

Abstract
Cryptococcal regulators of G protein signaling (CRG) are important for growth, differentiation, and virulence of Cryptococcus neoformans. Disruption of CRG2 resulted in dysregulated cAMP signaling and attenuated virulence, whereas disruption of CRG1 increased pheromone responses and enhanced virulence in the archetypal H99 strain. In tests with newly constructed near congenic mutants, a distinction between crg2Δ and crg1Δ gene expression was not apparent during macrophage interaction. Intranasal inoculation indicated that crg2Δ, crg1Δ, and wild-type strains reached the lungs within 0.5 hours of infection. However, CFUs were significantly decreased for crg2Δ at 2, 7, and 14 days post-infection. In contrast, crg1Δ proliferated to the same extent as the wild type (WT). Lung edema was not apparent in mice infected with crg2Δ 0.5 hours post-infection, which showed little cellular infiltrate in comparison to WT. Alveolar septal thickening was most evident in mice infected with crg1Δ, while mice infected with WT exhibited decreased septal thickening at later time points. Consistent with these observations, crg2Δ was less efficient in the elicitation of Th2 immune responses in a multiplex cytokine assay. Our results suggest that Crg2 is critical for establishment of early pulmonary infection and for persistence of infection, Crg1 regulates virulence in a strain-specific manner, and crg2Δ, crg1Δ and WT can all be distinguished on the basis of host tissue responses.
Keywords: Cryptococcal regulators of G protein signaling, macrophage interaction, host response, cytokine production, and fungal virulence
Introduction
Cryptococcus neoformans is an opportunistic human fungal pathogen that causes fatal meningoencephalitis in the absence of treatment. C. neoformans infections are thought to begin in the lungs of the host, and dissemination from the lungs into the central nervous system is correlated with treatment failure in humans, which can be as high as 20% [1,2]. Prevalence of this disease has risen with an increased population of long-term immunosuppressed and immunocompromised individuals [3,4]. It is estimated that HIV-related cryptococcosis is the fourth leading cause of death in sub-Saharan Africa, behind only malaria, diarrheal diseases and childhood diseases, and C. neoformans causes higher mortality than tuberculosis in this population [5]. Though the initial entry route is thought to be through the lung, infection of the brain resulting in meningoencephalitis is the primary manifestation of the infection in HIV-positive individuals [4]. Limited treatment options combined with the potential for drug resistance [6] necessitate discovery of novel drug targets, which should arise from investigations on how C. neoformans survives and causes disease within mammalian hosts.
Previous studies have revealed several factors that contribute to virulence of C. neoformans [7]. Elaboration of a complex polysaccharide capsule [8], cell wall-associated melanin polymers [9], and enzymolytic proteins such as phospholipases [10] and ureases [11] provide this fungus with the ability to evade phagocytosis, withstand oxidative and nitrosative stress, and disseminate throughout the host. Surprisingly, the ability to produce these factors alone is not sufficient for virulence [12]. Other modes of controlling expression of virulence, as well as the elucidation of essential biological functions, are the subjects of intensive investigations for their roles in the pathogenesis of this fungus.
The guanine nucleotide binding (G-protein) mediated signal transduction pathways are regulators of growth, differentiation, and virulence. In C. neoformans, the G-alpha subunits Gpa2 and Gpa3 collectively regulate a pheromone-responsive mating pathway, whereas the G-alpha subunit Gpa1 governs a cAMP-dependent signaling pathway important for the elaboration of both melanin and capsule [13–16]. Additionally, the regulators of G-protein signaling proteins (RGS), which accelerate the hydrolysis of GTP to GDP at G-alpha subunits, also impact a range of cellular functions [17,18].
Functions of RGS proteins in virulence have been studied for pathogenic fungi including chestnut blight fungus Cryphonectria parasitica [19], rice blast fungus Magnaporthe grisea [20] and, notably, C. neoformans. The latter encodes at least two RGS proteins, Crg2 and Crg1. The affect of CRG gene disruption upon virulence was described through use of a murine model of cryptococcosis [18,21,22]. In these studies, deletion of the CRG2 gene encoding Crg2, increased the intracellular cAMP level and attenuated virulence [21,22], while deletion of the CRG1 gene encoding Crg1 resulted in a heightened response to pheromone stimulation during heterothallic mating and increased virulence in the F99 (H99 ura5) strain background [18,23]. Additionally, reconstitution of the crg1Δ mutant with the wild type CRG1 gene restored normal mating and virulence [18]. The clear distinction in virulence expression by mutants of the two RGS proteins and generally fragmentary knowledge of fungal virulence mechanisms warrant comparative studies of virulence and host responses by these two regulatory proteins.
Mammalian cellular responses are critical in controlling fungal infections but complex to study in the multicellular milieu of the host. However, cultured macrophages and macrophage-like cell lines offer an alternate approach to study such host responses and therefore have been widely utilized [24–28]. C. neoformans is able to replicate within and escape from macrophages, though the role this plays in disease is still unclear [24,27]. Gene expression studies of C. neoformans interacting with macrophage-like cell lines also revealed activation of several virulence and mating associated genes, including CAP10, LAC1, and PKA1 [25].
