

Highlights
► Fast and direct method is proposed for monitoring of trace VOCs emitted by human skin.
Keywords: Volatile organic compounds (VOC), Ion mobility spectrometry (IMS), Multi-capillary column (MCC), Gas chromatography–mass spectrometry (GC–MS), Skin emissions
Abstract
Volatile organic compounds (VOCs) released by humans through their skin were investigated in near real time using ion mobility spectrometry after gas chromatographic separation with a short multi-capillary column. VOCs typically found in a small nitrogen flow covering the skin are 3-methyl-2-butenal, 6-methylhept-5-en-2-one, sec-butyl acetate, benzaldehyde, octanal, 2-ethylhexanol, nonanal and decanal at volume fractions in the low part per billion-(ppb) range. The technique presented here may contribute to elucidating some physiological processes occurring in the human skin.
1. Introduction
Human skin is the largest organ of the body, comprising 15–20% of total body weight. The skin constitutes the first line of defence against dehydration, pathogens, injuries and temperature extremes. Human skin is covered by a continuous hydro-lipidic film, representing the actual interface between the viable epidermal layers and the outer environment [1]. The composition of human skin surface lipids varies over the body, but in sebaceous gland-enriched areas (such as forehead, upper chest) the secretion, so-called sebum, contains mainly cholesterol, cholesteryl esters, squalene, diglycerides and triglycerides, wax esters and fatty acids [2]. With regard to fatty acids, the sebum is especially rich in oleic, linoleic and myristic acids [3].
Various volatile organic substances, particularly aldehydes with various chain lengths, are produced from fatty acids by homolytic b-scission caused by UV radiation or bacterial peroxisomal lipid oxidation activity. The molecular structure depends on the oxidized precursor and on the localization of the scission resulting from the oxidative attack. These volatile substances are parts of the body odour. Personal body odour, however, is also influenced by consumed food, lifestyle, gender, environmental exposure, genetics and medication [3].
Scent identification has attracted the attention of researchers for a long time. Identification of individuals based on body odour using dogs is a potentially valuable tool in forensic processes. Searching for entrapped persons after earthquakes or explosions is also based on compounds emanated through breath, skin or urine [4], [5], [6]. As an example, a recent study focused on modelling the metabolite plume of trapped people in a simulated collapsed building confirmed CO2 ammonia and acetone as effective markers for the presence of trapped humans [7].
Due to the complexity of the samples, previous studies have applied different analytical technologies for screening VOCs emanated by the skin, such as gas chromatography coupled with mass spectrometry (GC–MS) [8]. More than 100 compounds including C8–C12 aldehydes, ketones, alcohols and acids were detected by Gallagher et al., showing a wide range of natural variations in volatile compounds depending on age and sampling location [8]. Zhang et al. monitored emanations of the human arm also by using GC–MS and determined seasonal characteristics of the emitted compounds [9]. Beside this, skin volatiles have been intensively studied as attractants of mosquitoes [10], [11].
Some efforts have been made to mimic canine olfactory capabilities by applying electronic noses for the detection of volatile organic compounds emitted from the skin. Various saturated and unsaturated fatty acids in skin vapours were analysed in real time using mass spectrometry with an atmospheric pressure ionization source [12]. Di Natale at al. detected a steroid, 5a-androst-16-en-3-one, in skin emissions when using a sensor system based on eight quartz microbalances [13]. This steroid is supposed to be a male pheromone.
Another technique that allows quick analysis of volatile organic substances is ion mobility spectrometry (IMS). This technique was developed in the early 1970s for military use, especially for the detection of explosives and chemical warfare agents. During the last ten years it has been applied not only for military and civilian protection, but also for monitoring tasks in the pharmaceutical industry [14], [15], toxicology [16], environmental analytics [17], [18] and medical diagnostics [19], [20], [21].
This paper will focus on the near real-time detection of VOCs released by the skin using IMS coupled with a short multi-capillary column (MCC). IMS allows detection with extremely good sensitivity for compounds showing high proton affinity, thus especially suited for analysis of aldehydes, ketones, and alcohols (detection limits in the low ppb-range). In comparison with the often privileged technology of GC–MS, no sample preconcentration is necessary before analysis to permit the reduction of artefacts arising from contaminations as well as faster testing cycles. Because of the complexity of the samples including high moisture levels, pre-separation of the analytes using gas chromatography is necessary to increase selectivity. Nevertheless, IMS technique suffers from some limitations in the identification of volatiles due to various instrumental set-ups and the absence of commercially available substance library; therefore parallel GC-MS analyses are invaluable to advance reliability of identification.
Such investigations of human skin emissions might prove to be a feasible and non-invasive tool for monitoring chemical exposure and could contribute to the diagnosis of diseases.
2. Materials and methods
2.1. Subjects and sampling of skin emissions
Seven members of our research team (one woman and six men; age range: 34–42 years) participated in the study. At least 12 samples were taken from each volunteer during the experiments, with a sampling rate of 5 min per sample. Measurements were usually performed at noon at the same time on at least three consecutive days to examine individual-related changes in skin emission. However for one selected volunteer they were repeated in the afternoon at an interval of several days or even weeks between measurements. For one volunteer measurements were performed in two different labs to study the influence of the room air to which the subject was exposed.
