

Abstract
Pompe disease (PD) is a metabolic myopathy due to the deficiency of the lysosomal enzyme α-glucosidase (GAA). The only approved treatment for this disorder, enzyme replacement with recombinant human GAA (rhGAA), has shown limited therapeutic efficacy in some PD patients. Pharmacological chaperone therapy (PCT), either alone or in combination with enzyme replacement, has been proposed as an alternative therapeutic strategy. However, the chaperones identified so far also are active site-directed molecules and potential inhibitors of target enzymes. We demonstrated that N-acetylcysteine (NAC) is a novel allosteric chaperone for GAA. NAC improved the stability of rhGAA as a function of pH and temperature without disrupting its catalytic activity. A computational analysis of NAC–GAA interactions confirmed that NAC does not interact with GAA catalytic domain. NAC enhanced the residual activity of mutated GAA in cultured PD fibroblasts and in COS7 cells overexpressing mutated GAA. NAC also enhanced rhGAA efficacy in PD fibroblasts. In cells incubated with NAC and rhGAA, GAA activities were 3.7–8.7-fold higher than those obtained in cells treated with rhGAA alone. In a PD mouse model the combination of NAC and rhGAA resulted in better correction of enzyme activity in liver, heart, diaphragm and gastrocnemia, compared to rhGAA alone.
Introduction
Pompe disease (PD, OMIM 232300) is an inborn metabolic disorder caused by the functional deficiency of α-glucosidase (GAA, acid maltase, E.C.3.2.1.20), an acid glycoside hydrolase involved in the lysosomal breakdown of glycogen. GAA deficiency results in glycogen accumulation in lysosomes and in secondary cellular damage, through mechanisms not fully understood.1,2 Although GAA deficiency in PD is generalized, muscles are particularly vulnerable to glycogen storage. The disease manifestations are thus predominantly related to the involvement of cardiac and skeletal muscles. The phenotypic spectrum of the disease is wide and varies from a devastating classical infantile-onset form, to attenuated late-onset phenotypes. The manifestations related to progressive muscle hypotonia, which cause severe motor impairment and eventually respiratory failure, impact severely on the health of PD patients.1,3
Like for several other lysosomal storage diseases, an enzyme replacement therapy (ERT) with recombinant human α-glucosidase (rhGAA) has become available for PD in the early 2000s. ERT was shown to improve patients' survival and function and to stabilize the disease course.4,5,6,7,8 However, despite treatment, some patients experience limited clinical benefit or show signs of disease progression and it is clear that reaching therapeutic concentrations of the recombinant enzyme in skeletal muscle is particularly challenging.9
Several factors concur in limiting therapeutic success of ERT, including the age at start of treatment,10,11 the immunological and cross-reactive material status of patients,12 the preferential uptake of rhGAA by liver and the insufficient targeting of the enzyme to muscles,13 the relative deficiency of the mannose-6-phosphate receptor in muscle cells,14 and the “build up” of the autophagic compartment observed in myocytes.15,16 In addition, studies in other lysosomal storage diseases treatable by ERT, such as Gaucher disease (due to the deficiency of β-glucocerebrosidase) and Fabry disease (due to α-galactosidase A deficiency), point to the role of factors intrinsically related to the recombinant enzymes used for ERT, and suggest that these enzymes may be relatively unstable when exposed to stresses, like non-acidic pH, during their transit to lysosomes.17,18
In the recent years, pharmacological chaperone therapy (PCT) has been proposed as a strategy to increase physical stability of recombinant enzymes and to enhance the therapeutic action of ERT. This approach, based on using small-molecule ligands that increase stability of mutated proteins and prevent their degradation, was first designed for the treatment of diseases due to protein misfolding.19 Recent studies, however, have shown that chaperones are not only able to rescue misfolded defective proteins, but may also potentiate the effects of the wild-type recombinant enzymes used for ERT.20 We and others have provided preclinical evidence supporting this concept in two relatively prevalent lysosomal disorders, PD,21 and Fabry disease.18,22 In both disorders, when recombinant enzymes were administered to mutant fibroblasts in combination with the chaperone molecules N-butyl-deoxynojirimycin (NB-DNJ) and 1-deoxy-galactonojiirimycin, respectively, the lysosomal trafficking, the maturation and the intracellular activity of the enzymes improved. A similar effect was also obtained in cultured macrophages treated with the recombinant β-glucocerebrosidase (the enzyme used for ERT in Gaucher disease), in the presence of the chaperone isofagomine.23
Although small-molecule chaperones have several advantages, compared to ERT, in terms of biodistribution, oral availability, reduced impact on patients' quality of life, a major reason of concern for the clinical use of these drugs is that the chaperones so far identified for the treatment of lysosomal storage diseases are active site-directed molecules and are reversible competitive inhibitors of the target enzymes.24 Therefore, the identification of second-generation chaperones that protect the enzymes from degradation without interfering with its activity may be advantageous. Extensive search for new chaperones is currently being done by high-throughput screenings with chemical libraries.25,26
Here we report on the identification of N-acetylcysteine (NAC), a known pharmaceutical drug, as a novel allosteric chaperone for GAA. The strategy used for the identification and characterization of NAC's effects was based on the combination of biochemical studies, both in cell-free and in cellular systems, and a computational analysis of NAC–GAA interactions. We found that this drug stabilizes wild-type GAA at nonacidic pH, enhances the residual activity of mutated GAA and improves the efficacy of rhGAA used for ERT in this disease. This novel chaperone does not interact with the catalytic domain of GAA, and consequently is not a competitive inhibitor of the enzyme.
Results
NAC improves rhGAA stability in vitro
Resistance of wild-type enzymes to physical stresses, such as modifications of temperature and pH, is commonly taken as an indicator to monitor the efficacy of pharmacological chaperones.24 rhGAA 6.8 µmol/l was stable at pH 5.0 for up to 24 hours while rapidly lost its activity at nonphysiological pH (3.0 and 7.0), representative of nonlysosomal cellular compartments (Figure 1a). Coincubation with 10 mmol/l NAC (compound 1 in Supplementary Figure S1) rescued rhGAA activity at pH 7.0 in a dose-dependent manner (Figure 1b), and persisted even after 48 hours of incubation (Figure 1c). The same concentration of NAC also thermally stabilized 0.95 µmol/l rhGAA increasing by 10.5 ± 0.5 °C its melting temperature (Tm) (Figure 1d).
Figure 1.

Characterization of recombinant human α-glucosidase (rhGAA) in vitro and analysis of pharmacological chaperones stabilization. (a) rhGAA was stable at pH 5.0 for up to 24 hours while rapidly lost its activity at pH 3.0 and 7.0. (b) Coincubation with 10 mmol/l N-acetylcysteine (NAC) rescued rhGAA activity at pH 7.0 in a dose-dependent manner, and (c) persisted even after 48 hours of incubation. (d) NAC thermally stabilized rhGAA increasing by 10.5 ± 0.5 °C its melting temperature.
