

Abstract
NADPH oxidase (Nox) is a unique, multi-protein, electron transport system that produces large amounts of superoxide via the reduction of molecular oxygen. Nox-derived reactive oxygen species (ROS) are known to be involved in a variety of physiological processes, including host defense and signal transduction. However, over the past decade, the involvement of (Nox)-dependent oxidative stress in the pathophysiology of several neurodegenerative diseases has been increasingly recognized. ROS produced by Nox proteins contribute to neurodegenerative diseases through distinct mechanisms, such as oxidation of DNA, proteins, lipids, amino acids and metals, in addition to activation of redox-sensitive signaling pathways. In this review, we discuss the recent literature on Nox involvement in neurodegeneration, focusing on Parkinson and Alzheimer diseases.
Keywords: Alzheimer´s disease, neuroprotection, Nox enzymes, Parkinson´s disease, reactive oxygen species.
INTRODUCTION
Extensive research over the past few decades indicates that reactive oxygen species (ROS), key mediators of cellular oxidative stress and redox dysregulation, contribute to many pathological events of aging and major disease processes, including cancer [1], cardiovascular disorders [2], diabetes [3] and neurodegenerative diseases [4]. Accordingly, as described in detail in recent reviews [1, 2] a broad range of studies have been developed in animal models and have provided important insight into the involvement of ROS in pathological events in virtually all tissues. Endothelial nitric oxide synthase uncoupling, xanthine oxidase activation, mitochondrial respiration, peroxisome oxidases, cytochrome P-450, among other cellular sources, produce ROS as a byproduct of their biological activity [2]. Indeed, considering the nervous system particular vulnerability to oxidative stress, the expression of NADPH oxidase (Nox) family of superoxide (O2·−) and hydrogen peroxide (H2O2)-producing proteins in the brain tissue has been considered unlikely for a long time; however, in the past 15 years Nox family members and the ROS they produce have been implicated in a variety of neurological diseases [5]. This review summarizes current research on Nox involvement in neurodegeneration, with a focus on Parkinson and Alzheimer diseases.
REACTIVE OXYGEN SPECIES AND OXIDATIVE STRESS
ROS is a general designation of chemical species arising from oxygen reduction and their related precursors and/or reactive reaction products. These species can be classified into 2 groups of compounds, namely radicals and nonradicals. Superoxide ion radical (O2·−), hydroxyl radical (OH.), peroxyl (ROO.) and alkoxyl radicals (RO.), and one form of singlet oxygen (1O2) are among the species classified into the radical group because they contain at least 1 unpaired electron in the shells around the atomic nucleus and are capable of independent existence. The nonradical group of oxygen derivatives contains a large variety of molecules, which include the hypochlorous acid (HClO), ozone (O3), hydrogen peroxide (H2O2) and organic peroxides [6, 7].
A very important notion is the fact that properties such as reactivity, solubility, and diffusibility all make the physiological consequences of each specific ROS very distinct [8, 9]. ROS generation involves a cascade of reactions that generally starts with the production of O2·−, a molecule with relatively low reaction rates with biological components and unable to cross biological membranes. Oxygen reduction in the presence of a free electron generates O2·−. Under physiological pH, most of the O2·− is in its charged form hydroperoxyl (HO.2), which has higher reducing capacity, in comparison to O2·−, and which is able to more easily cross biological membranes. In a hydrophilic environment, both O2·− or HO.2 can reduce ferric ions (Fe+3) to ferrous ions (Fe+2), enabling them to undergo the Fenton reaction (see later) [7].
Superoxide spontaneously dismutate to H2O2, but this reaction can be catalyzed by superoxide dismutase. H2O2, in turn, can easily penetrate biological membranes and react with other species, leading to the generation of more deleterious reactive species, such as OH. or HClO, in the presence of myeloperoxidase. The oxidizing activity of H2O2 can also lead to deleterious chemical effects such as degradation of haem proteins; iron release; inactivation of enzymes; and oxidation of DNA, lipids, SH groups, and keto acids [5, 7]. However, catalase, glutathione peroxidase, and peroxidases all convert H2O2 to water and other metabolites within cells, even when H2O2 is present in very low concentrations [10]. Additionally, Fe+2 interacts with H2O2 to generate OH. through Fenton’s reaction. The OH. radical is considered to be a powerful oxidizing agent with extremely short life-span that can react at a high rate with organic and inorganic molecules in the cell, including DNA, proteins, lipids, amino acids and metals [10].
As the importance of oxidative stress in the pathophysiology of several neurodegenerative diseases is increasingly recognized, an important question is whether cellular ROS generation represents redox signaling as opposed to oxidative imbalance. Under normal conditions, the intracellular redox state, which implies the relative contribution of oxidation and reduction processes to the cell function, is constantly monitored and kept within a narrow range. The redox state plays a significant role in the regulation of signaling pathways, including kinase and phosphatase activity and gene expression through modulation of transcription factor function [e.g., nuclear factor kB (NFkB) and activator protein 1 (AP-1)] [7, 11]. Accumulating experimental evidence demonstrated that protein trafficking, synthesis, degradation and folding are also redox-sensitive processes [12]. The mechanisms responsible for regulating and maintaining the cellular redox homeostasis are not yet fully understood; however, it is widely accepted that alterations in this state toward lower (redosis) or higher (oxidosis) values might lead to several cellular deleterious processes [13, 14].
Considering both the toxic and beneficial ROS effects, Helmut Sies [15] defined, in 1985, oxidative stress as “a disturbance in the prooxidant– antioxidant balance in favor of the former”. More recent studies also suggested that alterations of ROS production may be restricted to specific cell compartments such as endosomes, caveolae, nucleus and do not necessarily imply changing the redox status of the major intracellular reductants glutathione or thioredoxin, or even the overall redox state of the cell [16, 17].
In addition to this view, the notion that mechanisms of oxidative stress affect several signaling/enzymatic mechanisms and are not limited to free radical damage to macromolecules, provided the basis for the contemporary definition of oxidative stress, as a condition where there is a disruption in the normal function of redox networks with or without free radical-induced macromolecular damage [9, 17].