Candidates were asked to not use any cosmetics on the day of the experiments and to preferably shower at least 12 h before the experiment. No specific diet was imposed; however candidates had to abstain from alcohol consumption from at least the day before the measurements. The study was approved by the local ethics committee of Innsbruck Medical University.
2.2. Procedures for VOC collection by GC–IMS
Sampling was performed with a cylindrical stainless steel pan having an internal diameter of 102 mm and height of 40 mm (volume approx. 314 ml) equipped with a gas inlet and a gas outlet (Fig. 1). Prior to the experiment this pan was tightly fixed on the skin of the navel area. During the measurements the volunteer was seated. Next, nitrogen of purity grade 99.9999% was introduced into the container, first at a high flow rate for 2 min for rinsing and elimination of air contaminants, and then at a flow rate of 3 ml/min as set by a mass flow controller (red-y, Burde Co Praezisionsarmaturen GmbH, Vienna, Austria). The outlet of the pan was directly connected to the sample loop integrated in the GC–IMS device. Sample gas was sucked in by an internal pump at 3 ml/min and directed through the sample loop (V = 5 ml) of a six-port valve. Thus, the loop was permanently rinsed with the nitrogen flow containing the emitted skin VOCs.
Fig. 1.

Set-up of GC–IMS. The stainless steel pan used for skin VOC sampling was tightly fixed to the navel/umbilicus of the volunteer using a bandage. The pan was rinsed with nitrogen flow (3 ml/min) and its outlet was directly connected to the sample loop integrated in the GC–IMS device.
Samples were taken automatically with a sampling rate of 5 min. By switching the valve the sample was injected directly into the MCC for separation and further detection by IMS. Samples of indoor air were also taken before and after the measurements at the place where the volunteer was sitting.
2.2.1. Sample flow optimization
Beside the 3 ml/min flow rate, which was the lowest possible stable flow rate for the internal pump, higher sample flows of 8 ml/min and 20 ml/min were also used to rinse the container. At 3 ml/min a constant condition was achieved. This allowed nearly half of the sample volume in the container to be exchanged within the 1 h measurement time, thus enabling adequate reproducibility between consecutive measurements. By contrast, at higher flow rates a strong decrease in volumes of detected peaks occurred because the container's gas content was exchanged more quickly.
2.3. GC-MS sampling and sample separation
For three volunteers, in parallel to the GC–IMS measurements GC–MS analyses were performed. The main goal of this protocol was to confirm the IMS identification of analytes under study using a technique offering more sophisticated identification tools. Headspace skin samples for the GC–MS analyses were taken using a 20 ml gas-tight glass syringe (Carl Roth GMBH, Karlsruhe, Germany) equipped with a replaceable needle. Sampling was achieved by manually drawing a volume of 18 ml from the gas stream flowing through the chamber and subsequently injecting this volume into an evacuated SPME vial (volume: 20 ml, Gerstel GmbH, Mühlheim an der Ruhr, Germany) sealed with a 1.3 mm butyl/PTFE septum (Macherey-Nagel, Dueren, Germany). The SPME procedure was performed automatically using a multipurpose sampler MPS (Gerstel, Germany). SPME was achieved by inserting a 75 μm carboxen-polydimethylsiloxane (CAR-PDMS) fibre (Supelco, Canada) into the vial and exposing it to the content for ten minutes at 37 °C. Immediately after extraction the fibre was introduced into the inlet of the gas chromatograph, where the adsorbed VOCs were thermally desorbed at 290 °C.
2.4. VOC analysis by GC–IMS
A commercially available GC–IMS (BreathSpec, G.A.S. mbH, Dortmund, Germany) was used for the skin experiments. The IMS is described elsewhere [22], [23], and only a brief summary will be given in this paper.
The GC–IMS device is equipped with a multi-capillary column (MCC, OV-5, Novosibirsk, Russia) for gas-chromatographic separation. The 20 cm long multi-capillary column consists of 1000 capillaries with an inner diameter of 40 μm and a film thickness of 2 μm for each capillary. The MCC was maintained isothermal at 40 °C. Nitrogen (99.9999% purity grade) was used as drift and carrier gas, with flow rates of 200 ml/min and 100 ml/min. The IMS was heated to maintain a constant temperature of 45 °C.
After separation the analytes are introduced into the IMS ionization chamber, where ions are generated by means of a radioactive ionization source (tritium (3H), 300 MBq activity). During the ionization process the molecules continually undergo a series of ion–molecule and ion–ion reactions (mainly proton transfer) with the ionized drift gas (nitrogen). An ion swarm is periodically injected into the drift tube (length: 50 mm) every 100 ms using a shutter grid. There, the ions drift at ambient pressure under the influence of a uniform electric field (400 V/cm). Ions of various sizes achieve different velocities inversely related to their size (cross-section), mass and charge. Collection of these ions on the detector (Faraday plate) delivers a time-dependent signal that corresponds to ion mobility (Fig. 1).
2.4.1. Data evaluation
Ion mobility spectra were recorded via an integrated computer (400 MHz X scale processor). During one run (measurement time: 5 min) 650 spectra were recorded, containing 3000 sample points per spectrum. This gives a matrix with a total of 1.95 million sample points. For data visualization and analysis LAV software (version 1.5.1, GAS GmbH, Dortmund, Germany) was used. In addition to correct minimal variations in drift and retention time, the data were aligned using the LAV software. Peak volumes in defined areas of the identified compounds on the 3D topographic plots were calculated and used to determine calibration curves and compute concentrations.