The related amino acids N-acetylserine and N-acetylglycine (NAS and NAG, compounds 2 and 3 in Supplementary Figure S1) also behaved as NAC by inducing remarkable stabilization of rhGAA at pH 7.0 (Supplementary Figure S2a–b), while the nonacetylated homologs cysteine, serine, and glycine and 2-mercaptoethanol (4–7 in Supplementary Figure S1) did not prevent enzyme inactivation (Supplementary Figure S2c–f), suggesting that the stabilizing effect was due to the presence of the acetyl rather than to the sulfidryl group.
Modeling the interaction of NAC with the rhGAA protein
To rationalize the interaction of NAC with the enzyme, a homology model of rhGAA was built based on its amino acid sequence and the 3D-structure of the highly homologous human enzyme maltase-glucoamylase (pdb code 3L4Z) showing 44% identity over the aligned 868 residues of GAA. Molecular dynamics (MD) simulations in conditions mimicking pH 5.0 and 7.0 showed that the rhGAA structure was more stable at pH 5.0 than at neutral pH (Figure 2a) and the analysis of the protein's pH-dependent geometric strain27 further confirmed that rhGAA was, in general, more rigid at pH 5.0 than at pH 7.0 (Supplementary Figure S3a). Then, we searched for sites (hot spots) of the protein whose local flexibility and structural organization changed upon passing from pH 5.0 to 7.0 (Supplementary Figure S3b). Projecting the results on the 3D structural model, the region defined by residues Arg525, Gly793, Glu794, Ser795, Leu796, Glu797, and Gly802 emerged as a possible target (Figure 2b). This region, which is conserved among characterized GH31 α-glucosidases from humans and other mammals (Supplementary Figure S3c), is located at the boundary between two domains (one of which contains the active site) defining a stable pocket, which is at 35 å distance from the active site (Supplementary Figure S3d). Thus, a putative ligand, stabilizing the interdomain interface, might confer to rhGAA at neutral pH the same properties showed by the apoenzyme at acidic pH.
Figure 2.

Modeling the structural determinants of α-glucosidase (GAA) stability and mapping the N-acetylcysteine (NAC)-binding sites. (a) Comparison of the molecular dynamics (MD) simulations of the GAA model at pH 5.0 (red) and 7.0 (black). (b) Identification of the NAC clustering regions: the surfaces indicate the areas of favorable NAC contact calculated from the simulations of GAA in the presence of 10 copies of NAC. The green surface indicated the surface identified as a possible allosteric-binding site as explained in the results (combined analysis of NAC-binding regions, changes in the dynamics of the residues in response to pH, SiteMap screening to define the suitability of the pocket to host a small molecule). The cyan surfaces indicate alternative NAC contact sites. It is possible to appreciate that they are all superficial and do not define cavities suitable for small-molecule binding. The active site is indicated by yellow van der Waals representations of the atoms. (c) Detail of the GAA model with NAC bound to the hot spot: the sulfur atom is highlighted in yellow and the binding site is in red filled spaces. (d) The residue-based profile of average strain calculated over the simulation time for the three GAA conditions; from top to bottom: isolated protein at pH 5.0, 7.0, and 7.0 plus NAC.
NAC-binding mechanism and binding site
To identify the regions of the protein surface that may be most frequently in contact with NAC, we combined MD simulations with molecular docking experiments. In a first, “coarse-grained” exploration of binding spots on the protein surface, multiple copies of NAC were simulated in the presence of GAA. The resulting density maps report on the areas of GAA where NAC clusters most favorably. NAC was observed multiple times during the simulation to bind with favorable interactions on specific regions of the protein. A control simulation was carried out with nonacetylated Gly in the same conditions and the results showed that this amino acid, which has no chaperoning effect, had a much lower tendency to bind in the vicinity of the surface. The list of residues defining the surface on which NAC makes favorable contacts is reported in Supplementary Table S1. Interestingly, one of the identified areas of favorable contact between NAC and GAA partially overlapped with the hotspot region located at the boundary between the catalytic domains and the flanking one identified through geometric strain analysis (Figure 2b). The list of identified possible binding regions was further filtered by the application of the “Site Map” function of the Maestro suite of programs, which aims to identify possible sites on a protein that fulfill structural and functional requirements optimal for binding a small, drug-like molecule thus being identified as druggable sites. The application of this further filter on rhGAA surface returned the pH responsive hot spot region identified earlier as the only one showing the stereochemical properties needed by a putative receptor site to bind a small-molecule (Figure 2b).
A refined docking analysis using the Grid program from the Maestro suite was next carried (see Materials and Methods section) to investigate the pose and the energy of NAC in the putative allosteric site. Interestingly, the binding energies for NAC at this site were comparable to the binding energies calculated for the docking of maltose or DNJ to the active site of GAA and similar results were obtained also with NAS and NAG (Supplementary Table S2). Moreover, in the model of the best-ranked pose, the -SH group is directed toward the surface of the protein whereas the acetyl group interacts with the allosteric GAA pocket described above (Figure 2c), providing further indication that the SH group was not crucial for the binding of the molecule to the enzyme. A control docking run was carried out with DNJ. DNJ was left free to move on the whole surface of GAA, without imposing restraints. Importantly, the minimum free energy and most populated docking solution shows DNJ binding preferentially in the active site of GAA, with a lower energy than that measured for the pharmacological chaperone in the allosteric pocket, showing the high affinity of this ligand for the active site and supporting the validity of our model (see Supplementary Table S2).
Finally, the GAA–NAC complex was refined by MD simulations as described above for the apoenzyme at the two different pHs. Interestingly, the presence of NAC at pH 7.0 confers to GAA a local flexibility profile similar to the one of unbound GAA at pH 5.0, thereby suggesting that NAC may actually act as a pharmacological chaperone of the enzyme (Figure 2d). This effect is not limited to the immediate vicinity of the NAC-binding site, but extends to distal regions of the protein. As a control, the same procedure was carried out using Gly as a possible ligand, but the resulting maps did not show any specific feature and Gly appears to nonspecifically sample many regions of the protein surface with very low affinity.
NAC and related compounds, binding reversibly to allosteric sites remote from the catalytic site, may act by blocking conformational fluctuations leading to a destabilized state of GAA, thus rescuing its functional state. This suggests that these molecules would be the first pharmacological chaperones that do not work as competitive inhibitors of the enzyme, expanding the molecular diversity of GAA pharmacological chaperones beyond imino sugar molecules. The modeling data were further supported by in vitro experiments showing that different concentrations of NAC and derivatives (up to 10 mmol/l) did not affect rhGAA activity (Figure 3a). Instead DNJ acted as a competitive inhibitor with a Ki of 3.4 µmol/l (Figure 3b).
Figure 3.

N-acetylcysteine (NAC) and related compounds are not α-glucosidase (GAA) inhibitors. GAA activity was measured in the presence of different concentrations of NAC and of the related compounds NAS and NAG, ranging from 0.1 to 10 mmol/l. (a) None of the compounds tested inhibited recombinant human α-glucosidase (rhGAA) activity, indicating that these compounds are not competitive inhibitors of the enzyme. (b) Instead DNJ acted as a competitive inhibitor with a Ki of 3.4 µmol/l.
NAC rescues mutated GAA in PD fibroblasts and transfected COS7 cells
We investigated the effect of NAC in cultured fibroblasts from five PD patients that carry different mutations and with different phenotypes (Table 1). The concentration of 10 mmol/l used for this purpose was in the same range as that used in studies on the antioxidant effect of NAC.28,29
Table 1. PD fibroblast cell lines studied.