The established intranasal mouse model of cryptococcal infection, which begins in the lungs of the mice, mimics the accepted route of infection in humans. Dissemination of C. neoformans from the lungs is correlated with treatment failure in humans and is a crucial step in progression of disease [2,9,29]. Occurrence, diagnosis and treatment of pulmonary C. neoformans are rare, but have been reported in both immunocompetent and immunocompromised populations [4,30–32]. Historically, even with diagnosis of cryptococcal pulmonary disease, treatment was infrequently recommended for the immunocompetent, as it was believed that a robust Th1 response would be mounted to clear the infection [4,30,31]. For the crg2Δ and crg1Δ mutants, the distinct phenotypic differences in vitro, as well as the dramatically different outcomes of virulence in vivo provide an ideal opportunity to study disease progression within the mouse lung. Clarifying the role of the Crg proteins during pathogenesis will provide a basis for modulating the host response to more appropriately clear infections or targets for future drug development to keep C. neoformans confined within the lung of even immunocompromised individuals. Sequestering C. neoformans within the lung could prove a useful adjunct in prophylactic therapy. Herein, we assess crg2Δ and crg1Δ in the context of macrophage interaction and gene expression, as well as dissemination throughout the murine host, establishment of pulmonary infection, and host tissue reactions. We show that Crg2 and Crg1 elicit host tissue reactions and impact gene expression within macrophage-like cell lines to some extent. This study also provides evidence for a substantial role for Crg2 during pulmonary infection and dissemination as well as establishment of early infection.
Materials and methods
Strains and DNA plasmids
H99 is a well-described serotype A parental strain of C. neoformans and is used here as the wild type (WT) control strain. Two stocks of H99 were used in this study: H99 original (ori) is a lab stored stock strain [18] and H99 ATCC was obtained from the ATCC (#208821). Knockout mutants crg2Δ::NAT and crg1Δ::NAT were created in the H99 (ori) background. The CRG2 and CRG1 gene cassettes previously employed in complementing strains of similar genetic background, crg2Δ::NAT [33] and crg1Δ::NAT [18] in plasmid pGMC200, were restriction enzyme digested with Kpn I and Hind III to release the NAT marker, and replaced with the NEO marker. The plasmid constructs were biolistically transformed into crg2Δ and crg1Δ and the resulting complemented strains were verified by PCR amplification (Supplementary Fig. 1 –online only) and phenotype examination. No previously published strains created in an auxotrophic background were used in the current study, all mutant strains were newly generated. For all experiments, strains were freshly streaked onto yeast extract-peptone-dextrose (YPD) agar and grown in liquid YPD overnight at 30°C with shaking. Yeast cells were precipitated by centrifugation, washed three times in sterile phosphate buffered saline (PBS, Fisher), and utilized following hemacytometer-based counting. Yeast cells used in virulence studies were taken directly from YPD agar, washed, and counted as above. Inocula viability was verified by plating on YPD medium.
Macrophage cell lines
Macrophage-like J774A.1 and RAW264.7 cell lines were a gift of J. Cutler and maintained in high glucose Dulbecco’s modified Eagle’s medium (DMEM, Sigma) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 0.1 mg/ml streptomycin at 37°C, 5% CO2. Cells were passaged two to three times weekly and those with 4–15 passages were employed in the experiments.
In vitro studies
J774A.1 and RAW264.7 cell lines of the appropriate passage stage were collected and counted in a hemacytometer and viability assessed with trypan blue stain. Cells were then seeded into flasks or on cover glasses as appropriate, allowed to grow overnight, and treated with stimulating medium containing 50 U/ml mouse recombinant interferon gamma (R&D Systems), 0.3 μg/ml E. coli 0111:B4 LPS (Sigma), and 1 μg/ml capsule-specific monoclonal antibody 18B7 (gift of A. Casadevall) before exposure to washed cryptococcal cells at a 2:1 multiplicity of infection, as described elsewhere [25]. Cells were incubated under 5% CO2 at 37°C for 2 h in stimulating media and observed using an inverted microscope. Unattached cryptococcal cells were removed by washing three times in 37°C sterile PBS and cultures were examined microscopically to confirm removal of extracellular fungal cells. For phagocytic index determination, cover glasses were fixed in methanol, Giemsa (Sigma) stained, and three fields per cover glass photographed for counts of macrophages with attached/ingested cryptococcal cells and macrophages without attached/ingested cryptococcal cells. Phagocytic index was calculated as previously described [28]. At minimum, 480 macrophages were observed. For gene expression studies, stimulated macrophage-like cell lines grown in flasks interacted with C. neoformans were washed three times with 37 ° C sterile PBS to remove unattached cryptococcal cells. The macrophage cells were then suspended in ice cold water containing 0.05% SDS to release cryptococcal cells. Cells were collected and RNA was extracted for gene expression studies.
Measurement of gene expression
Cells released from macrophages were collected by centrifugation and RNA was extracted by standard methods [34]. Briefly, cells were disrupted in Trizol with 0.4 mm glass beads and a Bio 101 bead beater following the manufacturer’s protocol. RNA was DNase treated, annealed with Oligo dT primers and reverse transcription was carried out using the SuperScript II kit (Invitrogen). Gene specific primers were designed from published sequences and by use of Primer3 software [35], across an intron where possible, and reverse transcribed real-time PCR was performed (Table 1). Efficiency of primer pairs was determined as specified by the manufacturer conditions with SYBR Green master mix (Agilent) and a BioRad iCycler. Melt curve analysis was performed for all reactions. Samples were assayed for gene expression, data were normalized to actin and efficiency corrected [36]. Gene expression was calculated as the fold change in comparison to the expression of wild type strains.
Table 1.
Primers used in qRT-PCR.