NOX FAMILY MEMBERS AND SUBUNITIES
Early Nox research was carried out in polymorphonuclear neutrophils. Since Sbarra and Karnovsky first suggested the existence of such an enzyme in neutrophils, a great deal has been learned about the leukocyte oxidase [18]. Following stimulation, neutrophils undergo a respiratory burst characterized by a 20-fold increase in oxygen consumption, which is accompanied by an increase of glucose utilization and production of reduced NADPH by the pentose phosphate pathway [19]. In parallel with the knowledge produced by understanding the respiratory burst, Nox isoforms have been ascribed an important role in the pathology of cardiovascular disorders such as hypertension and atherosclerosis [20]. On the other hand, despite agreement about the potential importance of Nox enzymes in the pathogenesis of many neurodegenerative diseases, comparatively less is known about mechanisms underlying the regulation of Nox complex activity and expression in brain tissue.
The NADPH oxidase is a multi-subunit enzyme that transfers electrons across biological membranes. The subunits are localized both in the cell membrane (cytochrome b558, comprised of p22phox and gp91phox) and in the cytoplasm (p40phox, p47phox, and p67phox) [21]. Upon stimulation, activation of a low-molecular weight G protein (Rac1 or Rac2) and phosphorylation of p47phox initiates migration of the cytoplasmic elements to the plasma membrane [22]. The catalytic component gp91phox facilitates electron transfer. The electron from cytoplasmic NADPH travels first to flavin adenine dinucleotide (FAD), then through the Nox heme groups, and finally across the membrane and it is transferred to oxygen. Superoxide is the primary product of the electron transfer, but other downstream ROS can also be generated [5]. Seven Nox isoforms have been identified so far: Nox1, gp91phox (Nox2), Nox3, Nox4, Nox5, and Dual Oxidase 1 and 2 (Duox1 and Duox2). Nox 1–5 are known to produce O2·−, whereas Duox enzymes are able to release H2O2 without forming a detectable amount of O2·−, because they contain an extracellular peroxidase-like domain in addition to the EF-hand Ca2+ binding domains and gp91phox homology domain [23]. However, as elegantly demonstrated by Serrander and col. [24], in cell lines expressing Nox4 the type of ROS released was predominantly H2O2, whereas O2·− was almost undetectable. Although close structural and functional similarities exist between the different Nox homologues, each isoform seems to be differentially expressed and regulated across distinct tissue and cell types [25]. Specifically in the central nervous system, the presence of Nox1, Nox2, Nox3, and Nox4 isoforms has been identified in several structures (Table 1) [26]. However, very little is known about the role of Nox5 and Duox1 and 2 in the nervous tissue. Detailed mechanisms of activation for individual Nox enzymes described in the nervous system are discussed below.
Table 1.
NADPH Oxidases in the Brain
| Brain Region | Specie | RNA | Protein | References | |
|---|---|---|---|---|---|
| Nox1 | Medulla | Human | + | [85] | |
| Superior colliculus | Rat | + | [26] | ||
| Hippocampus | Rat | + | [84] | ||
| Cerebellum | Rat | + | [83] | ||
| Forebrain | Mice | + | [25] | ||
| Midbrain | Mice | + | [25] | ||
| Hindbrain | Mice | + | [25] | ||
| Dorsal root ganglion | Mice | + | [86] | ||
| Hypothalamus | Mice | + | [87] | ||
| Nox2 | Medulla | Human/Rat | + | [74,83] | |
| Superior colliculus | Rat | + | [26] | ||
| Thalamus | Mice/Rat | + | + | [26, 78] | |
| Hippocampus | Mice/Rat/Human | + | + | [58, 69, 70, 88, 90] | |
| Cerebellum | Mice | + | [80] | ||
| Forebrain | Mice | + | [25] | ||
| Midbrain | Mice | + | [25] | ||
| Hindbrain | Mice | + | [25] | ||
| Hypothalamus | Mice/Rat | + | + | [73, 74, 76] | |
| Substantia nigra | Mice/Rat | + | + | [ 75, 77] | |
| Amygdala | Mice | + | [78] | ||
| Nucleus of the solitary tract | Rat | + | [79] | ||
| Striatum | Mice/Rat | + | + | [71, 72,75] | |
| Cortex | Mice/Rat/Human | + | + | [24, 69, 89, 90] | |
| Brainstem | Mice | + | [70] | ||
| Nox3 | Cerebellum | Rat | + | [81, 83] | |
| Hypothalamus | Rat | + | [81] | ||
| Cortex | Rat | + | [81] | ||
| Nox4 | Superior colliculus | Rat | + | [26] | |
| Hypothalamus | Mice | + | [73] | ||
| Cortex | Mice | + | + | [61, 82] | |
| Forebrain | Mice | + | [25] | ||
| Midbrain | Mice | + | [25] | ||
| Hindbrain | Mice | + | [25] | ||
| Hippocampus | Mice | + | + | [82] | |
| Cerebellum | Mice | + | + | [82] |
Observation: several studies that reported Nox protein occurrence by immunostaining have not actually used the appropriate negative controls, and therefore the data must be interpreted with caution.
Nox isoforms have distinct activation mechanisms. Nox1 associates with the membrane subunit p22phox, and mostly with the cytosolic subunits, NoxO1 (p47phox homologue) and NoxA1 (p67phox homologue) and Rac [27, 28]. Nox2 interacts with p22phox, phosphorylated p47phox, p67phox, and Rac. Nox2 has also been found to complex with p40phox, but the functional consequences of this interaction are not clear [2]. Nox3 activation is less well defined, but it is believed to be similar to Nox1, involving Rac, p47phox, and NoxA1. Nox4 is unique among the catalytic Nox subunits in that it only interacts with p22phox, and it is thought to be constitutively active [2]. Nox1 mRNA was found to be expressed in neurons, astrocytes and microglia, whereas Nox4 was found in neurons. In activated microglia, ROS production is frequently associated with Nox2 expression. However, Nox1 and Nox4 might also play a role in that process [2].
NOX AND PARKINSON’S DISEASE
Parkinson’s disease (PD) is a neurodegenerative disorder characterized by a progressive loss of dopaminergic neurons in the nigrostriatal pathway of the brain, which triggers complex functional modifications within the basal ganglia circuitry, leading primarily to motor dysfunctions. Although the etiology of PD is unknown, a common element of most theories is the involvement of oxidative stress, either as a primary or secondary event of the disease [4, 29-31]. Research on the pathogenesis of PD suggested that mitochondrial dysfunction is the major source of oxidative stress in this disease [32]; however, increasing evidence has been also found for a role of Nox enzymes in the process.