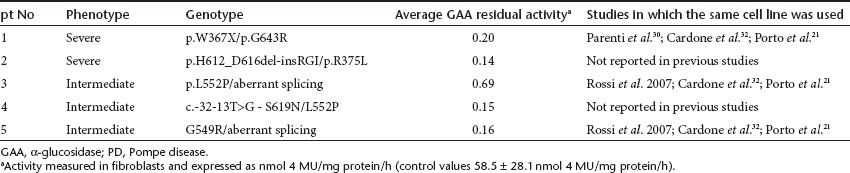
NAC enhanced the residual activity of mutated GAA in fibroblasts from patients 3 and 4 (Figure 4a). Both patients carried on one allele the mutation p.L552P, previously reported to be responsive to NB-DNJ and DNJ.30,31
Figure 4.

Effect of N-acetylcysteine (NAC) on the residual activity of mutated α-glucosidase (GAA) in fibroblasts and COS7 cells. (a) Incubation of Pompe disease (PD) fibroblast with 10 mmol/l NAC resulted in enhanced residual GAA activity in 2 of the 5 cell lines (from patients 3 and 4). The response of mutated GAA to NAC was also evaluated by expressing mutated GAA gene constructs in COS7 cells. (b) The mutations p.L552P, p.A445P, and p.Y455F, showed increases of GAA activity in the presence of NAC, and increased amounts of 76 and 70 kDa active GAA isoforms by western blot (c). These results indicate that NAC (10 mmol/l) has a different chaperoning profile, in terms of enhancement of GAA activity, compared to the imino sugar chaperone N-butyl-deoxynojirimycin (NB-DNJ) (20–100 µmol/l).31
The response of individual mutations to NAC was further evaluated by expressing a panel of mutated GAA gene constructs in COS7 cells (Figure 4b). The mutated constructs were chosen to be representative of both imino sugar-responsive and non-responsive mutations, in order to compare the chaperoning profile of NAC with that of imino sugars. The mutations p.L552P, p.A445P, and p.Y455F showed significant enhancement of GAA activity in the presence of 10 mmol/l NAC. The enhancement of enzyme activity for responsive mutations paralleled the increase in the amounts of the 76 and 70 kDa active isoforms of GAA on western blot analysis. Figure 4c shows a western blot analysis of COS7 cells overexpressing two of the responsive (p.L552P, p.A445P) and one non-responsive (p.G549R) mutation. For this latter mutation no change was seen in the amounts of the GAA active isoforms, already detectable in the absence of NAC, as previously reported.31 These results suggest that NAC has a different chaperoning profile compared to the active site-directed chaperones DNJ and NB-DNJ (Figure 4d).
NAC enhances rhGAA efficacy in PD fibroblasts and in the mouse model of PD
We tested whether NAC is able to synergistically enhance the efficacy of rhGAA. In fibroblasts from patient 3 coadministration of rhGAA and NAC (0.02–10 mmol/l) resulted in improved GAA activity with a dose-dependent effect (Figure 5a). Increases of 1.3-, 1.7- and 2.0-fold were already observed at NAC concentrations of 0.1, 1, and 5 mmol/l, respectively, with a maximal increase of 3.3-fold at 10 mmol/l. We then incubated five PD fibroblast cell lines with 50 µmol/l rhGAA in the absence and in the presence of 10 mmol/l NAC. The efficacy of rhGAA in correcting the enzymatic deficiency varied among the different cell lines, as already reported in previous papers,22,32 possibly due to individual factors implicated in the uptake and intracellular trafficking of the recombinant enzyme in each cell line. However, in all cell lines tested, coincubation of rhGAA with NAC resulted in an improved correction of GAA deficiency, with increases in GAA activity ranging from ~3.7–8.7-fold compared to the activity of cells treated with rhGAA alone (Figure 5b).
Figure 5.

Synergy between N-acetylcysteine (NAC) and recombinant human α-glucosidase (rhGAA) in Pompe disease (PD) fibroblasts. (a) The efficacy of rhGAA was enhanced by different concentrations (0.02–10 mmol/l) of NAC in patient 3, showing a dose-dependent effect. Five PD fibroblast cell lines were incubated with 50 µmol/l rhGAA in the absence and presence of 10 mmol/l NAC. (b) In all cell lines coincubation of rhGAA with the chaperone resulted in an improved correction of GAA deficiency, with increases in GAA activity ranging from ~3.7–8.7-fold the activity of cells treated with rhGAA alone. (c) In the presence of NAC also the amount of fluorochrome-labeled GAA increased, compared to cells incubated with fluorescent GAA alone. (d) The amounts of GAA polypeptides in the cells treated with NAC and rhGAA were also increased, compared to cells treated with the recombinant enzyme alone. Density scans of GAA bands show a relative increase of mature 76 and 70 kDa GAA isoforms in the presence of NAC (e).
The enhancing effect largely exceeded that due to the rescue of the native mutated enzyme (patients 3 and 4) and was observed also in non-responsive cell lines (patients 1, 2, and 5). We also observed an increase in the amounts of fluorochrome-labeled GAA in the presence of the chaperone NAC, compared to cells incubated with fluorescent GAA alone (Figure 5c). Using this approach only the fluorescent exogenous enzyme is detectable and variations in the intensity of fluorescence only reflect the effects on the recombinant enzyme. The combination of these results supports the concept that the enhancing effect of chaperones is directed towards the wild-type recombinant enzyme.
A western blot analysis (Figure 5d) and the quantitative analysis of each band (Figure 5e) showed increased amounts of GAA-related polypeptides in the cells treated with NAC and rhGAA, compared to cells treated with the recombinant enzyme alone. The processing of rhGAA into the active isoforms was also improved in the presence of NAC with a relative increase of the intermediate (95 kDa) and mature (76–70 kDa) GAA molecular forms, compared to the 110 kDa precursor. Since the GAA precursor is converted into the active forms in the late-endosomal/lysosomal compartment,33 this indicates improved lysosomal trafficking of the enzyme.
Other antioxidant drugs (resveratrol, epigallo chatechingallate) did not enhance rhGAA in PD cultured fibroblasts (Supplementary Figure S4). These results, together with the analysis of NAC–GAA interaction, with the data in cell-free systems, and with the lack of effect on another lysosomal glycosidase, α-galactosidase A that is defective in Fabry disease and belongs, like GAA, to the GH-D superfamily (Supplementary Figure S5), exclude that the effect of NAC is due to its antioxidant properties.
We also tested the combination of NAC and rhGAA in a mouse model of PD.34 Mice were treated with a single injection of rhGAA at high doses (100 mg/kg) in combination with oral NAC for 5 days (Figure 6a). Mice treated with the recombinant enzyme alone were used as controls. Forty-eight hours after rhGAA injection the animals were euthanized and GAA activity was assayed in different tissues. In all tissues examined (liver, heart, diaphragm and gastrocnemium) the combination of NAC and rhGAA resulted in higher levels of GAA compared to rhGAA alone (Figure 6b).
Figure 6.