| Gene | Primer | Sequence |
|---|---|---|
| ACT1 | PW566 | 5′-CGCCCAACATGTCTATGGAAGAAGAAG-3′ |
| PW567 | 5′-TGAGAAGACTGGGCCGCAGTCTGGAGCTCCTG-3′ | |
| STE12 | PW570 | 5′-CGCTCTTTGTTGAAGGGAAG-3′ |
| PW571 | 5′-CGGTTCCTGAGGTTGACTGT-3′ | |
| URE1 | PW572 | 5′-ACTGTCAAGCTATGGGTCGT-3′ |
| PW573 | 5′-ACATATCGCTTCACCCTGTT-3′ | |
| LAC1 | PW574 | 5′-CGGTACATCCCTTCATTCTC-3′ |
| PW575 | 5′-ACATTATCCGTTCGACCAGT-3′ | |
| CAP10 | PW605 | 5′-AAGCCCTTTTGGCTCTTCTC-3′ |
| PW606 | 5′-TTTTCGTTGATGGGGAAATC-3′ | |
| CAP60 | PW607 | 5′-CAAGTGATGCGGTCAGTCAT-3′ |
| PW608 | 5′-ACCAAGTCGACGAGCAAGTT-3′ | |
| PKA1 | JH2366 | 5′-CAAGTATGTACCTGATATCA-3′ |
| JH2484 | 5′-GTATCGCTTAGTGAGATC-3′ |
Mouse model of infection
Six-week-old female A/JCr mice (NCI) were anesthetized via peritoneal injection of a ketamine/xylazine cocktail and 5 × 104 cryptococcal cells suspended in sterile PBS were applied to the nostrils directly with a pipette tip [18]. Mice were monitored daily and euthanized via CO2 inhalation when moribund or at a predetermined time post inoculation [18]. Experiments involving murine models were approved by the Institutional Animal Care and Use Committee (IACUC).
To assess dissemination during infection, mice inoculated with crg2Δ::NAT, crg1Δ::NAT, H99 (WT) were sacrificed 2, 7, and 14 days post-infection or when moribund. Upon sacrifice, brain, lungs and both kidneys were collected. Organs were weighed, homogenized in a glass homogenizer in 1 ml sterile water containing 1 mg/ml chloramphenicol, diluted serially in sterile water containing 100 U/ml penicillin/0.1 mg/ml streptomycin, and plated on YPD. Plates were incubated at 30°C for 3–5 days and CFU counted. Early infection studies were performed similarly, however, mice inoculated with crg2Δ::NAT, crg2Δ CRG2, crg1Δ::NAT, crg1Δ CRG, and H99 (WT) were euthanized 0.5, 2, 8 and 48 h post-infection, and only lungs were assayed for CFU.
Standard histological procedures were followed for assessment of infection in mouse tissues. Briefly, whole lungs from mice infected with crg2Δ::NAT, crg1Δ::NAT or H99 (WT) were removed at 0.5 h, 2 h, 8 h, 2 day, 7 day, and 14 day post-infection or when moribund. Lungs were fixed in 4% neutral buffered formalin, processed in a Leica TP 1020 (Leica Microsystems, Inc., Bannockburn, IL), and embedded in paraffin. Blocks were sectioned at 5 μm thickness, adhered to glass slides, and stained according to standard methods with hematoxylin and eosin (H&E, Sigma) or periodic acid Schiff (PAS, Sigma) [37]. Staining was assessed in a blinded manner and photographed using an Olympus BX51 equipped with a digital camera. Representative photomicrographs are shown.
Cytokine analysis
Mice infected per the intranasal route (see above) with crg2Δ, crg1Δ or WT were observed for 2, 7, and 14 days. Lungs were collected and placed immediately on ice, weighed, and homogenized as above. Aliquots were removed for CFU determination and the remainder of the homogenate was suspended in protease inhibitor cocktail (Sigma) and immediately stored at −80°C until time of assay. Thawed samples were clarified by centrifugation and assayed in the Milliplex Map mouse cytokine 13-plex according to the manufacturer’s instructions (Millipore).
Statistics
All data were graphed and analyzed by use of GraphPad Prism. Error bars shown represent standard deviation. Significant differences were identified and isolated by one way ANOVA followed by a Bonferroni post test. P-values < 0.05 were considered significant.
Results
Cryptococcal cell and macrophage interactions
Host responses to fungal infection are critical in determining disease outcomes, but their study is often difficult because of the complex nature of multi-cellular mammalian tissues. We therefore utilized in vitro macrophage models to examine possible innate immune responses to crg2Δ and crg1Δ infection. Previous studies have shown that, when properly stimulated, macrophages exhibit measurable parameters in uptake, ingestion, and clearance or lysis of fungal cells that correlate with in vivo infection outcomes for C. neoformans [28,38] and C. gattii [39]. Moreover, in vitro gene expression profiles during this interaction can be used to further examine possible fungal virulence mechanisms [25].
When stimulated with mouse recombinant interferon gamma, LPS, and monoclonal antibody 18B7, J774A.1, a macrophage-like cell line, will ingest cryptococcal cells. At a ratio of 2:1 (yeast: macrophage), uptake of the fungal cells was 8.6%, 9% and 13.3% for crg2Δ, crg1Δ, and WT strains respectively. The viability of both macrophages and cryptococcal cells during the interaction was examined, along with the time of fungal escape from macrophages (8–10 h). However, no significant differences were observed between the strains during these interactions, suggesting that there were no differences in either the ability of the strains to evade phagocytes, or their susceptibility to macrophage killing, though more subtle responses to macrophage interaction could provide some insight into virulence differences (data not shown).
Because a more detailed analysis of the response of C. neoformans to macrophage interaction could provide greater insight into potential virulence differences, we next examined the expression of several genes encoding proteins important in the establishment of virulence determinants, such as melanin and capsule formation, during macrophage interaction. Using the J774A.1 cell line, we used quantitative real-time RT-PCR to assess C. neoformans gene expression. Genes assayed, shown in Table 1, were previously shown to be involved in macrophage interaction or virulence [25]. At 2 h of in vitro growth, there was no apparent change in the expression of CAP10, CAP60, or URE1 among three strains. The expression of LAC1 and STE12 appeared to be decreased for crg2Δ only, while PKA1 was decreased for both crg2Δ and crg1Δ strains as compared to WT during the macrophage interaction (Fig. 1A). No significant changes were seen in the expression of LAC1, PKA1, URE1 and STE12 for either crg2Δ or crg1Δ when grown in medium alone (Fig. 1B). Overall gene expression for crg2Δ and crg1Δ mutants did not differ significantly from WT during macrophage interactions at 2 h. Continued interaction at 8 h revealed that the expression of CAP10 increased four-fold in crg1Δ (Fig. 1C), and there was also a slight increase of expression in the related CAP60 gene. These expression changes were deemed specific as they were not observed in macrophage grown in medium alone (Fig. 1D). This suggests that, despite the absence of an observable capsular phenotype in vitro, the absence of Crg1 function resulted in an up-regulation of capsule-related genes during contacts with host immune cells, a possible pathogenic advantage. However, crg1Δ cells released from J774A.1 cells 2 and 8 h post interaction showed no increased capsular material as visualized with India ink staining (Supplementary Fig. 2 – online only). During J774A.1 interactions, crg2Δ displayed WT levels of expression for all genes assayed except for a drop in the expression of both LAC1 and PKA1 (Figs. 1A, 1C), indicating that crg2Δ may exhibit specific defects in virulence traits. The down regulation was seen for all genes without macrophage interaction with the exception of PKA1 (Fig. 1D).
