. J Biol Chem. 2009;284(5):3170–3182. doi: 10.1074/jbc.M804900200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiao L, Naganawa T, Lorenzo J, Carpenter TO, Coffin JD, Hurley MM. Nuclear isoforms of fibroblast growth factor 2 are novel inducers of hypophosphatemia via modulation of FGF23 and KLOTHO. J Biol Chem. 2010;285(4):2834–2846. doi: 10.1074/jbc.M109.030577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Larsson T, Marsell R, Schipani E, Ohlsson C, Ljunggren O, Tenenhouse HS, Juppner H, Jonsson KB. Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology. 2004;145(7):3087–3094. doi: 10.1210/en.2003-1768. [DOI] [PubMed] [Google Scholar]
- 9.Eicher EM, Southard JL, Scriver CR, Glorieux FH. Hypophosphatemia: mouse model for human familial hypophosphatemic (vitamin D-resistant) rickets. Proc Natl Acad Sci U S A. 1976;73(12):4667–4671. doi: 10.1073/pnas.73.12.4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu Y, Feng JQ. FGF23 in skeletal modeling and remodeling. Curr Osteoporos Rep. 2011;9(2):103–108. doi: 10.1007/s11914-011-0053-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao L, Sobue T, Esliger A, Kronenberg MS, Coffin JD, Doetschman T, Hurley MM. Disruption of the Fgf2 gene activates the adipogenic and suppresses the osteogenic program in mesenchymal marrow stromal stem cells. Bone. 2010;47(2):360–370. doi: 10.1016/j.bone.2010.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sheehan D HB. Theory and Practice of Histotechnology. 2nd (ed.) Ohil: Battelle Press; 1980. [Google Scholar]
- 13.Keila S, Pitaru S, Grosskopf A, Weinreb M. Bone marrow from mechanically unloaded rat bones expresses reduced osteogenic capacity in vitro. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 1994;9(3):321–327. doi: 10.1002/jbmr.5650090306. [DOI] [PubMed] [Google Scholar]
- 14.Gregory CA, Gunn WG, Peister A, Prockop DJ. An Alizarin red-based assay of mineralization by adherent cells in culture: comparison with cetylpyridinium chloride extraction. Analytical biochemistry. 2004;329(1):77–84. doi: 10.1016/j.ab.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 15.Pfaffl MW. A new mathematical model for relative quantification in real-time RTPCR. Nucleic Acids Res. 2001;29(9):e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang H, Yoshiko Y, Yamamoto R, Minamizaki T, Kozai K, Tanne K, Aubin JE, Maeda N. Overexpression of fibroblast growth factor 23 suppresses osteoblast differentiation and matrix mineralization in vitro. J Bone Miner Res. 2008;23(6):939–948. doi: 10.1359/jbmr.080220. [DOI] [PubMed] [Google Scholar]
- 17.Stachowiak MK, Fang X, Myers JM, Dunham SM, Berezney R, Maher PA, Stachowiak EK. Integrative nuclear FGFR1 signaling (INFS) as a part of a universal "feedforward- and-gate" signaling module that controls cell growth and differentiation. J Cell Biochem. 2003;90(4):662–691. doi: 10.1002/jcb.10606. [DOI] [PubMed] [Google Scholar]
- 18.Dunham-Ems SM, Lee YW, Stachowiak EK, Pudavar H, Claus P, Prasad PN, Stachowiak MK. Fibroblast growth factor receptor-1 (FGFR1) nuclear dynamics reveal a novel mechanism in transcription control. Mol Biol Cell. 2009;20(9):2401–2412. doi: 10.1091/mbc.E08-06-0600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier F, Gao G, Goldfarb M. Receptor specificity of the fibroblast growth factor family. J Biol Chem. 1996;271(25):15292–15297. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- 20.Yu X, White KE. Fibroblast growth factor 23 and its receptors. Ther Apher Dial. 2005;9(4):308–312. doi: 10.1111/j.1744-9987.2005.00287.x. [DOI] [PubMed] [Google Scholar]