Synergy between N-acetylcysteine (NAC) and recombinant human α-glucosidase (rhGAA) in vivo. (a) Mice were treated with oral NAC for 5 days and received an rhGAA injection on day 3. Animal treated with rhGAA alone were used as controls. In all tissues examined (liver, heart, diaphragm, and gastrocnemium) the combination of NAC and rhGAA (black bars) resulted in higher GAA enzyme activity compared to (b) rhGAA alone (grey bars).
Combined effect of NAC and imino sugar chaperones
A corollary of the fact that NAC and imino sugar chaperones interact with different protein domains, is that their effect may be cumulated. This hypothesis was supported by the results of thermal denaturation of rhGAA performed in the presence of NAC, of the imino sugar DNJ, and of both molecules. Both NAC and DNJ increased rhGAA thermal stability, with DNJ causing the best shift in Tm (65.9 ± 0.3 °C). The combination of NAC and DNJ (Tm = 75.9 ± 0.3), however, resulted in the highest stability of the enzyme (Figure 7a).
Figure 7.

Combination of the effect of N-acetylcysteine (NAC) with imino sugar chaperones. Thermal stability scans of recombinant human α-glucosidase (rhGAA) were performed in the absence and in the presence of NAC or DNJ. Both chaperones increased thermal stability of rhGAA, with N-butyl-deoxynojirimycin (NB-DNJ) resulting in the best shift in Tm of the enzyme. (a) The combination of DNJ and NAC resulted in further increase of the enzyme stability. Pompe disease (PD) fibroblasts from patients 2 and 4 were treated with rhGAA, with rhGAA plus either NAC or NB-DNJ, and with rhGAA plus the combination of the two chaperones. In both cell lines the combination of NAC and NB-DNJ resulted in the highest enhancement of GAA activity by rhGAA (b).
A similar cumulative effect was also seen in PD fibroblasts from patients 2 and 4 incubated with rhGAA. The cells were incubated with rhGAA, with rhGAA plus either NAC or NB-DNJ, and with rhGAA plus the combination of the two chaperones. In both cell lines the combination of NAC and NB-DNJ resulted in the greater enhancement of GAA activity by rhGAA (Figure 7b). This might represent an additional advantage for the treatment of patients, in order to obtain the best stabilization of rhGAA and the highest synergy with ERT.
Discussion
PCT is an attractive strategy to improve enzyme stability and is rapidly evolving towards clinical applications. However, concerns have been raised on its clinical translation. First, known chaperones are active site-directed molecules and thus potential inhibitors of target enzymes.24 In addition, it has been shown that chaperones are effective in rescuing only some disease-causing missense mutations, mainly located in specific enzyme domains, and are thus potentially effective in a limited number of patients. For PD, it is possible to speculate that ~10–15% patients may be amenable to PCT.31
These problems can be addressed by the identification of novel and allosteric noninhibitory chaperones. In this study, we have shown that NAC, and the related compounds NAS and NAG, are able to stabilize GAA without interfering with its activity and have a different chaperoning profile, compared to known chaperones. NAC is a known antioxidant that was evaluated in our laboratory, together with other related drugs (resveratrol, epigallo catechingallate) in PD fibroblasts for possible effects on rhGAA intracellular trafficking. The characterization of NAC's mechanism of action on rhGAA, however, indicated that molecular interactions with the enzyme, rather than the antioxidant effect, were responsible for rhGAA stabilization and that the other antioxidants studied did not stabilize the enzyme. This was somewhat surprising because NAC is structurally very different from the imino sugars, the only known pharmacological chaperones of GAA so far, that resemble the natural substrates/products of the enzyme.
We showed that NAC improved stability of GAA in response to physical stresses such as temperature and pH changes. Resistance to pH variations is of interest, as neutral pH may be representative of some of the environmental conditions encountered by recombinant enzymes in plasma and in certain cellular compartments. It has been shown that pH induces conformational changes in other lysosomal enzymes, such as β-glucocerebrosidase,35 and that binding with pharmacological chaperones may favor the most stable conformations of the enzyme.
Here, NAC, and the structurally related amino acids NAS and NAG, prevented the loss of GAA activity as a function of pH and increased the enzyme thermal stability with no inhibition, while the nonacetylated counterparts had no effect. To rationalize these data at molecular level, we analyzed the conformational dynamics of a 3D model of GAA through MD simulations. Although a high-resolution 3D-structure would certainly be the most accurate tool to perform these studies, structural models based on X-ray crystallography are not available for many glycosidases similar to GAA. According to these simulations, interaction with NAC favored a more stable conformation of the enzyme. Interestingly, the integrated analysis of the results of GAA simulations carried out in different conditions showed that a possible binding site for NAC could be located at 35 å distance from the active site, thereby validating the allosteric stabilization observed in vitro. The binding site was defined based on the combined characterization of the variations in the local dynamics of specific protein sites in response to different pH conditions and to the presence/absence of possible small-molecule ligands. Such analyses were next combined to the investigation of the druggability of possible identified binding pockets. The integrated analysis of the data eventually returned one solution that we defined as the putative allosteric pocket. Docking of NAC and its analogues to the allosteric pocket helped define the most favorable pose for the chaperone in complex with the protein. A control docking run with DNJ proved that this molecule strongly favors binding to the active site (Supplementary Table S2).
These observations were largely confirmed by the analyses performed in cellular systems (fibroblasts from PD patients and COS7 cells overexpressing mutated GAA constructs). In COS7 cells that overexpress mutated GAA incubation with NAC resulted in significantly increased residual GAA activity for three of the seven mutations studied. Remarkably, the chaperoning profile of NAC showed differences compared to that of NB-DNJ and DNJ. This may translate into an expansion of the number of chaperone-responsive mutations, and should be further investigated in large-scale studies. It would be tempting to envisage that preliminary screenings in vitro on a number of chaperones would allow personalization of treatment protocols aimed at obtaining the greatest beneficial effect in different PD patients.
NAC also increased the efficacy of rhGAA in correcting the enzyme defect in PD fibroblasts. Compared to the effect of NAC, and of chaperones in general, on the mutated enzymes, this effect holds greater promise for the cure of patients affected by PD, and possibly of other lysosomal disorders. It should be considered that, while the enhancement of endogenous defective enzymes by chaperones in most cases resulted in minor changes in terms of residual activity (likely with a modest impact on patients' outcome), the synergy of these drugs with ERT induced in PD fibroblasts remarkable increases in specific activity. In this study, coadministration of NAC and rhGAA resulted in complete correction of the enzymatic defect. An additional advantage of the combination of PCT and ERT is that the effect of chaperones is directed towards the wild-type recombinant enzyme and does not depend on the type of mutation carried by patients. Thus, any patient on ERT may benefit from the coadministration of PCT.