Fig. 1.

Gene expression in an in vitro macrophage interaction assay. Expression of several virulence-related genes was measured by reverse transcribed real time PCR after 2 h of interaction with J774A.1 cells (A) or medium alone (B), or after 8 h of interaction with J774A.1 cells (C) or medium alone (D). Gene expression is reported as a relative fold change of the expression of crg2Δ and crg1Δ mutant strains in comparison to wild type (WT) and is efficiency-corrected. Increase of 2 fold and over was considered significant [25]. Error bars represent standard error of the means, based on three replications (n = 3).
Survival in the murine model
Studies using the in vitro macrophage model provided only limited information on the virulence of crg2Δ and crg1Δ, and the modest change of only one family of virulence-related genes insufficiently differentiates the two mutant strains. Therefore, we turned to the more complex murine inhalational model. Though survival data have been previously published for crg2Δ and crg1Δ in separate studies, crg1Δ studies were carried out in an auxotrophic strain background [18,33]. We wanted to confirm results using strains with dominant marker genes to ensure that they would be genetically near isogenic. We then compared the survival and dissemination of these newly generated strains using the intranasal mouse model. Consistent with previous virulence studies, survival of mice infected with crg2Δ was significantly longer than mice infected with WT and the crg2Δ mutant complemented with the wild-type CRG2 gene. Survival times for mice infected with crg2Δ, WT, and crg2Δ CRG2 were 69–90 days, 26–29 days, and 28–35 days respectively (Fig. 2). Unexpectedly, no statistically significant difference was found in the survival of mice infected with crg1Δ::NAT (27 to 28 days), crg1Δ::NAT CRG1::NEO (26 to 31 days), and WT (26 to 29 days) (Fig. 2). This test, which was also repeated once with similar results, indicated that the previously observed enhancement of virulence with the crg1Δ::URA5 strain might indeed be strain-specific [18].
Fig. 2.
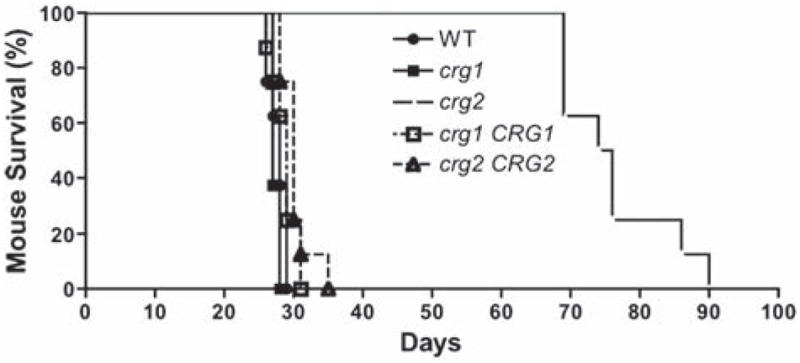
Crg2 is required for robust fungal virulence. The alleles for crg2Δ::NAT and crg1Δ::NAT genes were newly generated and mutants obtained by biolistic transformation. The wild type CRG2 and CRG1 genes were linked to a neomycin resistance marker (NEO) and used to complement the respective mutant strains. This test was repeated once with no significant variance shown, n = 8.
Additionally, the examination of fungal burden in mouse tissues such as lung, brain and kidney revealed that they were all heavily colonized by all strains once moribund. CFUs in mice infected with crg2Δ were lower than those infected with WT, particularly in the lung and brain. No significant difference was found between WT and crg1Δ in these tissues. The mean CFUs for crg2Δ, crg1Δ, and WT moribund animals in the lung were 7.4 × 106, 2.3 × 107, and 3.8 × 107, respectively (Fig. 3A). In the kidney, the mean CFUs for crg2Δ, crg1Δ, and WT were 2.9 × 105, 1.2 × 105, and 1.9 × 105 (Fig. 3B), and the mean CFUs of the brain in moribund animals for crg2Δ, crg1Δ, and WT were 7.4 × 106, 5.2 × 107, and 3.2 × 107, respectively (Fig. 3C). The approximately one order of magnitude reduction in the CFUs of crg2Δ infected lungs and brains was verified by a second independent test (Fig. 3D, Supplementary Fig. 3 – online only). CFUs in the brains of crg2Δ-infected mice were consistently and significantly lower throughout multiple studies and consistently lower than those in the lungs of infected mice. To resolve the ambiguity of lower CFUs in the crg2Δ- infected lungs, we promptly repeated the test, which again revealed lower CFUs in the crg2Δ- infected lungs. Decreased CFUs in crg2Δ-infected mice was noted only for the classic cryptococcal target organs, lung and brain, though not systemically, as kidney CFU were not significantly different from WT. The findings suggest that Crg2 function is necessary for efficient lung colonization and subsequent dissemination to the CNS.
Fig. 3.