Reduced ion mobilities of analytes were calculated from drift times using the normalization factor FIMS according to the following formula [24]:
whereby K0 RIP is the reference reduced ion mobility for reaction ion peak (RIP) [cm2 V−1 s−1], tD RIP is the drift time of RIP [ms], K0 analyte [cm2 V−1 s−1] and tD analyte is the drift time of analyte.
The normalization factor represents all variables that can change according to variations in environmental parameters, and thus was calculated for every measurement.
Reference reduced ion mobility for reaction ion peak (RIP) was calculated according to the following equation determined as 1.342 cm2 V−1 s−1.
where tD is the drift time [ms]; P is the pressure of the drift gas [h Pa]; P0 is the normal pressure = 1013.2 [h Pa]; T is the temperature of the drift gas [K]; T0 is the normal temperature = 273.2 [K].
2.4.2. Calibrations
Test gases for 3-methyl-2-butenal, 6-methylhept-5-en-2-one, sec-butyl acetate, benzaldehyde, octanal, 2-ethylhexanol, nonanal and decanal were prepared in zero air (hydrocarbon impurities < 10 ppb) with 100% relative humidity at 20 °C using a test gas generator (GASLAB, Breitfuss Messtechnik GmbH, Harpstedt, Germany). All chemicals were purchased from Sigma–Aldrich (Vienna, Austria). In the gas generator the liquid substances were introduced into a vaporizer with determined flow rate and evaporated at 100 °C. Next, their vapours were diluted with zero air by means of integrated mass flow controllers producing gas standards of the selected volatiles within the range of 0.2 and 30 ppb.
Calibration curves (Fig. 2a–h) were calculated using the peak volumes over areas determined by the characteristic drift time and retention time for each substance. The same areas were used to quantify the compounds in the skin samples. Instead of straight regression lines, polynomial or exponential calibration functions are commonly used for ion mobility spectrometry to model the saturation effect occurring due to the maximal amount of reaction ions (H3O+)n available [23]. These are the main proton donors during the ionization process. However, in the lower concentration ranges from 0.2 to 5 ppb linear function is a good approximation of the IMS response. Detection limits were evaluated from the calibration curves using t-distribution with 95% probability and amounted to 1.0 ppb for 2-ethyl-hexanol, 0.6 ppb for 3-methyl-2-butenal, 0.3 ppb for decanal, 0.5 ppb for nonanal, 0.6 ppb for octanal, 0.7 ppb for 6-methylhept-5-en-2-one, 0.3 ppb for benzaldehyde and 0.2 ppb with a signal-to-noise ratio S/N = 3.
Fig. 2.

(a–h) Calibration curves of the selected compounds measured with GC–IMS.
2.5. VOC analysis by SPME/GC–MS
The GC–MS analyses were performed using an Agilent 7890 A/5975 C GC–MS system (Agilent, Santa Clara, CA, USA). During the fibre desorption, the split/splitless inlet operated in the splitless mode (1 min), followed by the split mode at ratio 1:35. The analytes under study were separated using a PoraBond Q column (25 m × 0.32 mm, film thickness 5 μm, Varian, Palo Alto, CA, USA) working in a constant flow mode (helium at 1.4 ml/min). The column temperature programme was as follows: 40 °C for 2 min, increase to 260 °C at a rate of 7 °C/min, hold at 260 °C for 7 min. The mass spectrometer worked in a SCAN mode with an associated m/z range set between 20 and 200. The quadrupole, ion source and transfer line were maintained at 150 °C, 230 °C and 280 °C, respectively.
2.5.1. Data evaluation
Compounds were identified in two steps. First, the peak spectrum was checked against the NIST mass spectral library. Next, the NIST identification was confirmed using the retention times obtained from the standards prepared from pure compounds.
2.5.2. Calibrations
Calibration standards were produced by injecting of 1 μl of pure compound into an evacuated 1-l glass bulb (Supelco, Oakville, Ontario, Canada). Next, the bulb was heated to 80 °C for 15 min in order to ensure evaporation and subsequently balanced with high-purity nitrogen. This primary standard was used to prepare six secondary calibration mixtures. This was accomplished by transferring appropriate amounts of primary standard into 3-l Tedlar bags (SKC Inc., Houston, USA) already pre-filled with 2500 ml of nitrogen. Effectively, gas mixtures with volume fractions ranging from 0.5 to 30 ppb were used during calibration and validation of the analytical method.
Limits of detection (LODs) were calculated using the mean value of the blank responses and their standard deviations obtained on the basis of seven blank measurements. The limit of quantification (LOQ) was defined as three times the LOD. The relative standard deviations (RSDs) were calculated from five consecutive analyses of standard mixtures. The validation parameters, retention times (Rt), LODs (ppb), RSDs (%), correlation coefficients (R2) and linear ranges (ppb) obtained for compounds under study are presented in Table 1.
Table 1.