The results that support a synergy between chaperones and recombinant enzymes have important clinical implications and may translate into improved clinical efficacy of ERT. The data obtained in our proof-of-concept in vivo experiments in PD mice appear promising in this respect, although treatment protocols with NAC need to be carefully optimized in studies on a larger number of animals. The concentrations used in our studies may appear relatively high and difficult to translate into human therapy. However, toxicity of NAC is reported to be low and plasma concentrations in the range of 2-3 mmol/l can be reached for the treatment of paracetamol overdose.36 Long-term treatment with NAC doses of 5 g/kg/day (higher than those used in the in vivo experiments our study) has been used in the mouse model of ethylmalonic encephalopathy and resulted in improved survival of treated animals.37 NAC therapy was also used in patients affected by this metabolic disorder, albeit at lower doses, and again resulted in improved clinical and neurological course and in improvement of biochemical markers. In addition it must be taken into account that, for a possible clinical translation of our data, a therapeutic protocol would be based on the administration of the chaperone at the time of ERT infusion, and not on a long-term daily administration of the chaperone. By using such an approach the risk of adverse events would likely be reduced. It is also possible that modifications to NAC might be required to have the same stabilizing effect at lower concentrations.
It should be mentioned that clinical trials based on the combination of imino sugar competitive chaperones and ERT are already in progress (see trials NCT00214500 and NCT01196871 at http://clinicaltrials.gov; Telethon foundation trial GUP09017, http://www.telethon.it/ricerca-progetti/progetti-finanziati).
The identification of NAC and derivatives, which are structurally very different from the other known pharmacological chaperones identified in PD is promising. In fact, other molecules, whose chaperoning activity cannot be simply inferred from their structure, may be effective in several lysosomal storage disease, thereby opening new and wider opportunities for the identification of novel therapeutic drugs.
Materials and Methods
Fibroblast cultures. Fibroblasts from PD and Fabry disease patients were derived from skin biopsies performed for diagnostic purposes after obtaining the informed consent from patients or from their parents. All cell lines were grown at 37 °C with 5% CO2 in Dulbecco's modified Eagle's medium (Invitrogen, Grand Island, NY) and 10% fetal bovine serum (Sigma-Aldrich, St Louis, MO), supplemented with 100 U/ml penicillin and 100 mg/ml streptomycin.
Reagents. rhGAA (algucosidase, Myozyme) and rh-α-Gal A (agalsidase-β, Fabrazyme, for the experiments shown in Supplementary Figure S5) were from Genzyme, Cambridge, MA. As source of enzyme we used the residual amounts of the reconstituted recombinant enzymes prepared for the treatment of PD and Fabry patients at the Department of Pediatrics, Federico II University, Naples, Italy. NAC, NAS, NAG, cysteine, serine, glycine, 2-mercaptoethanol, 4-nitrophenyl-α-glucopyranoside (4NP-Glc), DNJ, NB-DNJ, and 1-deoxy-galactonojiirimycin were from Sigma-Aldrich.
The rabbit anti-GAA primary antibody used for immunofluorescence and western blot analysis was a gift from Dr Bruno Bembi and Dr Andrea Dardis, Centro di Coordinamento Regionale per le Malattie Rare, Udine, Italy; the anti-β-actin mouse monoclonal antibody was from Sigma-Aldrich. The anti-rabbit secondary antiserum were from Molecular Probes, Eugene, OR; horseradish peroxidase-conjugated anti-rabbit or anti-mouse immunoglobulin G were from Amersham, Freiburg, Germany. Labeling of rhGAA was performed using the Alexa Fluor 546 labeling kit (Molecular Probes) according to the manufacturer instructions.
Enzyme characterization. The standard activity assay of rhGAA was performed in 200 µl by using 5 µg of enzyme at 37 °C in 100 mmol/l sodium acetate pH 4.0 and 20 mmol/l 4NP-Glc. The reaction was started by adding the enzyme; after suitable incubation time (1-2 minutes) the reaction was blocked by adding 800 µl of 1 mol/l sodium carbonate pH 10.2. Absorbance was measured at 420 nm at room temperature, the extinction coefficient to calculate enzymatic units was 17.2 mmol/l–1 × cm–1. One enzymatic unit is defined as the amount of enzyme catalyzing the conversion of 1 µmol substrate into product in 1 minute, under the indicated conditions.
Effect of pH on rhGAA. The effect of different pHs on the rhGAA stability was measured by preparing reaction mixtures containing 0.75 mg ml–1 of enzyme in the presence of 50 mmol/l citrate/phosphate (pH 3.0–7.0) at a certain pH. rhGAA mixture was kept at 37 °C and aliquots were withdrawn at the times indicated (0–24 hours) and the residual GAA activity was measured with the standard assay.
To test the effect on the pH stability of rhGAA of pharmacological chaperones and other molecules and to test the effect of chaperones on rhGAA activity, experiments were performed as described above by adding to the reaction mixtures 0.1, 1, 10 mmol/l of the different compounds indicated in the text.
Analysis of thermal stability of rhGAA. Thermal stability scans of rhGAA were performed as described in ref. 38; briefly, 2.5 µg of enzyme were incubated in the absence and in the presence of NAC and DNJ, 10 and 0.1 mmol/l, respectively, SYPRO Orange dye, and sodium phosphate 25 mmol/l buffer, 150 mmol/l NaCl, pH 7.4 or sodium acetate 25 mmol/l buffer, 150 mmol/l NaCl, pH 5.2. Thermal stability scans were performed at 1 °C/minute in the range 25–95 °C in a Real-Time Light Cycler (Biorad, Milan, Italy). SYPRO Orange fluorescence was normalized to maximum fluorescence value within each scan to obtain relative fluorescence.
Modeling and molecular dynamics studies of GAA. The atomic structural model of GAA was constructed via homology modeling. The homology module of the ICM software (Molsoft, San Diego, CA; www.molsoft.com) was used for this purpose. The structure of the human maltase-glucoamylase,39 showing 44% identity over the aligned 868 residues of GAA, was used as a template. After alignment of the target sequence with the related 3D structure the modeling proceeded through the following steps: (i) a starting model was created based on the homologous structure with the conserved portion fixed and the nonconserved portion having standard covalent geometry and free torsion angles; (ii) the Biased Probability Monte Carlo procedure was applied to search the subspaces of either all the nonconservative side-chain torsion angles or torsion angles in a loop backbone and surrounding side chains. The Biased Probability Monte Carlo procedure globally optimizes the energy function consisting of ECEPP/3 and solvation energy terms. Next, the structure was refined through energy minimization with the Gromos96 force-field and the good quality of the model was assessed using ICM, the WHAT_CHECK module of the program WHAT_IF and the program PROCHECK. This model was compared with previous modeling studies and the results appeared to be consistent with what reported by other authors.31,40
The structure obtained after the modeling step was subjected to MD simulation analysis. Three sets of MD simulations were run using the same protocol: one simulation for the isolated apo-GAA, one for GAA bound to NAC, and one for GAA surrounded by multiple copies of NAC or NAG. pH variations were mimicked by protonating ionizable groups according to their pKa values.
In each MD run, the system was solvated in a tetrahedral solvation box with a size allowing the presence of a 1.2-nm layer of water on each side of the protein. All simulations and the analysis of the trajectories were performed using the 4.0.3 version of the GROMACS software package41 using the GROMOS96 force field42,43 and the SPC water model.44 In the case of the simulation with multiple copies of NAC or NAG, 10 molecules of co-solute were randomly placed in the simulation box at a distance of 1.0 nm from GAA.