Assessment of fungal burdens during infection of murine models. Lung (A and D), kidney (B), and brain (C) of moribund animals infected with crg2Δ, crg1Δ, and WT were dissected and CFUs counted. Asterisks indicate that the differences are statistically significant (P < 0.05). All strains reach similar levels in the kidneys of moribund animals, but significantly fewer crg2Δ in the brains and the lungs of a repeat test (P < 0.05, one way ANOVA and Bonferroni post test). Bars represent standard error, A–C, n = 6; D, n = 5.
Dynamics of cryptococcal dissemination during infection
Studies of fungal dissemination throughout the course of disease may help to reveal distinctions in virulence between crg2Δ and crg1Δ infected mice. Therefore, the mouse intra-nasal model was used and animals were sacrificed at predetermined time points throughout infection (2, 7, and 14 days), tissues collected, and fungal CFUs assessed. Consistent with previous work [11,40], no CFUs were detectable in the brains or kidneys of animals at 2–14 days post-infection, and cryptococcal cells were mostly found in the lungs. The lung mass was not significantly different between crg2Δ infected animals and WT or crg1Δ infected animals and weighed approximately 2/3 of the controls after 2 weeks of infection (Fig. 4A). As the CFU data reflects, the yeast burden in lungs of mice infected with crg2Δ was significantly less than the burden in WT or crg1Δ strains (Fig. 4B). No cryptococcal growth was observed in any blood cultures obtained 2, 7, or 14 days post infection.
Fig. 4.

Mutation of Crg2 but not Crg1 impacts the mass of the infected mouse lung tissue (A) and fungal burden throughout the disease process (B). Mice infected through the intranasal route were monitored for 2, 7 and 14 days following infection. Mice were sacrificed at indicated time points and CFU assay performed on brain, kidney and lung. No viable C. neoformans was recovered from the brain or kidneys of infected animals during this period of time. Large variations in mass of the lung suggest varying degrees of inflammation, and a steady decline in CFU over 2–14 days indicates the necessity of Crg2 for robust infection in the lung, n = 5.
Importantly, while the presence of viable C. neoformans could be detected in the lungs at all time points for all strains, significantly fewer fungal cells were detected in mice infected with crg2Δ as early as 2 days post-infection and up to 14 days post-infection (Fig. 4B). Mice infected with crg2Δ exhibited more than one log reduction in CFUs at day two, which progressed to over two orders of magnitude less at time of death (Figs. 3A, 3D). In both WT and crg1Δ infections, the initial inocula was maintained, with an increase of more than one log by two days post-infection, which continued to increase steadily throughout infection, reaching numbers approximately 103 times the initial inoculum (5 × 104) (Figs. 3A, 4B). Collectively, these findings indicate that, despite the lack of significant distinctions between the mutants and wild-type strains in the in vitro macrophage work, crg2Δ does not grow nearly as well as crg1Δ or WT in lungs during the first weeks of infection, which could well explain the low virulence of crg2Δ.
Early stages of infection and dissemination
To further examine this early infection and also verify that the lower number of crg2Δ cells in the lungs of infected animals at 2 days post-infection was not due to a mechanical barrier preventing the mutants from reaching the lungs, mice were again inoculated by the intranasal route with equal amounts of crg2Δ, crg1Δ or WT, and sacrificed at 0.5, 2, 8, and 48 h post inoculation, with subsequent tissue examination. All strains reached the mouse lung by 0.5 h post-infection, and WT and crg1Δ were closely matched in CFU counts, which also steadily increased from 8–48 h post-infection, with CFUs at 48 h nearly double that of the 0.5 h values (Fig. 5). CFU for crg2Δ, however, decreased at every time point after 0.5 h and significantly fewer crg2Δ cells, less than one-third of the 0.5 h levels, were detected 48 h post-infection (Fig. 5). A repeat of this experiment yielded similar results, indicating that early establishment of infection, within 0.5 h, is critical for pathogenesis and that Crg2 is necessary for the robust establishment of early infection within the lungs. This defect does not appear to be attributable to any growth defect at 37°C in either YPD or tissue culture medium (Supplementary Fig. 4 –online only).
Fig. 5.

Crg2 is critical for early infection of the murine lungs. Lungs of mice infected via the intranasal model were collected 0.5, 2, 8, and 48 h post-infection, and CFUs assessed. All strains reached the lungs in equivalent numbers at 0.5 h, but crg2Δ decreased significantly at 48 h post-infection (P < 0.05, one way ANOVA, Bonferroni post test). The asterisk indicates that the difference is statistically significant. Bars represent standard error, n = 3 and repeated with similar results.
To test possible early host responses to these infections, histological examination was performed over the same time course. In general, WT infection uniquely resulted in severe edema of the lung 0.5 h post-infection, which cleared by 2–8 h post-infection resulting in thickening of the alveolar septal membranes, and showing marked cellular infiltrate 48 h post-infection (Fig. 6, top row). In comparison, edema was not apparent in crg2Δ-infected mice, whose lung tissues exhibited septal thickening at 2 h post-infection but with little cellular infiltrate (Fig. 6, middle row). The crg1Δ infected lungs did not show severe edema at any time assayed, and instead showed moderate septal thickening from 0.5–8 h post-infection with mild cellular infiltrate at 48 h post-infection (Fig. 6, bottom row). These findings indicate that Crg2 may impact very early host responses to infection, and, although WT and crg1Δ cause quantitatively indistinguishable infections, Crg1 may also have a role at the early stage of infection.
Fig. 6.

Comparison of early host responses to infection. Mutation of CRG2 elicits less edema in the lungs of infected mice. Lungs of mice intranasally infected with crg2Δ, crg1Δ, and WT were collected 0.5, 2, 8 and 48 h post-infection, fixed in formalin, processed and H&E stained. Edema was noted in all time points for crg1Δ and WT-infected animals, and in crg2Δ infected animals 2 h post-infection, n = 3. Black arrows indicate thickened alveolar septa, asterisks indicate areas of RBCs, PMNs and/or macrophages.