The validation parameters; retention times (Rt), LODs (ppb), RSDs (%), correlation coefficients (R2) and linear ranges (ppb) obtained for compounds under study using GC–MS.
| VOC | CAS | Rt (min) | RSD (%) | LOD (ppb) | R2 | Linear range (ppb) |
|---|---|---|---|---|---|---|
| n-Butyl acetate | 123-86-4 | 26.17 | 3 | 0.1 | 0.999 | 0.3–30 |
| Benzaldehyde | 100-52-7 | 28.89 | 12 | 0.2 | 0.985 | 0.6–31 |
| 6-Methylhept-5-en-2-one | 110-93-0 | 31.12 | 7 | 0.3 | 0.986 | 0.9–40 |
| Octanal | 124-13-0 | 31.86 | 11 | 0.3 | 0.978 | 0.9–17 |
3. Results
Combination of ion mobilities and retention time of the analytes permits 3D visualization of the measured data. For such a plot characteristic drift time and retention time of the analytes are presented on the X and Y axes, respectively, with both parameters being used for compound identification. Peak height enabling quantification of the compounds is illustrated by the colour scale. The 3D GC–IMS chromatogram, as shown in Fig. 3, displays the pattern of VOCs emitted by the skin. More than 20 peaks were detected in the seven individuals.
Fig. 3.

3D-visualization of IMS-chromatogram of emitted skin VOCs. Drift time and retention time are presented in X- and Y-axis, respectively, and peak height is displayed using colour-scale.
For peak identification a library containing both retention time and drift time of measured standards was set up based on previous GC–MS investigations [8], [9], which were compared with the peak parameters detected in skin samples. The measured unknown peak and a library peak were considered to match if the difference in the measured drift times for standards and skin samples was less than 2%, and for retention times less than 5%. The eight compounds mentioned above were identified and detected in almost every skin gas sample. Table 2 shows the average retention times and calculated mobilities with standard deviations (SD) of the compounds under study.
Table 2.
Average retention times and calculated reduced ion mobilities (K0) with standard deviations, numbers of replications (n) for identified compounds measured in gas standards and in skin samples using GC–IMS.
| Substance | CAS | Retention time (gas standards) n = 14 (s) |
Reduced mobilities K0 (gas standards) n = 14 (cm2 V−1 s−1) |
Retention time (sample) n = 30 (s) |
Reduced mobilities K0 (sample) n = 30 (cm2 V−1 s−1) |
|---|---|---|---|---|---|
| 3-Methyl-2-butenal | 598-75-4 | 10.7 ± 0.4 | 1.201 ± 0.018 | 10.8 ± 0.3 | 1.220 ± 0.010 |
| Sec-butylacetate | 105-46-4 | 12.7 ± 0.1 | 1.058 ± 0.022 | 12.8 ± 0.4 | 1.081 ± 0.024 |
| Benzaldehyde | 100-52-7 | 26.5 ± 0.7 | 1.149 ± 0.005 | 27.1 ± 0.3 | 1.145 ± 0.002 |
| 6-Methylhept-5-en-2-one | 110-93-0 | 31.4 ± 0.3 | 1.135 ± 0.007 | 31.6 ± 0.3 | 1.142 ± 0.006 |
| Octanal | 124-13-0 | 33.53 ± 0.8 | 0.949 ± 0.011 | 34.1 ± 0.2 | 0.952 ± 0.015 |
| 2-Ethyl-hexanol | 104-76-7 | 42.3 ± 0.8 | 0.936 ± 0.008 | 43.9 ± 0.7 | 0.951 ± 0.011 |
| Nonanal | 124-19-6 | 72.4 ± 0.5 | 0.904 ± 0.008 | 73.4 ± 0.4 | 0.909 ± 0.008 |
| Decanal | 112-31-2 | 172.7 ± 1.3 | 0.866 ± 0.009 | 174.4 ± 1.2 | 0.873 ± 0.008 |
| Ammonia | 7664-41-7 | 6.5 ± 0.4 | 1.953 ± 0.005 | 6.4 ± 0.3 | 1.955 ± 0.004 |
| Ethanol | 64-17-5 | 6.2 ± 0.3 | 1.526 ± 0.008 | 6.2 ± 0.2 | 1.516 ± 0.007 |
3.1. Observed VOCs in skin samples
Five of the eight compounds identified belong to the group of aldehydes. One of the possible sources of aldehyde production is lipid peroxidation, in which process they are formed as secondary products alongside simple hydrocarbons. As an example, decanal may be formed by decomposition of oleic acid [25], which is a common fatty acid contained in skin sebum. In this process a fatty acid radical is first produced by the reaction of a free radical with the fatty acid (2). This reaction involves hydrogen atom abstraction. The fatty acid radical reacts with oxygen, forming a fatty acid hydro-peroxide (2) [26].
| CH3—(CH2)7—CH CH—(CH2)7COOH → CH3—(CH2)7—CH CH—CH•—(CH2)6COOH + H• | (1) |
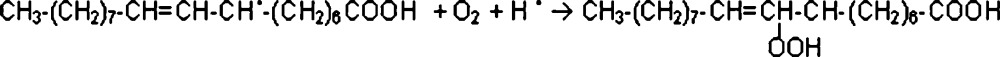 |
(2) |
After that, the most probable process is the production of an alkoxy radical from hydro-peroxide, which results in an additional hydroxyl radical according to the following reaction (3):
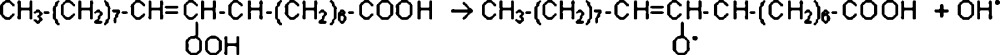 |
(3) |
The alkoxy radical may then cleave at the position beside the double carbon-carbon bond (next to the hydro-peroxide group) (4). There, an alkyl radical containing ten carbon atoms may be produced at the methyl end of the molecule. This radical can then combine with a hydroxyl radical to produce an alcohol, from which an aldehyde (decanal) can be formed through tautomerization (5) [27].