Each system was first energy relaxed with 2,000 steps of steepest descent energy minimization followed by another 2,000 steps of conjugate gradient energy minimization, in order to remove possible bad contacts from the initial structures. Next, the systems were first equilibrated by 50 ps of MD runs with position restraints on the protein and ligand to allow relaxation of the solvent molecules. These first equilibration runs were followed by other 50 ps runs without position restraints on the solute. The first 20 ns of each trajectory were not used in the subsequent analysis in order to minimize convergence artifacts. Equilibration of the trajectories was checked by monitoring the equilibration of the root mean square deviation with respect to the initial structure, and of the internal protein energy.
Production runs span 100 ns each system studied. The electrostatic term was described by using the particle mesh Ewald algorithm. The LINCS45 algorithm was used to constrain all bond lengths. For the water molecules the SETTLE algorithm46 was used. A dielectric permittivity, є = 1, and a time step of 2 fs were used. All atoms were given an initial velocity obtained from a Maxwellian distribution at the desired initial temperature of 300K. The density of the system was adjusted performing the first equilibration runs at NPT condition by weak coupling to a bath of constant pressure (P0 = 1 bar, coupling time ôp = 0.5 ps).47 In all simulations the temperature was maintained close to the intended values by weak coupling to an external temperature bath with a coupling constant of 0.1 ps. The proteins and the rest of the system were coupled separately to the temperature bath.
The structural stability was initially monitored by analyzing the time evolution of structural parameters such as the root mean square deviation of each conformation visited from the starting structure.
The mechanistic determinants of GAA structural stability at different pHs was obtained by analyzing the protein's pH-dependent geometric strain.27 This quantity reports on the variations of the local deformations of the residues' contact networks as a consequence of the changes in their respective local structural environment. This parameter thus reflects the extent of conformational changes throughout the protein structure in response to specific perturbations.
Analysis of internal dynamic properties from MD trajectories. For each MD trajectory we computed the regions in which the geometric deformation accumulates. Such quantity, defined as geometric strain accumulates can be characterized by defining the strain of a given amino acid, i, as
where j runs over all protein amino acids, and f is a sigmoidal function that restricts the contribution to the sum to amino acids that are within about 5 å from amino acid i: f(x) = [1−tanh(x−5)]/2, where x is expressed in ångstroms. Regions that respond differently to a certain perturbation (such as the pH change, the presence of a certain ligand) are characterized by different values of p because in the course of the dynamical evolution their local network of contacts appreciably changes due to the relative motion of neighboring substructures.
To identify the regions of the protein surface that may be most frequently in contact with NAC, we combined MD simulations with molecular docking experiments. The density maps report the areas on the protein where NAC molecules cluster for most of the time. It is worth mentioning that this simulation should be intended as a coarse-grained exploration of the surface rather than a docking calculation. We ran an additional 100 ns MD simulation with one copy of GAA surrounded by 10 copies of NAC molecules, initially placed at random positions in the simulation box.
In order to generate a refined and optimized model of the possible NAC–GAA complex, the surface of the protein was further scanned for possible small-molecule binding sites with the “Site Map” function of the Maestro suite of programs. This function identifies possible sites on a protein that have the structural and functional requirements that are optimal for binding a small molecule. The pH responsive hot spot region identified earlier was thus used as a target for a set of unrestrained docking calculations, using a large receptor 3-D grid of 20 å of length on each side around the hot spot, which also included the residues that resulted to contact NAC in the aforementioned MD exploration.
Effect of NAC in COS7 cells and PD fibroblasts. Cultured PD fibroblasts and COS7 cells were treated with 10 mmol/l NAC for 24 hours before being harvested and used for GAA assay. COS cells were transfected with mutated GAA gene constructs as indicated in refs. 30,31.
GAA activity was assayed by using the fluorogenic substrate 4-methylumbelliferyl-α-D-glucopyranoside (Sigma-Aldrich) as described in ref. 22. Briefly, 25 µg of protein were incubated with the fluorogenic substrate (2 mmol/l) in 0.2 mol/l acetate buffer, pH 4.0, for 60 minutes in incubation mixtures of 100 µl. The reaction was stopped by adding 700 µl of glycine-carbonate buffer, pH 10.7. Fluorescence was read at 365 nm (excitation) and 450 nm (emission) on a Turner Biosystems Modulus fluorometer. Protein concentration in cell homogenates was measured by the Bradford assay (Biorad, Hercules, CA).
Western blot analysis. To study GAA immunoreactive material, fibroblast extracts were subjected to western blot analysis as described in ref. 21. The cells were harvested, washed in phosphate-buffered saline, resuspended in water, and disrupted by five cycles of freeze-thawing. Equal amounts (20 µg protein) of fibroblast extracts were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis and proteins were transferred to PVD membrane (Millipore, Billerica, MA).An antihuman GAA antiserum was used as primary antibody to detect GAA polypeptides; to detect β-actin, a monoclonal mouse antibody was used. Immunoreactive proteins were detected by chemiluminescence (ECL; Amersham). To quantitate the amounts of GAA-related bands western blots were analyzed by ImageJ.
Immunofluorescence analysis and confocal microscopy. To study the distribution of AlexaFluor546-labeled GAA, PD fibroblasts grown on coverslips were fixed using methanol, permeabilized using 0.1% saponin and locked with 0.01% saponin, 1% fetal bovine serum diluted in phosphate-buffered saline for 1 hour. The cells were incubated with the primary antibodies, with secondary antibodies in blocking solution and then mounted with vectashield mounting medium (Vector Laboratories, Burlingame, CA). Samples were examined with a Zeiss LSM 5–10 laser scanning confocal microscope.
In vivo studies. Animal studies were performed according to the European Union Directive 86/609, regarding the protection of animals used for experimental purposes. PD mice were housed in the TIGEM animal facility. Every procedure on the mice was performed with the aim of ensuring that discomfort, distress, pain, and injury would be minimal. Mice were euthanized following avertin anesthesia by cervical dislocation. To study the effects of the combination of NAC and rhGAA the animals were allowed to drink 138 mmol/l NAC in water ad libitum (4.2 g/kg/day) for 5 days. On day 3 the animals received a single rhGAA injection. On day 5 the animals were euthanized and the tissues were homogenized with a tissue lyser and GAA activity was measured as described in ref. 21.
SUPPLEMENTARY MATERIAL Figure S1. Molecular structures of the compounds analyzed in this study. Figure S2. Effect of NAS, NAG, and nonacetylated amino acids on rhGAA at different concentrations (0, 0.1, 1, 10 mmol/l) and up to 48 hours of incubation. Figure S3. The analysis of the root mean square fluctuation (RMSF) per residue showed that the protein residues experience lower structural fluctuations at acidic pH than at neutral pH, suggesting a higher structural stability at the lower pH condition (data not shown). Figure S4. Effect of the antioxidants epigallo catechingallate (EGCG) and resveratrol on the efficacy of rhGAA in cultured PD fibroblasts (patient 3). Figure S5. Molecular modeling data suggest that the interactions of NAC occur at a specific protein domain that shows sequence homology within other glycosidases belonging to family GH31. Table S1. The lists of residues reported in the table have been calculated by running a simulation of GAA in the presence of 10 NAC molecules starting from random positions in the simulation box. Table S2. Grid Docking scores of acetylated compounds to the allosteric site, and of DNJ to the active site.