The research on the mechanisms involved in PD disease has relied on the development of animal models that reproduce the pathological and behavioral characteristics of the disease. Classically, these models are based on the systemic or intracerebral administration of neurotoxins capable of selectively degenerate the nigrostriatal system. A very useful model is based on systemic administration or striatal injection of 1-methyl-4-phenyl-1,2,3,6-tetrahydro-pyridine (MPTP), which causes a PD-like syndrome highly similar to the human disease [33-35]. As demonstrated under in vivo conditions, translocation of p67phox was induced by MPTP in mouse brain and prevented by the tetracycline derivative minocycline [36]. More recently, it has been demonstrated that p47phox phosphorylation and p47phox–gp91phox complexes are significantly increased in mice substantia nigra (SN) after systemic injections of MPTP [37]. In addition, MPTP induced increases of both gp91phox and 3-nitrotyrosine in the SN of ageing mice, which were inhibited by oral treatment with the NO-donating derivative of flurbiprofen [2-fluoro-α-methyl (1,1'-biphenyl)-4-acetic-4-(nitrooxy)butyl ester (HCT1026)] [38]. In line with these findings, degeneration of dopaminergic neurons induced by MPTP was attenuated in gp91phox-/- mice as compared to Wt littermates [39]. In the same PD model, gp91phox immunoreactivity colocalizes with microglial cell markers but not with astrocyte markers, confirming a microglial origin for Nox [40]. In vitro overnight MPP+ (a MPTP metabolite) treatment of N27 rat dopaminergic cells was able to induce Nox2 protein expression and O2·− generation, as measured by flow cytometric detection. This effect was inhibited by siRNA silencing of p22phox [41]. Thus, it appears that activation of Nox2 plays a relevant role in the loss of dopaminergic neurons in the MPTP-induced PD model.
Another commonly used procedure for obtaining experimental nigrostriatal lesion in rodents is based on local infusion of 6-hydroxydopamine (6-OHDA) (reviewed in [42]). The biological effects of 6-OHDA are mainly related to the massive oxidative stress caused by the toxin that, once accumulated in the cytosol, seems to be auto-oxidated, promoting a high rate of free radical generation [43]. As detected by dihydroethidium fluorescence, the treatment of primary mesencephalic cultures with 6-OHDA induced a significant increase of the intracellular generation of O2·− in dopaminergic neurons, as well as in microglial cells [44]. There is evidence to implicate Nox-derived ROS in this process, but the mechanisms involved are poorly understood. Important advances in this regard were provided by a series of recent studies. For instance, data from our laboratory suggest a relevant role for Nox2 in 6-OHDA-induced PD. In this study, the membrane protein levels of p67phox were markedly elevated in the SN of 6-OHDA lesioned mice, suggesting that the p67phox subunit translocated from the cytosol to the plasma membrane, thus forming a Nox entity capable of producing superoxide after 6-OHDA injection. Tyrosine hydroxylase immunolabeling indicated that gp91phox-/- mice appear to be protected from dopaminergic cell loss in the SN and from dopaminergic terminal loss in the striatum. Moreover, wild type mice treated with apocynin, a Nox inhibitor [45], and gp91phox-/- mice all exhibited significantly ameliorated apomorphine-induced rotational behavior after 6-OHDA lesion (Hernandes et al., submitted). These results are corroborated by some in vitro observations. In rat primary mesencephalic cultures, 6-OHDA induced a significant increase of gp91phox and p47phox immunolabeling. Confocal microscopy revealed that both gp91phox and p47phox were intensely expressed in microglia cells. Microglial activation and O2·− generation in dopaminergic neurons were significantly reduced by apocynin [46]. Six-OHDA also induced increased expression of gp91phox in human dopaminergic neuroblastoma cells [47]. In addition, it has been recently reported that striatal injection of 6-OHDA increased Nox1 expression in dopaminergic neurons of the rat SN. Rac1, a key regulator in the Nox1 system, was also activated. Nox1 was localized into the nucleus, and immunostaining for a DNA oxidative stress marker, 8-oxo-dG, was increased. Adeno-associated virus-mediated Nox1 knockdown and Rac1 inhibition were both able to reduce 6-OHDA-induced oxidative DNA damage and dopaminergic neuronal degeneration [48].
Nevertheless, the role of Nox enzymes in 6-OHDA-induced PD might not be only limited to the Nox2 isoform. As recently demonstrated, striatal administration of 6-OHDA increased Nox1 expression in dopaminergic neurons of the SN. Furthermore, adeno-associated virus-mediated Nox1 knockdown reduced 6-OHDA-induced oxidative DNA damage and dopaminergic neuronal degeneration in the rat SN [48].
The involvement of Nox in PD has also been revealed through other, unrelated PD models. In mesencephalic primary cultures, activated microglia generated Noxderived superoxide and enhanced lipopolysaccharide-elicited dopaminergic neurodegeneration [49]. Furthermore, microglial Nox but not neuronal Nox, renders dopaminergic neurons more sensitive to rotenone, an herbicide able to reproduce features of PD in rats [50]. In mesencephalic neuron-glia cultures from gp91phox-/- mice the deleterious effect of microglia induced by substance P on tyrosine hydroxylase-positive neurons was significantly attenuated [51]. The Nox involvement in the cytotoxic action of paraquat, another widely used parkinsonism inducing agent, has also been recently described. Apocynin attenuated paraquat-induced dopaminergic degeneration, Nox activation, cytochrome c release and caspases-9/-3 and microglia activation. According to the authors, paraquat induces oxidative stress through Nox activation and depletion of glutathione, which in turn activate the apoptotic machinery leading to dopaminergic neurodegeneration [52]. It has been also reported that the Nox inhibitor diphenyleneiodonium (DPI) blocked the paraquat-induced ROS production and subsequent dopaminergic neurodegeneration [53].
NOX AND ALZHEIMER’S DISEASE
Alzheimer’s disease (AD) is characterized by an initial mild cognitive impairment that progressively develops into a loss of higher cognitive functions, resulting in dementia. Accumulation of amyloid-β peptide (Aβ) in the brain is considered one of the main pathological features of AD. Other AD microscopic hallmarks include abnormal protein folding, exacerbated activation of glial cells, and synaptic and neuronal loss [54]. Importantly, a growing body of evidence supports a role for abnormal Nox activation in this pathology. Activation of Nox2 in the brain of AD subjects has been demonstrated, as evaluated by the translocation of Nox2 subunits [55]. In addition, analysis of frontal lobe tissue of AD patients demonstrated significantly increased levels of Nox1 and Nox3 mRNAs, suggesting that other isoforms beyond Nox2 can contribute to that neuropathology [56]. There is also evidence that Nox-associated redox pathways might participate in the early pathogenesis of AD. By using a luminescent assay to detect Nox-dependent ROS production, it was shown that Nox activity is increased in the superior/middle temporal gyri over control levels at the earliest clinical manifestations of disease, but not in late-stage AD. The observed increases of Nox activity were associated with increased expression of p47phox and gp91phox in both microglia and neurons [57].