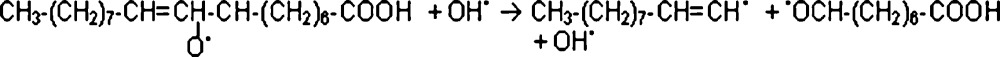 |
(4) |
| CH3—(CH2)7—CH CH• + •OH → CH3—(CH2)7—CH CHOH ↔ CH3—(CH2)7—CH2CHO (decanal) | (5) |
In addition to decanal, other aldehydes in the same homolog series such as octanal and nonanal can be formed in a similar process, e.g. from the C9 alkyl radical or the C8 alkyl radical.
The above-mentioned three aldehydes were detected in almost every skin sample. They show similar trend with regard to mean concentrations for individual volunteers (Fig. 4, Fig. 5). Thus, octanal, nonanal and decanal can each be found at comparatively higher levels with a mean value of 4.3 ppb, 7.1 ppb and 4.7 ppb, respectively, in samples from Volunteer 2. The lowest levels were found for Volunteer 5, in whose samples octanal was not detected, nonanal reached a mean value of 0.6 ppb and decanal 1.5 ppb. For these concentrations the IMS response can be well modelled by the linear function. Thus, it could be assumed that a sufficient number of reaction ions is present for ionization and that quantification is therefore precise. Octanal was confirmed by means of GC–MS analysis in two of the three volunteers examined. Although the calculated concentrations of GC–MS measurements are tenfold those of IMS values, they stay in the low ppb range. This difference between the results of the two independent techniques might be attributed to different sample preparation, calibration procedures and sensitivity for particular compounds.
Fig. 4.

(a and b) Concentration profile of octanal and nonanal detected in gas samples collected above skin.
Fig. 5.

(a) Concentration profile of decanal detected in skin samples of seven volunteers; (b) increasing emitted concentration values for decanal during skin experiments.
During the 1 h sampling time yielding 12 consecutive analyses the concentration profiles of these three aldehydes were found to increase for almost every subject, as shown for decanal in Fig. 5a. This evidences the suggestion that these compounds are emanated by the skin. The production rate can be estimated as about 0.12–1.82 μg/(h m2) skin surface for decanal, 0.16–1.96 μg/(h m2) for nonanal, and 0.87–1.53 μg/(h m2) for octanal. Although one possible process for the production of aldehydes could be lipid peroxidation as deduced above, their biological origin remains unclear and should be confirmed in further investigations.
For the two other aldehydes, namely benzaldehyde and 3-methyl-2-butenal, a similar trend in the seven persons’ skin samples was observed: they were in the same range of approximately 0–2.6 ppb for each compound. For 3-methyl-2-butenal Volunteer 3 showed the highest mean value (1.8 ppb), for benzaldehyde Volunteer 5 the highest (1.5 ppb), exhibiting high RSDs (56% and 86%, respectively) in calculated concentrations (Fig. 6a and b). No significant increase in concentrations was found during the 1 h experiment times for these compounds. Moreover, benzaldehyde and 3-methyl-2-butenal were also measured in room air samples at a mean level of 0.5 ppb and 0.7 ppb, respectively. The GC–MS analyses detected benzaldehyde, but not 3-methyl-butenal. Benzaldehyde concentrations were computed in the samples of Volunteers 6 and 7 as 10.1 ppb and 3.8 ppb, respectively. The great difference in concentration of up to one order of magnitude as compared to IMS measurements may result from the high blank signals obtained with GC–MS. Thus, these compounds may either stem from the environment or be emitted by the skin due to the influence of food and cosmetics as well as frequently used flavouring agents. Benzaldehyde is an aroma compound used particularly in artificial cherry and almond flavours and is also a constituent of many other kinds of food like sausage and wine [28], [29]. 3-Methyl-2-butenal occurs naturally in several plants like the blackberry (up to 0.34%) [30] and wild ginger (0.05%) [31] and additionally in raw beef (0.36%) [32] and dry fermented sausage [33]. With regard to the other aldehydes, nonanal is a constituent of tea [34] and cooked black rice [35]. Octanal has been detected in orange juice [36] and orange essence oil [37], while decanal may contribute to the aroma of ham [38]. The fact that these compounds could not be detected in room air samples – with the exception of nonanal at concentrations below 0.5 ppb – may confirm the assumption that these three aldehydes might originate from peroxidation of skin lipids.
Fig. 6.

(a and b) Concentration profile of 3-methyl-2-butenal and benzaldehyde detected in skin gas samples.