- 21.Fukumoto S. [Fibroblast growth factor (FGF) 23 works as a phosphate-regulating hormone and is involved in the pathogenesis of several disorders of phosphate metabolism] Rinsho Byori. 2007;55(6):555–559. [PubMed] [Google Scholar]
- 22.Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M, Mohammadi M, Sirkis R, Naveh-Many T, Silver J. The parathyroid is a target organ for FGF23 in rats. J Clin Invest. 2007;117(12):4003–4008. doi: 10.1172/JCI32409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamashita T, Konishi M, Miyake A, Inui K, Itoh N. Fibroblast growth factor (FGF) 23 inhibits renal phosphate reabsorption by activation of the mitogen-activated protein kinase pathway. J Biol Chem. 2002;277(31):28265–28270. doi: 10.1074/jbc.M202527200. [DOI] [PubMed] [Google Scholar]
- 24.Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, Kuro-o M. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. 2006;281(10):6120–6123. doi: 10.1074/jbc.C500457200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444(7120):770–774. doi: 10.1038/nature05315. [DOI] [PubMed] [Google Scholar]
- 26.Shalhoub V, Ward SC, Sun B, Stevens J, Renshaw L, Hawkins N, Richards WG. Fibroblast growth factor 23 (FGF23) and alpha-klotho stimulate osteoblastic MC3T3.E1 cell proliferation and inhibit mineralization. Calcif Tissue Int. 2011;89(2):140–150. doi: 10.1007/s00223-011-9501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390(6655):45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 28.Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108(1):17–29. doi: 10.1016/s0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- 29.Nishio Y, Dong Y, Paris M, O’Keefe RJ, Schwarz EM, Drissi H. Runx2-mediated regulation of the zinc finger Osterix/Sp7 gene. Gene. 2006;372:62–70. doi: 10.1016/j.gene.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 30.Pregizer S, Baniwal SK, Yan X, Borok Z, Frenkel B. Progressive recruitment of Runx2 to genomic targets despite decreasing expression during osteoblast differentiation. J Cell Biochem. 2008;105(4):965–970. doi: 10.1002/jcb.21900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gopalakrishnan R, Suttamanatwong S, Carlson AE, Franceschi RT. Role of matrix Gla protein in parathyroid hormone inhibition of osteoblast mineralization. Cells Tissues Organs. 2005;181(3–4):166–175. doi: 10.1159/000091378. [DOI] [PubMed] [Google Scholar]
- 32.Xiao L, Liu P, Sobue T, Lichtler A, Coffin JD, Hurley MM. Effect of overexpressing fibroblast growth factor 2 protein isoforms in osteoblastic ROS 17/2.8 cells. J Cell Biochem. 2003;89(6):1291–1301. doi: 10.1002/jcb.10589. [DOI] [PubMed] [Google Scholar]
- 33.Liu S, Tang W, Fang J, Ren J, Li H, Xiao Z, Quarles LD. Novel regulators of Fgf23 expression and mineralization in Hyp bone. Mol Endocrinol. 2009;23(9):1505–1518. doi: 10.1210/me.2009-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kyono A, Avishai N, Ouyang Z, Landreth GE, Murakami S. FGF and ERK signaling coordinately regulate mineralization-related genes and play essential roles in osteocyte differentiation. J Bone Miner Metab. 2011 doi: 10.1007/s00774-011-0288-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng H, Myers J, Fang X, Stachowiak EK, Maher PA, Martins GG, Popescu G, Berezney R, Stachowiak MK. Integrative nuclear FGFR1 signaling (INFS) pathway mediates activation of the tyrosine hydroxylase gene by angiotensin II, depolarization and protein kinase C. J Neurochem. 2002;81(3):506–524. doi: 10.1046/j.1471-4159.2002.00833.x. [DOI] [PubMed] [Google Scholar]
- 36.Hoang QQ, Sicheri F, Howard AJ, Yang DS. Bone recognition mechanism of porcine osteocalcin from crystal structure. Nature. 2003;425(6961):977–980. doi: 10.1038/nature02079. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.