Examination of host responses to infection
To assess host responses to the infection, freshly harvested mouse tissues were homogenized and smeared on slides for microscopic examination. Encapsulated fungal cells of larger sizes (up to 20 μm in diameter) were found in lung tissue homogenates for all infecting strains (crg2Δ, crg1Δ and WT) 2, 7, and 14 days post-infection and in moribund animals, as reported previously in whole mouse lung tissue or homogenates [8,21], and infected human lung tissue [41]. Examination of H&E stained formalin fixed lung tissues revealed the majority of yeast cells were located extracellularly within the lung after 2 days of infection, which is consistent with a previous observation for a shift from intracellular to extracellular by 24 h post-infection [26]. Additionally, given that lung mass is an indicator of inflammatory response [42], histological examination revealed that the increased lung mass in these samples (Fig. 4A) could be attributed both to influx of host cells and proliferation of cryptococcal cells (Fig. 7).
Fig. 7.

Comparison of host responses to infection. Lungs of intranasally-infected mice were preserved in formalin, processed and embedded in paraffin. Five μm sections were adhered to glass slides and H&E stained. Bar equals 20 μm. Representative sections are shown for 2, 7, and 14 days post-infection as indicated, and moribund mice occurred 68–89 days (crg2Δ), 28–33 days (crg1Δ), and 33–35 days (WT) post-infection. Consistent with the CFU assessment, cryptococcal cells were visualized at all time points. The majority of cryptococcal cells were extracellular, and a notable population had cellular diameters in excess of 20 μm for all tested strains. Black arrows indicate enlarged cryprococcal cells.
To compare the abilities of crg2Δ, crg1Δ, and WT strains to elicit host immune responses, staining of infected tissues was performed. Presence of red blood cells and host cellular influx consisting of macrophages and neutrophils within the interstitium of the alveolar septa was noted 2 days post-infection for WT-infected mouse lungs (Fig. 7, top row), 14 days post-infection for crg2Δ (Fig. 7, middle row), and 7 days post-infection for crg1Δ (Fig. 7, bottom row). Hematoxylin and eosin (H&E) staining of the lungs from moribund animals showed that both crg2Δ and crg1Δ infected mice exhibited pathologies similar to WT-infected mice where lung architecture was severely damaged in all moribund mice, with an obvious profusion of cryptococcal cells of varying diameters (Fig. 7). Differences were noted primarily in the time at which fungal cells reached tissues outside of the lungs, such as the kidney and the brain (89 days for crg2Δ, 32 days for crg1Δ, Figs. 3B, 3C). In addition, differences in the timing of host cellular infiltrates in histological sections were observed (Figs. 6, 7). Periodic acid-Schiff (PAS) staining was also performed, no significant differences in cryptococcal encapsulation were observed (data not shown). Together, these data indicate that the early pathologies noted in the lung, including septal thickening and edema, worsened throughout infection. However, this host reaction is delayed until 14 days post-infection for crg2Δ-infected animals.
Furthermore, in an effort to gain further insights about the host cell influx seen in the lungs of infected mice at 2, 7 and 14 days post-infection, a cytokine/chemokine assay panel was performed. Overall, cytokine levels were low to undetectable with all infecting strains at 2 days post-infection, despite the appearance of cellular infiltrates at this time in WT histological specimens. At 7 days post-infection, IL-4, IL-5, IL-6, and IL-13 were increased beyond levels at 2 days post-infection for WT and crg1Δ, but not crg2Δ, infected animals (Table 2). This Th2-biased response was seen 7 days post-infection for both crg1Δ and WT infected mice, with high levels of macrophage recruitment (MCP-1, 2427 pg/ml for WT and 1263 pg/ml for crg1Δ, contrasting with 187 pg/ml for crg2Δ homogenates), consistent with histological observations of interstitial macrophage influx within the alveolar septa. Despite this overall Th2 bias, IL-10 levels were undetectable 7 and 14 days post-infection for all strains. All cytokine levels were reduced by 14 days post-infection for WT and crg1Δ. As predicted by histological evaluation and the low cryptococcal burden, most cytokine levels were less than 100 pg/ml to undetectable for crg2Δ infected animals at any time. Th2 cytokines were detected at 14 days post-infection in these animals, consistent with cellular influx seen in the 14 day post-infection histological sections (Table 2).
Table 2.
Chemokine and cytokine profiles.
| 2 days
|
7 days
|
14 days
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| WT | crg1Δ | crg2Δ | WT | crg1Δ | crg2Δ | WT | crg1Δ | crg2Δ | |
| GM-CSF | 59.37a ± 9.23b | 41.82 ± 32.97 | 71.80 ± 16.55 | 94.27 ± 40.35 | 78.29 ± 42.76 | UDc | UD | UD | UD |
| IFN-γ | UD | UD | UD | 97.45 ± 41.51 | 84.46 ± 42.43 | UD | UD | UD | UD |
| IL-12 p70 | UD | UD | UD | UD | UD | UD | UD | UD | 21.58 ± 14.30 |
| IL-2 | UD | UD | UD | UD | UD | UD | UD | UD | UD |
| TNFα | UD | UD | 12.83 ± 5.994 | 30.98 ± 13.54 | 22.84 ± 5.307 | UD | UD | UD | UD |
| IL-1β | 37.24 ± 30.38 | 41.97 ± 15.81 | 30.26 ± 21.27 | 119.7 ± 65.62 | 78.19 ± 47.41 | UD | 61.44 ± 29.98 | 73.94 ± 42.64 | 41.03 ± 18.65 |
| IL-6 | 75.71 ± 37.32 | 62.88 ± 22.98 | 44.38 ± 17.08 | 2103 ± 1033 | 992.3 ± 655.9 | 99.48 ± 6.180 | 89.61 ± 70.20 | 115.8 ± 79.33 | 69.80 ± 61.19 |
| IL-7 | UD | UD | UD | UD | UD | UD | UD | UD | UD |
| MCP-1 | 164.5 ± 57.20 | 152.0 ± 25.80 | 181.9 ± 36.09 | 2427 ± 1626 | 1263 ± 930.6 | 187.0 ± 114.9 | UD | 112.5 ± 58.27 | 158.1 ± 54.56 |
| IL-10 | 33.48 ± 25.65 | 32.19 ± 30.57 | UD | UD | UD | UD | UD | UD | UD |
| IL-13 | UD | UD | UD | 1832 ± 1393 | 562.2 ± 543.3 | UD | 408.2 ± 56.45 | 563.6 ± 388.0 | 187.5 ± 90.50 |
| IL-4 | UD | UD | UD | 338.6 ± 252.4 | 106.5 ± 66.74 | UD | 181.0 ± 45.63 | 174.6 ± 80.95 | 78.75 ± 44.44 |
| IL-5 | 30.37 ± 9.853 | 34.59 ± 25.85 | 53.48 ± 39.60 | 1438 ± 990.6 | 506.6 ± 462.0 | 71.25 ± 63.48 | 108.4 ± 43.83 | 146.7 ± 91.68 | 159.3 ± 92.45 |
Note:
All values expressed as pg/ml,
Standard deviation,
Undetectable, < 3.2 pg/ml.