The remaining compounds, 2-ethyl-hexanol, 6-methyl-5-hept-2-one and sec-butyl acetate, show no coherence in the skin profile of the different persons. Each of these compounds was also detected in the room air samples using IMS. Their mean concentrations were 0.9 ppb for 2-ethyl-hexanol, 1.3 ppb for 6-methyl-5-hept-2-one and 0.9 ppb for sec-butyl acetate. The latter was not found in the samples of Volunteers 6 or 7 and its maximal observed value amounted to 1.6 ppb (Volunteer 3). During the 1 h measurement time it shows a decreasing profile in almost every volunteer (see Fig. 7b). Thus, it is probable that sec-butyl acetate arises from the environment and the decrease in its concentration can be explained by the dilution of the nitrogen stream (3 ml/min) used for rinsing.
Fig. 7.

(a) Concentration profile of sec-butylacetate detected in gas samples collected above skin; (b) decrease of sec-butylacetate concentration during skin measurements.
Furthermore, 2-ethyl-hexanol and 6-methylhept-5-en-2-one can be exogenous contaminants, since they occur naturally in air are emitted by plants, moulds [39], several herbs and as a fragrance contained in cleaning agents [40]. In addition, 6-methylhept-5-en-2-one as an ingredient in essential oils extracted from lemon grass can be found in various cosmetics and foods [40]. Accordingly, the difference between the mean values of 2-ethyl-hexanol measured in skin samples is rather small at 1.2 ppb and 1.8 ppb (Fig. 8). In the case of 6-methylhept-5-en-2-one comparatively high concentration values with high SD (mean 4.1 ppb) were measured for Volunteer 7. From this it can be assumed that this compound is adsorbed on the skin surface and probably arises from cosmetics. While the experiments were performed in two labs, room air contamination would influence measurements in other volunteers and also result in higher concentration differences.
Fig. 8.

(a and b) Concentration profile of 2-ethyl-hexanol and 6-methylhept-5-en-2-one detected in skin samples.
Detection of 6-methylhept-5-en-2-one was confirmed by GC–MS analysis. In agreement with the IMS results the highest concentrations were observed in Volunteer 7 and the lowest in Volunteer 6. Although the data obtained with the various techniques show a tenfold difference (Table 3), concentration values show good agreement with regard to low concentration levels and the different blank concentration values.
Table 3.
Mean concentrations of compounds detected in comparative GC–MS and GC–IMS analyses of skin samples in case of three volunteers. Blank values of the particular substances are subtracted.
| Compound | CAS | Volunteer 5 mean |
Volunteer 6 mean |
Volunteer 7 mean |
|||
|---|---|---|---|---|---|---|---|
| GC–MS n = 2 |
GC–IMS n = 18 |
GC–MS n = 2 |
GC–IMS n = 12 |
GC–MS n = 2 |
GC–IMS n = 14 |
||
| Benzaldehyde | 100-52-7 | 0.0 | 0.8 | 10.1 | 0.9 | 3.8 | 1.5 |
| 5-Hepten-2-one, 6-methyl- | 110-93-0 | 5.2 | 0.5 | 2.0 | 0.2 | 11.5 | 4.1 |
| Octanal | 124-13-0 | 5.3 | 0.6 | 0.0 | 0.8 | 11.4 | 1.5 |
3.2. Reproducibility of skin measurements
Since the emission of volatile organic compounds is influenced by various factors, different tests for the determination of reproducibility were performed. Here, we were interested in investigating daily changes in compounds emanated by the skin as well as the influence of sampling place, and especially the effects of room air contaminants occurring at various concentrations levels. The first column in Table 4 shows standard errors of mean values of the detected compounds calculated from experiments performed in the same volunteer at the same time, but on different days. The highest standard error (36.66%) was calculated for 3-methyl-2-butenal, the smallest for benzaldehyde (9.37%). For octanal, nonanal and decanal, mean SE values were determined to be 16.73%, 12.44% and 11.02%, respectively.
Table 4.
Mean values and relative standard errors of means (RSE) for skin gas compounds in reproducibility tests. Experiments belonging to different tests were carried out by the same volunteer.
| Substance | Daily reproducibility (at the same time on different days) n = 38 |
Reproducibility of measurements at different times on different days n = 37 |
Reproducibility of measurements investigated in two different labs n = 25 |
|||
|---|---|---|---|---|---|---|
| Mean | RSE | Mean | RSE | Mean | RSE | |
| 3-Methyl-2-Butenal | 0.130 | 36.66% | 2.567 | 74.59% | 0.141 | 56.54% |
| Sec-butylacetate | 0.234 | 25.44% | 0.365 | 37.34% | 0.265 | 19.56% |
| Benzaldehyde | 0.344 | 9.37% | 0.685 | 29.07% | 0.369 | 9.64% |
| 6-Methylhept-5-en-2-one | 0.960 | 19.95% | 1.008 | 14.12% | 1.152 | 2.87% |
| Octanal | 0.579 | 16.73% | 2.899 | 10.74% | 0.667 | 10.94% |
| 2-Ethyl-hexanol | 0.515 | 9.40% | 1.094 | 4.04% | 0.482 | 12.72% |
| Nonanal | 1.386 | 12.44% | 7.133 | 15.08% | 1.558 | 1.17% |
| Decanal | 2.414 | 11.02% | 4.9293 | 6.28% | 2.176 | 9.43% |
Performing the tests at different times on different days revealed a higher SE for each of the compounds except 6-methylhept-5-en-2-one, 2-ethyl-hexanol and decanal. One reason for this could be the daily variation in room temperature at the sampling place. This variation influences the transpiration rate and thus the amount of VOCs released.