However, in vitro and in vivo studies have generated most of the information related to the role of Nox enzymes in AD pathogenesis. It has been recently shown that the cholesterol oxidation product, 24-hydroxycholesterol, markedly potentiates the pro-apoptotic and pro-necrogenic effects of Aβ. This effect depends on its strong enhancement of the intracellular generation of Nox-derived ROS, mainly H2O2, and the consequent impairment of the neuronal redox state, measured in terms of the GSSG/GSH ratio [58]. Gp91ds-tat, a Nox2 inhibitory peptide, decreased both oxidative stress and AD pathology in aged mice [59]. Also, It has been recently shown that feeding AβPP/PS1 double transgenic mice, a mouse model of AD, with a diet containing phenolic antioxidant tert-butylhydroquinone, inhibits Nox2 protein expression and suppressed lipid peroxidation in the cerebral cortex and hippocampus [60]. It was also found that age-dependent increases of Aβ had a significant linear relationship with both Nox4 activity and cognitive performance in “humanized” APP×PS1 knock-in mice [61]. Apocynin treatment reduces Aβ deposition and the number of microglial cells in the cortex and hippocampus of aged transgenic mice overexpressing the human amyloid precursor protein (hAPP (751)(SL), but it failed to inhibit cytosolic p67PHOX translocation to the membrane and to reduce the levels of TNFα [62]. Similarly, apocynin did not improve cognitive and synaptic deficits, and did not decrease Aβ deposition, microgliosis and hyperphosphorylated tau in transgenic AD mice [63]. In vitro exposure of hippocampal neuronal/glial co-cultures to Aβ peptides resulted in activation of glial Nox, followed by neurodegeneration [64]. In another in vitro study using a co-culture of microglia and neuroblastoma cells over-expressing the Aβ precursor protein (APP), ROS generated by microglia induced neurodegeneration. This effect was attenuated by ROS-scavengers and was dose-dependently inhibited by DPI, suggesting that APP-dependent microglia activation and subsequent ROS generation by Nox play a crucial role in neuronal degeneration [65]. In addition, some studies demontrated that Aβ-induced Nox2 activation in astrocytes contributes to neurodegeneration [66, 67]. Aside from its involvement in neurodegeneration, production of H2O2 from Nox2 regulated microglial proliferation induced by Aβ, as demonstrated in a primary mixed glial culture obtained from rat cerebral cortex. This effect was prevented by apocynin and catalase, a H2O2-degrading enzyme [68]. In summary, strong evidence indicates that oxidative stress in AD involves ROS generation by Nox enzymes, in particular Nox2.
CONCLUSIONS
Regardless of the general agreement on the potential importance of ROS-generating Nox enzymes in the pathogenesis of many neurodegenerative diseases, our knowledge on the specific molecular mechanisms of activation and subsequent functional consequences of activating specific Nox enzymes in the brain tissue is limited. In addition to examining the local expression and activation of Nox enzymes under neurodegenerative conditions, the cellular consequences of a chronically dysregulated oxidative environment must also be taken into account. For instance, it is necessary to evaluate the ROS-dependent activation of inducible transcription factors and modulation of gene expression. Indeed, as nonspecific scavenging may prevent ROS from acting in essential biochemical pathways, that knowledge is essential to allow and improve the development of specific novel therapies targeting Nox proteins, therefore reducing the pathological consequences of oxidative stress.
GRANT INFO
Work in the author’s laboratory is supported by grants from FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo), CAPES (Coordenação de Aperfeiçoamento de Pessoal de Ensino Superior), University of São Paulo-Núcleo de Apoio a Pesquisa em Neurociência Aplicada - and CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico). M.S.H. is the recipient of a fellowship from FAPESP.
Fig. (1).
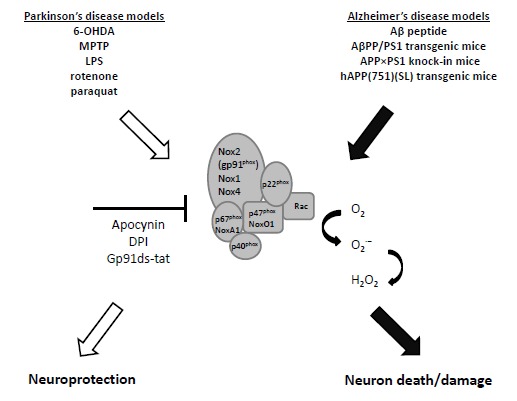
Nox activation, neurodegeneration and neuroprotection. In Parkinson´s and Alzheimer´s disease rodent models, increased activation of the Nox family of ROS-producing proteins contributes to neurodegeneration. Blockade of Nox generates neuroprotection in some instances. Abbreviations: 6-hydroxydopamine (6-OHDA); 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine (MPTP); Lipopolysaccharide (LPS); amyloid-β peptide (Aβ); superoxide (O2·−); hydrogen peroxide (H2O2) and diphenyleneiodonium (DPI).
ACKNOWLEDGEMENTS
Declared none.
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflict of interest.