Discussion
Cryptococcus neoformans infects primarily immunodeficient individuals causing meningoencephalitis. It has been suggested that the fungus utilizes a plethora of mechanisms to invade and evade the host immunodefensive mechanism and to proliferate within hostile host tissues. While macrophages are known to play a critical role in cryptococcal infection, the utility of this host cell in defending against the pathogen or aiding in the spread of the pathogenic fungus is unclear [38,43]. Nevertheless, the in vitro model of macrophage interaction has been employed in numerous studies in an attempt to better understand the capabilities of pathogenic fungi, such as C. neoformans, to elude host defenses. We carried out analysis of the ability of macrophage-like cell lines to interact with the crg2Δ and crg1Δ mutant strains. Interestingly, our data indicates that regulatory proteins of fungal origin, such as the RGS proteins that are integrated components of signaling transduction pathways demonstrated to play important regulatory roles in growth and pathobiology of C. neoformans, have little to no impact upon the efficiency of cryptococcal uptake by macrophage-like cell lines. Therefore, the possibility that phenotypic virulence differences can be attributed to interactions with this innate immune cell is not supported by our findings.
When examining fungal gene expression, however, it is clear that phagocytosis by macrophage-like cell lines impacts the expression of a select number of virulence-related genes in crg mutants. The ability to produce laccase required for melanin formation is thought to play a role in dissemination from the lung for C. neoformans [9,44], and the previously identified virulence enhancement for crg1Δ was attributed to increased melanization at body temperature [18]. However, no change was seen in melanization of cells among crg2Δ, crg1Δ, and WT unless 5% CO2 pressure was applied in which crg1Δ exhibited greater melanin formation (data not shown). As laccase expression was not found to be increased upon gene expression analysis, it can be presumed that while the ability to produce laccase is important for dissemination from the lungs, it does not seem to be the explanation for the disparity between the observed virulence of crg2Δ and crg1Δ.
On the whole, genes assayed in our expression studies revealed few upregulated genes under in vitro conditions. The expression of capsule-related CAP10 and CAP60 genes was elevated in crg1Δ after 8 hours of macrophage interaction, but the influence this has on virulence is not clear, as no differences were seen when comparing the capsules of the crg1Δ mutant to WT. There was no decreased gene expression in crg2Δ, which showed no deficiencies in its interaction with the macrophage. Suppressed PKA1 gene transcription was noted after 8 hours of macrophage interaction, and since crg2Δ shares a decreased virulence phenotype with pka1Δ [45], this would indicate that the virulence differences noted between the crg2Δ and crg1Δ mutant strains are not regulated at the transcriptional level. However, this scenario is unproven and unlikely, based on studies of RGS protein over a wide range of eukaryotic organisms. On the other hand, the lack of any distinction in macrophage interaction or in vitro gene expression studies may indicate that in vivo models remain the only eligible approach to study gene expression and virulence mechanisms.
In mice infected with crg2Δ, lower CFUs within the lungs, both initially and throughout the infection, indicate that Crg2 is required for effective establishment of infection, survival, and robust proliferation within the lungs. The persistently lower CFUs of crg2Δ in the lung and the brain would indicate that the immunocompetent host, even when confronted with a strain unable to rapidly and proficiently establish colonization, is unable to efficiently clear the infection. The absence of cellular infiltrate up to 14 days post-infection seems to indicate that, although the crg2Δ cells were not cleared by the host, they were largely unable to proliferate in the lung environment. Cytokine levels were low to undetectable in the lungs of crg2Δ infected-mice, suggesting that either insufficient numbers of fungal cells were present or there was an insufficient immunoreactive host response. Lack of a pronounced Th2 host response may occur later in infection, when crg2Δ population in the lung increases – as it must, given the fungal burden in moribund animals – though the timing of this event and the subsequent response of the host is a subject for future investigation. Overall, the current in vivo data suggest that both Crg2 and Crg1 are dispensable for fulminant infection of the lung as well as systemic dissemination, and that Crg2 is required for efficient fungal colonization and likely dissemination to target organs.