Acknowledgments
This work was supported by the Telethon Foundation, Rome, Italy [grant TGPMT4TELD to G.P.], by Programma Operativo Nazionale (PON) 01_00862, by the Agenzia Spaziale Italiana [project MoMa n.1/014/06/0] and by the project “Nuove glicosidasi d'interesse terapeutico nella salute umana” within the exchange programme Galileo 2009-10 of the Università Italo-Francese (M.C.F., B.C.-P., and M.M.). We thank Dr Graciana Diez-Roux (Telethon Institute of Genetics and Medicine) for critically reviewing the manuscript and for helpful discussion. The study was performed in Naples, Italy.
Supplementary Material
Molecular structures of the compounds analyzed in this study.
Effect of NAS, NAG, and nonacetylated amino acids on rhGAA at different concentrations (0, 0.1, 1, 10 mmol/l) and up to 48 hours of incubation.
The analysis of the root mean square fluctuation (RMSF) per residue showed that the protein residues experience lower structural fluctuations at acidic pH than at neutral pH, suggesting a higher structural stability at the lower pH condition (data not shown).
Effect of the antioxidants epigallo catechingallate (EGCG) and resveratrol on the efficacy of rhGAA in cultured PD fibroblasts (patient 3).
Molecular modeling data suggest that the interactions of NAC occur at a specific protein domain that shows sequence homology within other glycosidases belonging to family GH31.
The lists of residues reported in the table have been calculated by running a simulation of GAA in the presence of 10 NAC molecules starting from random positions in the simulation box.
Grid Docking scores of acetylated compounds to the allosteric site, and of DNJ to the active site.
REFERENCES
- van der Ploeg AT., and, Reuser AJ. Pompe's disease. Lancet. 2008;372:1342–1353. doi: 10.1016/S0140-6736(08)61555-X. [DOI] [PubMed] [Google Scholar]
- Shea L., and, Raben N. Autophagy in skeletal muscle: implications for Pompe disease. Int J Clin Pharmacol Ther. 2009;47 Suppl 1:S42–S47. doi: 10.5414/cpp47042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parenti G., and, Andria G. Pompe disease: from new views on pathophysiology to innovative therapeutic strategies. Curr Pharm Biotechnol. 2011;12:902–915. doi: 10.2174/138920111795542606. [DOI] [PubMed] [Google Scholar]
- Van den Hout JM, Kamphoven JH, Winkel LP, Arts WF, De Klerk JB, Loonen MC.et al. (2004Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk Pediatrics 113e448–e457. [DOI] [PubMed] [Google Scholar]
- Van den Hout H, Reuser AJ, Vulto AG, Loonen MC, Cromme-Dijkhuis A., and, Van der Ploeg AT. Recombinant human alpha-glucosidase from rabbit milk in Pompe patients. Lancet. 2000;356:397–398. doi: 10.1016/s0140-6736(00)02533-2. [DOI] [PubMed] [Google Scholar]
- Kishnani PS, Corzo D, Nicolino M, Byrne B, Mandel H, Hwu WL.et al. (2007Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease Neurology 6899–109. [DOI] [PubMed] [Google Scholar]
- Strothotte S, Strigl-Pill N, Grunert B, Kornblum C, Eger K, Wessig C.et al. (2010Enzyme replacement therapy with alglucosidase alfa in 44 patients with late-onset glycogen storage disease type 2: 12-month results of an observational clinical trial J Neurol 25791–97. [DOI] [PubMed] [Google Scholar]
- van der Ploeg AT, Clemens PR, Corzo D, Escolar DM, Florence J, Groeneveld GJ.et al. (2010A randomized study of alglucosidase alfa in late-onset Pompe's disease N Engl J Med 3621396–1406. [DOI] [PubMed] [Google Scholar]
- Schoser B, Hill V., and, Raben N. Therapeutic approaches in glycogen storage disease type II/Pompe Disease. Neurotherapeutics. 2008;5:569–578. doi: 10.1016/j.nurt.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien YH, Lee NC, Thurberg BL, Chiang SC, Zhang XK, Keutzer J.et al. (2009Pompe disease in infants: improving the prognosis by newborn screening and early treatment Pediatrics 124e1116–e1125. [DOI] [PubMed] [Google Scholar]
- Kishnani PS, Corzo D, Leslie ND, Gruskin D, Van der Ploeg A, Clancy JP.et al. (2009Early treatment with alglucosidase alpha prolongs long-term survival of infants with Pompe disease Pediatr Res 66329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani PS, Goldenberg PC, DeArmey SL, Heller J, Benjamin D, Young S.et al. (2010Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants Mol Genet Metab 9926–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N, Danon M, Gilbert AL, Dwivedi S, Collins B, Thurberg BL.et al. (2003Enzyme replacement therapy in the mouse model of Pompe disease Mol Genet Metab 80159–169. [DOI] [PubMed] [Google Scholar]
- Wenk J, Hille A., and, von Figura K. Quantitation of Mr 46000 and Mr 300000 mannose 6-phosphate receptors in human cells and tissues. Biochem Int. 1991;23:723–731. [PubMed] [Google Scholar]
- Fukuda T, Ahearn M, Roberts A, Mattaliano RJ, Zaal K, Ralston E.et al. (2006Autophagy and mistargeting of therapeutic enzyme in skeletal muscle in Pompe disease Mol Ther 14831–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N, Baum R, Schreiner C, Takikita S, Mizushima N, Ralston E.et al. (2009When more is less: excess and deficiency of autophagy coexist in skeletal muscle in Pompe disease Autophagy 5111–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YH, Ponce E, Sun Y, Leonova T, Bove K, Witte D.et al. (1996Turnover and distribution of intravenously administered mannose-terminated human acid beta-glucosidase in murine and human tissues Pediatr Res 39313–322. [DOI] [PubMed] [Google Scholar]
- Benjamin ER, Khanna R, Schilling A, Flanagan JJ, Pellegrino LJ, Brignol N.et al. (2012Co-administration with the pharmacological chaperone AT1001 increases recombinant human a-galactosidase A tissue uptake and improves substrate reduction in Fabry mice Mol Ther 20717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan JQ. A counterintuitive approach to treat enzyme deficiencies: use of enzyme inhibitors for restoring mutant enzyme activity. Biol Chem. 2008;389:1–11. doi: 10.1515/BC.2008.009. [DOI] [PubMed] [Google Scholar]
- Parenti G. Treating lysosomal storage diseases with pharmacological chaperones: from concept to clinics. EMBO Mol Med. 2009;1:268–279. doi: 10.1002/emmm.200900036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porto C, Cardone M, Fontana F, Rossi B, Tuzzi MR, Tarallo A.et al. (2009The pharmacological chaperone N-butyldeoxynojirimycin enhances enzyme replacement therapy in Pompe disease fibroblasts Mol Ther 17964–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porto C, Pisani A, Rosa M, Acampora E, Avolio V, Tuzzi MR.et al. (2012Synergy between the pharmacological chaperone 1-deoxygalactonojirimycin and the human recombinant alpha-galactosidase A in cultured fibroblasts from patients with Fabry disease J Inherit Metab Dis 35513–520. [DOI] [PubMed] [Google Scholar]