Interestingly, tests performed in the same volunteer but in two different labs showed a smaller SE. This confirms the necessity of purging of the metal pan prior to measurements in order to eliminate the majority of room air contaminants.
4. Conclusions
The present paper demonstrates that MCC–IMS can be used for near real-time monitoring of human skin emissions. The method is based on the high sensitivity of IMS for compounds from numerous chemical classes, like ketones, aldehydes, alcohols and esters. From more than twenty detected analytes, eight identified and quantified in samples taken from the skin head-space of 7 volunteers. Three of them octanal, nonanal and decanal, may originate as volatile metabolites produced by the skin. In this study a narrow demographic range with regard to age was set when examining the applicability of the method developed. Forthcoming studies will aim at investigations of age dependence of skin emission, changes in released VOC profiles after stress situations such as UV irradiation, skin rash or select skin diseases.
These preliminary results should encourage further research for a better understanding of the constitution and role of human scent and its basic biochemistry.
Acknowledgements
The research leading to these results has received funding from the European Community's Seventh Framework Program (FP7/2007-13) under grant agreement No. 217967 (“SGL for USaR” project, Second Generation Locator for Urban Search and Rescue Operations, www.sgl-eu.org). We appreciate funding from the Austrian Federal Ministry for Transport, Innovation and Technology (BMVIT/BMWA, Project No. 836308, KIRAS) and from the Austrian Agency for International Cooperation in Education and Research (OeAD) under Grant Agreement No. SPA/02-87/FEM_TRACE. We thank Elisabeth Niederstetter, Pascalle Maier, Tabea Halmschlager, Hannah-Sophia Feuerstein, Ann-Cathrine Sassmann, Julia Lovasz, Lilli-Ruth Fidler, Therese Sperlich for their work on the FEM_TRACE project. Dr. Veronika Ruzsanyi gratefully acknowledges a Lise-Meitner Fellowship from the Austrian Science Fund (FWF, Project No.: M1213).
We greatly appreciate the generous support of the Government of Vorarlbergand its Governor Dr Herbert Sausgruber.
Contributor Information
Veronika Ruzsanyi, Email: Veronika.Ruzsanyi@i-med.ac.at.
Anton Amann, Email: Anton.Amann@i-med.ac.at.
References
- 1.De Luca C., Valacchi G. Mediators Inflamm. 2010;2010:321494. doi: 10.1155/2010/321494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith K.R., Thiboutot D.M. J. Lipid Res. 2008;49:271. doi: 10.1194/jlr.R700015-JLR200. [DOI] [PubMed] [Google Scholar]
- 3.Gurr M.I. Blackwell Science Ltd; Oxford: 2002. Lipid Biochemistry. [Google Scholar]
- 4.Jezierski T., Sobczynska M., Walczak M., Gorecka-Bruzda A., Ensminger J. J. Forensic Sci. 2012 doi: 10.1111/j.1556-4029.2011.02029.x. [DOI] [PubMed] [Google Scholar]
- 5.Mochalski P., Agapiou A., Statheropoulos M., Amann A. Analyst. 2012;137:3278. doi: 10.1039/c2an35214a. [DOI] [PubMed] [Google Scholar]
- 6.Agapiou A., Mochalski P., Schmid A., Amann A. Elsevier; Amsterdam: 2012. Volatile Biomarkers: Non-invasive Diagnosis in Physiology and Medicine. [Google Scholar]
- 7.Huo R., Agapiou A., Bocos-Bintintan V., Brown L.J., Burns C., Creaser C.S., Devenport N.A., Gao-Lau B., Guallar-Hoyas C., Hildebrand L., Malkar A., Martin H.J., Moll V.H., Patel P., Ratiu A., Reynolds J.C., Sielemann S., Slodzynski R., Statheropoulos M., Turner M.A., Vautz W., Wright V.E., Thomas C.L.P. J. Breath Res. 2011;5 doi: 10.1088/1752-7155/5/4/046006. [DOI] [PubMed] [Google Scholar]
- 8.Gallagher M., Wysocki J., Leyden J.J., Spielman A.I., Sun X., Preti G. Brit. J. Dermatol. 2008;159:780. doi: 10.1111/j.1365-2133.2008.08748.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Z.M., Cai J.J., Ruan G.H., Li G.K. J. Chromatogr. B. 2005;822:244. doi: 10.1016/j.jchromb.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 10.Bernier U.R., Booth M.M., Yost R.A. Anal. Chem. 1999;71:1. doi: 10.1021/ac980990v. [DOI] [PubMed] [Google Scholar]