REFERENCES
- 1.Wondrak GT. Redox-directed cancer therapeutics: molecular mechanisms and opportunities. Antioxid. Redox Signal. 2009;11(12):3013–69. doi: 10.1089/ars.2009.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic. Biol. Med. 2009;47(9):1239–53. doi: 10.1016/j.freeradbiomed.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gobbo MG, Ribeiro DL, Taboga SR, de Almeida EA, Góes RM. Oxidative stress markers and apoptosis in the prostate of diabetic rats and the influence of vitamin C treatment. J Cell Biochem. 2012;113(7):2223–33. doi: 10.1002/jcb.24092. [DOI] [PubMed] [Google Scholar]
- 4.Boll MC, Alcaraz-Zubeldia M, Rios C. Medical management of Parkinson's disease: focus on neuroprotection. Curr. Neuropharmacol. 2011;9(2):350–9. doi: 10.2174/157015911795596577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 6.Halliwell B.B. GJMC. Free Radicals in Biology and Medicine. 3rd. Oxford: Clarenton Press; 1999. [Google Scholar]
- 7.Kohen R, Nyska A. Oxidation of biological systems: oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol. Pathol. 2002;30(6):620–50. doi: 10.1080/01926230290166724. [DOI] [PubMed] [Google Scholar]
- 8.Santos CX, Anaka LY, Wosniak J, Laurindo FR. Mechanisms and Implications of Reactive Oxygen Species Generation During the Unfolded Protein Response: Roles of Endoplasmic Reticulum Oxidoreductases, Mitochondrial Electron Transport and NADPH Oxidase. Antioxid. Redox Signal. 2009;10:2409–27. doi: 10.1089/ars.2009.2625. [DOI] [PubMed] [Google Scholar]
- 9.Jones DP. Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol. 2008;295(4):849–68. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fridovich I. Superoxide anion radical (O2-.), superoxide dismutases, and related matters. J. Biol. Chem. 1997;272(30):18515–7. doi: 10.1074/jbc.272.30.18515. [DOI] [PubMed] [Google Scholar]
- 11.Trachootham D, Lu W, Ogasawara MA, Nilsa RD, Huang P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008;10(8):1343–74. doi: 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bardwell JC. The dance of disulfide formation. Nat. Struct. Mol. Biol. 2004;11(7):582–3. doi: 10.1038/nsmb0704-582. [DOI] [PubMed] [Google Scholar]
- 13.Chen J, Ji X, Hu X, Luo Y, Zhang L, Li P, Liu X, Yan F, Vosler P, Gao Y, Stetler RA, Chen J. Transgenic overexpression of peroxiredoxin-2 attenuates ischemic neuronal injury via suppression of a redox-sensitive pro-death signaling pathway. Antioxid. Redox Signal. 2012;17(5):719–32. doi: 10.1089/ars.2011.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rouillon JD, Bernard C, Robert G, Magnin P. Oxidation-reduction potential studies: a new method in pharmacology principles, materials, and methods. J. Pharmacol. Methods. 1987;17(2):179–84. doi: 10.1016/0160-5402(87)90029-5. [DOI] [PubMed] [Google Scholar]
- 15.Sies H. Oxidative Stress: Introductory Remarks. London: Academic Press; 1985. [Google Scholar]
- 16.Babior BM. NADPH oxidase: an update. Blood. 1999;93(5):1464–76. [PubMed] [Google Scholar]
- 17.Jones DP. Redefining oxidative stress. Antioxid. Redox Signal. 2006;8(9-10):1865–79. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- 18.Karnovsky MBL. Phagocytosis—Past and Future. New York: Academic; 1982. [Google Scholar]
- 19.Babior BM, Lambeth JD, Nauseef W. The neutrophil NADPH oxidase. Arch. Biochem. Biophys. 2002;397(2):342–4. doi: 10.1006/abbi.2001.2642. [DOI] [PubMed] [Google Scholar]
- 20.Laurindo FR, Fernandes DC, Amanso AM, Lopes LR, Santos CX. Novel role of protein disulfide isomerase in the regulation of NADPH oxidase activity: pathophysiological implications in vascular diseases. Antioxid. Redox Signal. 2008;10(6):1101–13. doi: 10.1089/ars.2007.2011. [DOI] [PubMed] [Google Scholar]
- 21.Lopes LR, Dagher MC, Gutierrez A, Young B, Bouin AP, Fuchs A, Babior BM. Phosphorylated p40PHOX as a negative regulator of NADPH oxidase. Biochemistry. 2004;43(12):3723–30. doi: 10.1021/bi035636s. [DOI] [PubMed] [Google Scholar]
- 22.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004;4(3):181–9. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 23.Banfi B, Molnár G, Maturana A, Steger K, Hegedûs B, Demaurex N, Krause KH. A Ca(2+)-activated NADPH oxidase in testis, spleen, and lymph nodes. J. Biol. Chem. 2001;276(40):37594–601. doi: 10.1074/jbc.M103034200. [DOI] [PubMed] [Google Scholar]
- 24.Serrander L, Cartier L, Bedard K, Banfi B, Lardy B, Plastre O, Sienkiewicz A, Fórró L, Schlegel W, Krause KH. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem. J. 2007;406(1):105–14. doi: 10.1042/BJ20061903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Infanger DW, Sharma RV, Davisson RL. NADPH oxidases of the brain: distribution, regulation, and function. Antioxid Redox Signal. 2006. pp. 1583–96. [DOI] [PubMed]
- 26.Hernandes MS, Britto LR, Real CC, Martins DO, Lopes LR. Reactive oxygen species and the structural remodeling of the visual system after ocular enucleation. Neuroscience. 2010;170(4):1249–60. doi: 10.1016/j.neuroscience.2010.07.065. [DOI] [PubMed] [Google Scholar]
- 27.Banfi B, Clark RA, Steger K, Krause KH. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J. Biol. Chem. 2003;278(6):3510–3. doi: 10.1074/jbc.C200613200. [DOI] [PubMed] [Google Scholar]
- 28.Geiszt M, Lekstrom K, Brenner S, Hewitt SM, Dana R, Malech H L, Leto TL. NAD(P)H oxidase 1, a product of differentiated colon epithelial cells, can partially replace glycoprotein 91phox in the regulated production of superoxide by phagocytes. J. Immunol. 2003;171(1):299–306. doi: 10.4049/jimmunol.171.1.299. [DOI] [PubMed] [Google Scholar]
- 29.Greenamyre JT and Hastings TG, Hastings TG. Biomedicine. Parkinson's--divergent causes, convergent mechanisms. Science. 2004;304(5674):1120–2. doi: 10.1126/science.1098966. [DOI] [PubMed] [Google Scholar]