- Shen JS, Edwards NJ, Hong YB., and, Murray GJ. Isofagomine increases lysosomal delivery of exogenous glucocerebrosidase. Biochem Biophys Res Commun. 2008;369:1071–1075. doi: 10.1016/j.bbrc.2008.02.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzano KJ, Khanna R, Powe AC, Boyd R, Lee G, Flanagan JJ.et al. (2011Identification and characterization of pharmacological chaperones to correct enzyme deficiencies in lysosomal storage disorders Assay Drug Dev Technol 9213–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropak MB, Blanchard JE, Withers SG, Brown ED., and, Mahuran D. High-throughput screening for human lysosomal beta-N-Acetyl hexosaminidase inhibitors acting as pharmacological chaperones. Chem Biol. 2007;14:153–164. doi: 10.1016/j.chembiol.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Padia J, Urban DJ, Jadhav A, Goker-Alpan O, Simeonov A.et al. (2007Three classes of glucocerebrosidase inhibitors identified by quantitative high-throughput screening are chaperone leads for Gaucher disease Proc Natl Acad Sci USA 10413192–13197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morra G, Potestio R, Micheletti C., and, Colombo G. Corresponding functional dynamics across the Hsp90 Chaperone family: insights from a multiscale analysis of MD simulations. PLoS Comput Biol. 2012;8:e1002433. doi: 10.1371/journal.pcbi.1002433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers ML, Almeida ME, Vicente-Manzanares M, Horwitz AF., and, Santos MF. High glucose-mediated oxidative stress impairs cell migration. PLoS ONE. 2011;6:e22865. doi: 10.1371/journal.pone.0022865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieber M., and, Rieber MS. N-Acetylcysteine enhances UV-mediated caspase-3 activation, fragmentation of E2F-4, and apoptosis in human C8161 melanoma: inhibition by ectopic Bcl-2 expression. Biochem Pharmacol. 2003;65:1593–1601. doi: 10.1016/s0006-2952(03)00147-3. [DOI] [PubMed] [Google Scholar]
- Parenti G, Zuppaldi A, Gabriela Pittis M, Rosaria Tuzzi M, Annunziata I, Meroni G.et al. (2007Pharmacological enhancement of mutated alpha-glucosidase activity in fibroblasts from patients with Pompe disease Mol Ther 15508–514. [DOI] [PubMed] [Google Scholar]
- Flanagan JJ, Rossi B, Tang K, Wu X, Mascioli K, Donaudy F.et al. (2009The pharmacological chaperone 1-deoxynojirimycin increases the activity and lysosomal trafficking of multiple mutant forms of acid alpha-glucosidase Hum Mutat 301683–1692. [DOI] [PubMed] [Google Scholar]
- Cardone M, Porto C, Tarallo A, Vicinanza M, Rossi B, Polishchuk E.et al. (2008Abnormal mannose-6-phosphate receptor trafficking impairs recombinant alpha-glucosidase uptake in Pompe disease fibroblasts Pathogenetics 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisselaar HA, Kroos MA, Hermans MM, van Beeumen J., and, Reuser AJ. Structural and functional changes of lysosomal acid alpha-glucosidase during intracellular transport and maturation. J Biol Chem. 1993;268:2223–2231. [PubMed] [Google Scholar]
- Raben N, Nagaraju K, Lee E, Kessler P, Byrne B, Lee L.et al. (1998Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II J Biol Chem 27319086–19092. [DOI] [PubMed] [Google Scholar]
- Lieberman RL, Wustman BA, Huertas P, Powe AC, Jr, Pine CW, Khanna R.et al. (2007Structure of acid beta-glucosidase with pharmacological chaperone provides insight into Gaucher disease Nat Chem Biol 3101–107. [DOI] [PubMed] [Google Scholar]
- Walsh TS, Hopton P, Philips BJ, Mackenzie SJ., and, Lee A. The effect of N-acetylcysteine on oxygen transport and uptake in patients with fulminant hepatic failure. Hepatology. 1998;27:1332–1340. doi: 10.1002/hep.510270520. [DOI] [PubMed] [Google Scholar]
- Viscomi C, Burlina AB, Dweikat I, Savoiardo M, Lamperti C, Hildebrandt T.et al. (2010Combined treatment with oral metronidazole and N-acetylcysteine is effective in ethylmalonic encephalopathy Nat Med 16869–871. [DOI] [PubMed] [Google Scholar]
- Niesen FH, Berglund H., and, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- Sim L, Willemsma C, Mohan S, Naim HY, Pinto BM., and, Rose DR. Structural basis for substrate selectivity in human maltase-glucoamylase and sucrase-isomaltase N-terminal domains. J Biol Chem. 2010;285:17763–17770. doi: 10.1074/jbc.M109.078980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima Y, Matsuzawa F, Aikawa S, Okumiya T, Yoshimizu M, Tsukimura T.et al. (2007Structural and biochemical studies on Pompe disease and a “pseudodeficiency of acid alpha-glucosidase” J Hum Genet 52898–906. [DOI] [PubMed] [Google Scholar]
- Hess B, Kutzner C, Van Der Spoel D, Lindahl E. GROMACS 4: Algorithms for highly efficient load-balanced, scalable molecular simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- van Gunsteren WF, Daura WFX., and, Mark AE. GROMOS force field. Encyclopedia of Computational Chemistry. 1998;2:1211–1216. [Google Scholar]
- Scott WRP, Hünenberger PH, Tironi IG, Mark AE, Billeter SR, Fennen J.et al. (1999The GROMOS biomolecular simulation program package J Phys Chem A 1033596–3607. [Google Scholar]
- Berendsen HJ, Grigera JR, Straatsma TP. The missing term in effective pair potentials. J Phys Chem. 1987;91:6269–6271. [Google Scholar]
- Hess E, Bekker H, Berendsen HJC., and, Fraije JGEM. LINCS: A linear constraint solver. J Comput Chem. 1997;18:1463–1472. [Google Scholar]
- Miyamoto S, Kollmann PA. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J Comp Chem. 1992;13:952-962. [Google Scholar]
- Berendsen HJ, Postma JP, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Molecular structures of the compounds analyzed in this study.
Effect of NAS, NAG, and nonacetylated amino acids on rhGAA at different concentrations (0, 0.1, 1, 10 mmol/l) and up to 48 hours of incubation.
The analysis of the root mean square fluctuation (RMSF) per residue showed that the protein residues experience lower structural fluctuations at acidic pH than at neutral pH, suggesting a higher structural stability at the lower pH condition (data not shown).
Effect of the antioxidants epigallo catechingallate (EGCG) and resveratrol on the efficacy of rhGAA in cultured PD fibroblasts (patient 3).
Molecular modeling data suggest that the interactions of NAC occur at a specific protein domain that shows sequence homology within other glycosidases belonging to family GH31.
The lists of residues reported in the table have been calculated by running a simulation of GAA in the presence of 10 NAC molecules starting from random positions in the simulation box.
Grid Docking scores of acetylated compounds to the allosteric site, and of DNJ to the active site.