- 11.Bernier U.R., Kline D.L., Barnard D.R., Schreck C.E., Yost R.A. Anal. Chem. 2000;72:747. doi: 10.1021/ac990963k. [DOI] [PubMed] [Google Scholar]
- 12.Martinez-Lozano P., de la Mora J.F. J. Am. Soc. Mass Spectrom. 2009;20:1060. doi: 10.1016/j.jasms.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 13.Di Natale C., Macagnano A., Paolesse R., Tarizzo E., Mantini A., D’Amico A. Sens. Actuators B – Chem. 2000;65:216. [Google Scholar]
- 14.Ochoa M.L., Harrington P.B. Anal. Chem. 2004;76:985. doi: 10.1021/ac035123r. [DOI] [PubMed] [Google Scholar]
- 15.O’Donnell R.M., Sun X.B., Harrington P.D. TRAC – Trend Anal. Chem. 2008;27:44. [Google Scholar]
- 16.Mercer J., Shakleya D., Bell S. J. Anal. Toxicol. 2006;30:539. doi: 10.1093/jat/30.8.539. [DOI] [PubMed] [Google Scholar]
- 17.Borsdorf H., Rammler A. J. Chromatogr. A. 2005;1072:45. doi: 10.1016/j.chroma.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 18.Ruzsanyi V., Sielemann S., Baumbach J.I. J. Environ. Monit. 2007;9:61. doi: 10.1039/b613951e. [DOI] [PubMed] [Google Scholar]
- 19.Ruzsanyi V., Baumbach J.I., Sielemann S., Litterst P., Westhoff M., Fretag L. J. Chromatogr. A. 2005;1084:145. doi: 10.1016/j.chroma.2005.01.055. [DOI] [PubMed] [Google Scholar]
- 20.Westhoff M., Freitag P.L.L., Ruzsanyi V., Bader S., Urfer W., Baumbach J.I. Chest. 2005;128:155s. [Google Scholar]
- 21.Perl T., Carstens E., Hirn A., Quintel M., Vautz W., Nolte J., Junger M. Brit. J. Anaesth. 2009;103:822. doi: 10.1093/bja/aep312. [DOI] [PubMed] [Google Scholar]
- 22.Baumbach J.I., Eiceman G.A. Appl. Spectrosc. 1999;53:338A. doi: 10.1366/0003702991947847. [DOI] [PubMed] [Google Scholar]
- 23.Eiceman G.A.K., Karpas Z. 2nd edition. CRC Press; 2004. Ion Mobility Spectrometry. [Google Scholar]
- 24.Vautz W., Bödeker B., Baumbach J.I., Bader S., Westhoff M., Perl P. Int. J. Ion Mobil. Spectrom. 2009:47. [Google Scholar]
- 25.Fujisaki M., Endo Y., Fujimoto K. J. Am. Oil Chem. Soc. 2002;79:909. [Google Scholar]
- 26.Porter N.A., Weber B.A., Weenen H., Khan J.A. J. Am. Chem. Soc. 1980;102:5597. [Google Scholar]
- 27.C. Leray, http://www.cyberlipid.org/, 2011.
- 28.Genovese A., Gambuti A., Piombino P., Moio L. Food Chem. 2007;103:1228. doi: 10.1016/j.foodchem.2013.12.100. [DOI] [PubMed] [Google Scholar]
- 29.Zhao M.M., Sun W.Z., Zhao Q.Z., Zhao H.F., Yang B. Food Chem. 2010;121:319. [Google Scholar]
- 30.Georgilopoulos D.N., Gallois A.N. Z. Lebensm. Unters. For. 1987;185:299. doi: 10.1007/BF01123035. [DOI] [PubMed] [Google Scholar]
- 31.Motto M.G., Secord N.J. J. Agric. Food Chem. 1985;33:789. [Google Scholar]
- 32.King M.F., Hamilton B.L., Matthews M.A., Rule D.C., Field R.A. J. Agric. Food Chem. 1993;41:1974. [Google Scholar]
- 33.Marco A., Navarro J.L., Flores M. J. Agric. Food Chem. 2007;55:3058. doi: 10.1021/jf0631880. [DOI] [PubMed] [Google Scholar]
- 34.Qin P.Y., Ma T.J., Wu L., Shan F., Ren G.X. J. Food Sci. 2011;76:S401. doi: 10.1111/j.1750-3841.2011.02223.x. [DOI] [PubMed] [Google Scholar]
- 35.Yang D.S., Lee K.S., Jeong O.Y., Kim K.J., Kays S.J. J. Agric. Food Chem. 2008;56:235. doi: 10.1021/jf072360c. [DOI] [PubMed] [Google Scholar]
- 36.Qiao Y., Xie B.J., Zhang Y., Zhang Y., Fan G., Yao X.L., Pan S.Y. Molecules. 2008;13:1333. doi: 10.3390/molecules13061333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hognadottir A., Rouseff R.L. J. Chromatogr. A. 2003;998:201. doi: 10.1016/s0021-9673(03)00524-7. [DOI] [PubMed] [Google Scholar]
- 38.Song H., Cadwallader K.R. J. Food Sci. 2008;73:C29. doi: 10.1111/j.1750-3841.2007.00593.x. [DOI] [PubMed] [Google Scholar]
- 39.Van Lancker F., Adams A., Delmulle B., De Saeger S., Moretti A., Van Peteghem C., De Kimpe N. J. Environ. Monit. 2008;10:1127. doi: 10.1039/b808608g. [DOI] [PubMed] [Google Scholar]
- 40.Loumouamou A.N., Biassala E., Silou T. Adv. J. Food Sci. Technol. 2010;2:312. [Google Scholar]