- 30.Jenner P. Oxidative stress in Parkinson's disease. Ann. Neurol. 2003;53 Suppl 3:S26–36. doi: 10.1002/ana.10483. discussion S36-8. [DOI] [PubMed] [Google Scholar]
- 31.Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009;7(1):65–74. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mudo G, Mäkelä J, Di Liberto V, Tselykh TV, Olivieri M, Piepponen P, Eriksson O, Mälkiä A, Bonomo A, Kairisalo M, Aguirre JA, Korhonen L, Belluardo N, Lindholm D. Transgenic expression and activation of PGC-1alpha protect dopaminergic neurons in the MPTP mouse model of Parkinson's disease. Cell Mol. Life Sci. 2012;69(7):1153–65. doi: 10.1007/s00018-011-0850-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.George JL, Mok S, Moses D, Wilkins S, Bush AI, Cherny RA, Finkelstein DI. Targeting the progression of Parkinson's disease. Curr. Neuropharmacol. 2009;7(1):9–36. doi: 10.2174/157015909787602814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tian L, Karimi M, Loftin SK, Brown CA, Xia H, Xu J, Mach RH, Perlmutter JS. No differential regulation of dopamine transporter (DAT) and vesicular monoamine transporter 2 (VMAT2) binding in a primate model of Parkinson disease. PLoS One. 2012;7(2):e31439. doi: 10.1371/journal.pone.0031439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van den Berge SA, van Strien ME, Korecka JA, Dijkstra AA, Sluijs JA, Kooijman L, Eggers R, De Filippis L, Vescovi AL, Verhaagen J, van de Berg WD, Hol EM. The proliferative capacity of the subventricular zone is maintained in the parkinsonian brain. Brain. 2011;134(Pt 11):3249–63. doi: 10.1093/brain/awr256. [DOI] [PubMed] [Google Scholar]
- 36.Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski. S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J. Neurosci. 2002;22(5):1763–71. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huh SH, Chung YC, Piao Y, Jin MY, Son HJ, Yoon NS, Hong JY, Pak YK, Kim YS, Hong JK, Hwang O, Jin BK. Ethyl pyruvate rescues nigrostriatal dopaminergic neurons by regulating glial activation in a mouse model of Parkinson's disease. J. Immunol. 2011;187(2):960–9. doi: 10.4049/jimmunol.1100009. [DOI] [PubMed] [Google Scholar]
- 38.L'Episcopo F, Tirolo C, Caniglia S, Testa N, Serra PA, Impagnatiello F, Morale MC, Marchetti B. Combining nitric oxide release with anti-inflammatory activity preserves nigrostriatal dopaminergic innervation and prevents motor impairment in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. J. Neuroinflam. 2010;7:83. doi: 10.1186/1742-2094-7-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang W, Wang T, Qin L, Gao HM, Wilson B, Ali SF, Zhang W, Hong JS, Liu B. Neuroprotective effect of dextromethorphan in the MPTP Parkinson's disease model: role of NADPH oxidase. FASEB J. 2004;18(3):589–91. doi: 10.1096/fj.03-0983fje. [DOI] [PubMed] [Google Scholar]
- 40.Wu DC, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, Przedborski S. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Proc. Natl. Acad. Sci. U. S. A. 2003;100(10):6145–50. doi: 10.1073/pnas.0937239100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zawada WM, Banninger GP, Thornton J, Marriott B, Cantu D, Rachubinsk AL, Das M, Griffin WS, Jones SM. Generation of reactive oxygen species in 1-methyl-4-phenylpyridinium (MPP+) treated dopaminergic neurons occurs as an NADPH oxidase-dependent two-wave cascade. J. Neuroinflam. 2011;8:129. doi: 10.1186/1742-2094-8-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reiter RJ, Manchester LC, DX Tan. Neurotoxins: free radical mechanisms and melatonin protection. Curr. Neuropharmacol. 2010;8(3):194–210. doi: 10.2174/157015910792246236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schober A. Classic toxin-induced animal models of Parkinson's disease: 6-OHDA and MPTP. Cell Tissue Res. 2004;318(1):215–24. doi: 10.1007/s00441-004-0938-y. [DOI] [PubMed] [Google Scholar]
- 44.Rodriguez-Pallares J, Rey P, Parga JA, Muñoz A, Guerra MJ, Labandeira-Garcia JL. Brain angiotensin enhances dopaminergic cell death via microglial activation and NADPH-derived ROS. Neurobiol. Dis. 2008;31(1):58–73. doi: 10.1016/j.nbd.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 45.Pestana RR, Kinjo ER, Hernandes MS, Britto LR. Reactive oxygen species generated by NADPH oxidase are involved in neurodegeneration in the pilocarpine model of temporal lobe epilepsy. Neurosci. Lett. 2010;484(3):87–91. doi: 10.1016/j.neulet.2010.08.049. [DOI] [PubMed] [Google Scholar]
- 46.Rodriguez-Pallares J, Parga JA, Muñoz A, Rey P, Guerra MJ, Labandeira-Garcia JL. Mechanism of 6-hydroxydopamine neurotoxicity: the role of NADPH oxidase and microglial activation in 6-hydroxydopamine-induced degeneration of dopaminergic neurons. J. Neurochem. 2007;103(1):145–56. doi: 10.1111/j.1471-4159.2007.04699.x. [DOI] [PubMed] [Google Scholar]
- 47.Lin YC, Uang HW, Lin RJ, Chen IJ, Lo YC. Neuroprotective effects of glyceryl nonivamide against microglia-like cells and 6-hydroxydopamine-induced neurotoxicity in SH-SY5Y human dopaminergic neuroblastoma cells. J. Pharmacol. Exp. Ther. 2007;323(3):877–87. doi: 10.1124/jpet.107.125955. [DOI] [PubMed] [Google Scholar]
- 48.Choi DH, Cristóvão AC, Guhathakurta S, Lee J, Joh TH, Beal MF, Kim YS. NADPH oxidase 1-mediated oxidative stress leads to dopamine neuron death in Parkinson's disease. Antioxid. Redox Signal. 2012;16(10):1033–45. doi: 10.1089/ars.2011.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao HM, Jiang J, Wilson B, Zhang W, Hong JS, Liu B. Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: relevance to Parkinson's disease. J. Neurochem. 2002;81(6):1285–97. doi: 10.1046/j.1471-4159.2002.00928.x. [DOI] [PubMed] [Google Scholar]
- 50.Gao HM, Liu B, Hong JS. Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. J. Neurosci. 2003;23(15):6181–7. doi: 10.1523/JNEUROSCI.23-15-06181.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Block ML, Li G, Qin L, Wu X, Pei Z, Wang T, Wilson B, Yang J, Hong JS. Potent regulation of microglia-derived oxidative stress and dopaminergic neuron survival: substance P vs. dynorphin. FASEB J. 2006;20(2):251–8. doi: 10.1096/fj.05-4553com. [DOI] [PubMed] [Google Scholar]
- 52.Kumar A, Singh BK, Ahmad I, Shukla S, Patel DK, Srivastava G, Kumar V, Pandey HP, Singh C. Involvement of NADPH oxidase and glutathione in zinc-induced dopaminergic neurodegeneration in rats: similarity with paraquat neurotoxicity. Brain Res. 2012;1438:48–64. doi: 10.1016/j.brainres.2011.12.028. [DOI] [PubMed] [Google Scholar]
- 53.Kim S, Hwang J, Lee WH, Hwang DY, Suk K. Role of protein kinase Cdelta in paraquat-induced glial cell death. J. Neurosci. Res. 2008;86(9):2062–70. doi: 10.1002/jnr.21643. [DOI] [PubMed] [Google Scholar]
- 54.Graham RK, Ehrnhoefer DE, Hayden MR. Caspase-6 and neurodegeneration. Trends Neurosci. 2011;34(12):646–56. doi: 10.1016/j.tins.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 55.Shimohama S, Tanino H, Kawakami N, Okamura N, Kodama H, Yamaguchi T, Hayakawa T, Nunomura A, Chiba S, Perry G, Smith MA, Fujimoto S. Activation of NADPH oxidase in Alzheimer's disease brains. Biochem. Biophys. Res. Commun. 2000;273(1):5–9. doi: 10.1006/bbrc.2000.2897. [DOI] [PubMed] [Google Scholar]
- 56.de la Monte SM, Wands JR. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer's disease. J. Alzheimers Dis. 2006;9(2):167–81. doi: 10.3233/jad-2006-9209. [DOI] [PubMed] [Google Scholar]
- 57.Bruce-Keller AJ, Gupta S, Parrino TE, Knight AG, Ebenezer PJ, Weidner AM, LeVine H, 3rd, Keller JN, Markesbery WR. NOX activity is increased in mild cognitive impairment. Antioxid. Redox Signal. 2010;12(12):1371–82. doi: 10.1089/ars.2009.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gamba P, Leonarduzzi G, Tamagno E, Guglielmotto M, Testa G, Sottero B, Gargiulo S, Biasi F, Mauro A, Viña J, Poli G. Interaction between 24-hydroxycholesterol, oxidative stress, and amyloid-beta in amplifying neuronal damage in Alzheimer's disease: three partners in crime. Aging Cell. 2011;10(3):403–17. doi: 10.1111/j.1474-9726.2011.00681.x. [DOI] [PubMed] [Google Scholar]
- 59.Park L, Zhou P, Pitstick R, Capone C, Anrather J, Norris EH, Younkin L, Younkin S, Carlson G, McEwen BS, Iadecola C. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc. Natl. Acad. Sci. U. S. A. 2008;105(4):1347–52. doi: 10.1073/pnas.0711568105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Akhter H, Katre A, Li L, Liu X, Liu RM. Therapeutic potential and anti-amyloidosis mechanisms of tert-butylhydroquinone for Alzheimer's disease. J. Alzheimers Dis. 2011;26(4):767–78. doi: 10.3233/JAD-2011-110512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bruce-Keller AJ, Gupta S, Knight AG, Beckett TL, McMullen JM, Davis PR, Murphy MP, Van Eldik LJ, St Clair D, Keller JN. Cognitive impairment in humanized APPxPS1 mice is linked to Abeta(1-42) and NOX activation. Neurobiol. Dis. 2011;44(3):317–26. doi: 10.1016/j.nbd.2011.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lull ME, Lull ME, Levesque S, Surace MJ, Block ML. Chronic apocynin treatment attenuates beta amyloid plaque size and microglial number in hAPP(751)(SL) mice. PLoS One. 2011;6(5):e20153. doi: 10.1371/journal.pone.0020153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dumont M, Stack C, Elipenhali C, Calingasan NY, Wille E, Beal MF. Apocynin administration does not improve behavioral and neuropathological deficits in a transgenic mouse model of Alzheimer's disease. Neurosci. Lett. 2011;492(3):150–4. doi: 10.1016/j.neulet.2011.01.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Abeti R, Abramov AY, Duchen MR. Beta-amyloid activates PARP causing astrocytic metabolic failure and neuronal death. Brain. 2011;134(Pt 6):1658–72. doi: 10.1093/brain/awr104. [DOI] [PubMed] [Google Scholar]
- 65.Qin B, Cartier L, Dubois-Dauphin M, Li B, Serrander L, Krause KH. A key role for the microglial NADPH oxidase in APP-dependent killing of neurons. Neurobiol. Aging. 2006;27(11):1577–87. doi: 10.1016/j.neurobiolaging.2005.09.036. [DOI] [PubMed] [Google Scholar]
- 66.Abramov AY, Canevari L, Duchen MR. Calcium signals induced by amyloid beta peptide and their consequences in neurons and astrocytes in culture. Biochim. Biophys. Acta. 2004;1742(1-3):81–7. doi: 10.1016/j.bbamcr.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 67.Abramov AY, Duchen MR. The role of an astrocytic NADPH oxidase in the neurotoxicity of amyloid beta peptides. Philos. Trans. R Soc. Lond B Biol. Sci. 2005;360(1464):2309–14. doi: 10.1098/rstb.2005.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jekabsone A, Mander PK, Tickler A, Sharpe M, Brown GC. Fibrillar beta-amyloid peptide Abeta1-40 activates microglial proliferation via stimulating TNF-alpha release and H2O2 derived from NADPH oxidase: a cell culture study. J. Neuroinflamm. 2006;3:24. doi: 10.1186/1742-2094-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kim MJ, Shin K S, Chung Y B, Jung K W, Cha C I, Shin D H. Immunohistochemical study of p47Phox and gp91Phox distributions in rat brain. Brain Res. 2005;1040(1-2 ):178–86. doi: 10.1016/j.brainres.2005.01.066. [DOI] [PubMed] [Google Scholar]
- 70. Tejada-Simon MV, Serrano F, Villasana L E, Kanterewicz B I, Wu G.Y, Quinn MT, Klann E. Synaptic localization of a functional NADPH oxidase in the mouse hippocampus. Mol. Cell Neurosci. 2005;29(1 ):97–106. doi: 10.1016/j.mcn.2005.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kumar A, Singh B K, Ahmad I, Shukla S, Patel D K, Srivastava G, Kumar V, Pandey H P, Singh C. Involvement of NADPH oxidase and glutathione in zinc-induced dopaminergic neurodegeneration in rats: similarity with paraquat neurotoxicity. Brain Res. 2012;1438:48–64. doi: 10.1016/j.brainres.2011.12.028. [DOI] [PubMed] [Google Scholar]
- 72. Guemez-Gamboa A, Estrada-Sanchez A M, Montiel T, Paramo B, Massieu L, Moran J. Activation of NOX2 by the stimulation of ionotropic and metabotropic glutamate receptors contributes to glutamate neurotoxicity in vivo through the production of reactive oxygen species and calpain activation. J. Neuropathol. Exp. Neurol. 2011;70(11 ):1020–35. doi: 10.1097/NEN.0b013e3182358e4e. [DOI] [PubMed] [Google Scholar]
- 73. Xue B, Beltz T G, Johnson R F, Guo F, Hay M, Johnson A K. PVN adenovirus-siRNA injections silencing either NOX2 or NOX4 attenuate aldosterone/NaCl-induced hypertension in mice. Am. J. Physiol. Heart Circ. Physiol. 2012;302(3 ):H733–41. doi: 10.1152/ajpheart.00873.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Agarwal D, Welsch M A, Keller J N, Francis J. Chronic exercise modulates RAS components and improves balance between pro- and antiinflammatory cytokines in the brain of SHR. Basic Res. Cardiol. 2011;106(6 ):1069–85. doi: 10.1007/s00395-011-0231-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Binukumar BK, Bal A, Gill KD. Chronic dichlorvos exposure: microglial activation, proinflammatory cytokines and damage to nigrostriatal dopaminergic system. Neuromol.Med. 2011;13(4 ):251–65. doi: 10.1007/s12017-011-8156-8. [DOI] [PubMed] [Google Scholar]
- 76. Huang BS, Zheng H, Tan J, Patel K P, Leenen F H. Regulation of hypothalamic renin-angiotensin system and oxidative stress by aldosterone. Exp. Physiol. 2011;96(10 ): 1028–38. doi: 10.1113/expphysiol.2011.059840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mangano EN, Litteljohn D, So R, Nelson E, Peters S, Bethune C, Bobyn J, Hayley S. Interferon-gamma plays a role in paraquat-induced neurodegeneration involving oxidative and proinflammatory pathways. Neurobiol Aging. 2011. [DOI] [PubMed]
- 78. Serrano F, Kolluri N S, Wientjes F B, Card J P, Klann E. NADPH oxidase immunoreactivity in the mouse brain. Brain Res. 2003;988(1-2 ):193–8. doi: 10.1016/s0006-8993(03)03364-x. [DOI] [PubMed] [Google Scholar]
- 79. Wang G, Tompkins K D, Simonyi A, Korthuis R J, Sun A Y, Sun G Y. Nox2, Ca2+, and protein kinase C play a role in angiotensin II-induced free radical production in nucleus tractus solitarius. Hypertension. 2006;48(3 ):482–9. doi: 10.1161/01.HYP.0000236647.55200.07. [DOI] [PubMed] [Google Scholar]
- 80. Villani GR, Gargiulo N, Faraonio R, Castaldo S, Gonzalez Y, Reyero E, Di Natale P. Cytokines, neurotrophins, and oxidative stress in brain disease from mucopolysaccharidosis IIIB. J. Neurosci. Res. 2007;85(3 ):612–22. doi: 10.1002/jnr.21134. [DOI] [PubMed] [Google Scholar]
- 81. Cohen AC, Tong M, Wands J R, de la Monte S M. Insulin and insulinlike growth factor resistance with neurodegeneration in an adult chronic ethanol exposure model. Alcohol Clin. Exp. Res. 2007;31(9 ):1558–73. doi: 10.1111/j.1530-0277.2007.00450.x. [DOI] [PubMed] [Google Scholar]
- 82. Vallet P, Charnay Y, Steger K, Ogier-Denis E, Kovari E, Herrmann F, Michel J P, Szanto I. Neuronal expression of the NADPH oxidase NOX4, and its regulation in mouse experimental brain ischemia. Neuroscience. 2005;132(2 ):233–8. doi: 10.1016/j.neuroscience.2004.12.038. [DOI] [PubMed] [Google Scholar]
- 83. Chu J, Tong M, de la Monte SM. Chronic ethanol exposure causes mitochondrial dysfunction and oxidative stress in immature central nervous system neurons. Acta Neuropathol. 2007;113(6 ):659–73. doi: 10.1007/s00401-007-0199-4. [DOI] [PubMed] [Google Scholar]
- 84. Skurlova M, Stofkova A, Jurcovicova J. Anxiety-like behavior in the elevated-plus maze tests and enhanced IL-1beta, IL-6, NADPH oxidase-1, and iNOS mRNAs in the hippocampus during early stage of adjuvant arthritis in rats. Neurosci. Lett. 2011;487(2 ):250–4. doi: 10.1016/j.neulet.2010.10.032. [DOI] [PubMed] [Google Scholar]
- 85. Fischer MT, Sharma R, Lim J L, Haider L, Frischer J M, Drexhage J, Mahad D, Bradl M, van Horssen J, Lassmann H. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain. 2012;135(Pt 3):886–99. doi: 10.1093/brain/aws012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Cao X, Dai X, Parker L M, Kreulen D L. Differential regulation of NADPH oxidase in sympathetic and sensory Ganglia in deoxycorticosterone acetate salt hypertension. Hypertension. 2007;50(4 ):663–71. doi: 10.1161/HYPERTENSIONAHA.107.089748. [DOI] [PubMed] [Google Scholar]
- 87. Guggilam A, Haque M, Kerut E K, McIlwain E, Lucchesi P, Seghal I, Francis J. TNF-alpha blockade decreases oxidative stress in the paraventricular nucleus and attenuates sympathoexcitation in heart failure rats. Am. J. Physiol. Heart Circ. Physiol. 2007;293(1 ):H599–609. doi: 10.1152/ajpheart.00286.2007. [DOI] [PubMed] [Google Scholar]
- 88. Byrnes KR, Loane D J, Stoica B A, Zhang J, Faden A I. Delayed mGluR5 activation limits neuroinflammation and neurodegeneration after traumatic brain injury. J. Neuroinflam. 2012;9:43. doi: 10.1186/1742-2094-9-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yuan G, Khan S A, Luo W, Nanduri J, Semenza G L, Prabhakar N R. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J. Cell Physiol. 2011;226(11 ):2925–33. doi: 10.1002/jcp.22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Qin L, Crews FT. NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. J. Neuroinflam. 2012;9:5. doi: 10.1186/1742-2094-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]