The poor but apparent ability of crg2Δ to still disseminate systemically in infected mice remains poorly understood. We previously showed that crg2Δ mutant strains exhibited a swollen phenotype under in vitro conditions, and crg2Δ cells were also larger with thicker capsules than wild-type cells in capsule inducing medium [21,22]. As recent studies suggested that large sized cells might negatively affect the ability of the organism to effectively disseminate within the host [46,47], we actively identified such cells in lungs histology. The occurrence of large sized cells was apparent, but less frequent than wild type and crg1Δ mutant strains (Fig. 7, indicated by black arrows). This is consistent with the findings that crg2Δ mutants are dysregulated in cAMP signaling and normal cAMP signaling is required for the formation of large cells [21,22,47]. This finding suggested that a different mechanism is involved in the delayed dissemination and virulence attenuation of crg2Δ mutants. Regardless, the coordinated distribution of CFUs between lung and brain is consistent with previous studies suggesting that the lung serves as the primary reservoir for subsequent haematogenous transmission [3]. We can conclude, therefore, that Crg2 deletion uniquely impacts survival within and dissemination to target organs, reducing overall virulence and extending survival times in the mouse model.
The current observation using nearly isogenic strains failed to recapitulate the earlier observation that Crg1 deletion resulted in increased virulence [18], suggesting that the previous finding might be strain-specific. However, it is worth noting that in the original observation the crg1Δ mutant was restored to wild-type using the full-length CRG1 gene, and a recent finding showed that disruption of Znf2, a zinc-finger motif containing transcriptional factor, enhances not only mating responses but also virulence [48]. These studies indicated that the long-held association between mating responses and virulence remain strong despite its elusive nature. The crg1Δ mutant used in our studies, nevertheless, remains an important point of comparison for crg2Δ, whose mutation consistently results in attenuated virulence.
As underscored by a recent study of outcomes of human cryptococcal infections, dissemination of fungal cells from the lungs is an indicator of future treatment failure [2]. While much is known about the C. neoformans proteins, including laccase, phospholipases B and inositol phosphosphingolipid-phospholipase, that are required for escape from the lung [9], the process by which they facilitate tissue invasion is not fully characterized. Understanding the mechanism by which C. neoformans remains in the lung without causing disease in the human host may well lead to novel preventive approaches that delay or prevent dissemination. Our work clearly supports further studies into Crg2-related pathways. Given the many effects of Crg2 deletion on the survival and proliferation of C. neoformans within the murine lung, future study should be beyond the usual virulence factors to include the ability to respond to the host environment through signaling pathways.
Supplementary Material
Acknowledgments
The authors would like to thank J. Cutler, D. Fox, L. Freytag, J. Sturtevant, and S. Pincus for helpful suggestions and critical comments, and A. Casadevall for providing the monoclonal antibody 18B7. We also wish to thank G. Shen, S. Martin, P. Calmes, and R. Martinez for helpful discussions and technical assistance. This study was supported in part by NIH grants (AI054958 and AI074001) and a fund from the Research Institute for Children, New Orleans.
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Supplementary material available online
References
- 1.Charlier C, Dromer F, Lévêque C, et al. Cryptococcal neuroradiological lesions correlate with severity during cryptococcal meningoencephalitis in HIV-positive patients in the HAART era. PLoS ONE. 2008;3:e1950. doi: 10.1371/journal.pone.0001950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dromer F, Mathoulin-Pelissier S, Launay O, Lortholary O. Determinants of disease presentation and outcome during cryptococcosis: the CryptoA/D study. PLoS Med. 2007;4:e21. doi: 10.1371/journal.pmed.0040021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Casadevall A, Perfect JR. Cryptococcus neoformans. Washington, DC: ASM Press; 1998. [Google Scholar]
- 4.Huston SM, Mody CH. Cryptococcosis: an emerging respiratory mycosis. Clin Chest Med. 2009;30:253–264. doi: 10.1016/j.ccm.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 5.Park BJ, Wannemuehler KA, Marston BJ, et al. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS. 2009;23:525–530. doi: 10.1097/QAD.0b013e328322ffac. [DOI] [PubMed] [Google Scholar]
- 6.Sionov E, Lee H, Chang YC, Kwon-Chung KJ. Cryptococcus neoformans overcomes stress of azole drugs by formation of disomy in specific multiple chromosomes. PLoS Pathog. 2010;6:e1000848. doi: 10.1371/journal.ppat.1000848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buchanan KL, Murphy JW. What makes Cryptococcus neoformans a pathogen? Emerging Infect Dis. 1998;4:71–83. doi: 10.3201/eid0401.980109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feldmesser M, Kress Y, Casadevall A. Dynamic changes in the morphology of Cryptococcus neoformans during murine pulmonary infection. Microbiology. 2001;147:2355–2365. doi: 10.1099/00221287-147-8-2355. [DOI] [PubMed] [Google Scholar]
- 9.Eisenman HC, Casadevall A, McClelland EE. New insights on the pathogenesis of invasive Cryptococcus neoformans infection. Curr Infect Dis Rep. 2007;9:457–464. doi: 10.1007/s11908-007-0070-8. [DOI] [PubMed] [Google Scholar]
- 10.Santangelo R, Zoellner H, Sorrell T, et al. Role of extracellular phospholipases and mononuclear phagocytes in dissemination of cryptococcosis in a murine model. Infect Immun. 2004;72:2229–2239. doi: 10.1128/IAI.72.4.2229-2239.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cox GM, Mukherjee J, Cole GT, Casadevall A, Perfect JR. Urease as a virulence factor in experimental cryptococcosis. Infect Immun. 2000;68:443–448. doi: 10.1128/iai.68.2.443-448.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu OW, Chun CD, Chow ED, et al. Systematic genetic analysis of virulence in the human fungal pathogen Cryptococcus neoformans. Cell. 2008;135:174–188. doi: 10.1016/j.cell.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alspaugh JA, Perfect JR, Heitman J. Cryptococcus neoformans mating and virulence are regulated by the G-protein alpha subunit GPA1 and cAMP. Genes Dev. 1997;11:3206–3217. doi: 10.1101/gad.11.23.3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alspaugh JA, Pukkila-Worley R, Harashima T, et al. Adenylyl cyclase functions downstream of the Galpha protein Gpa1 and controls mating and pathogenicity of Cryptococcus neoformans. Eukaryot Cell. 2002;1:75–84. doi: 10.1128/EC.1.1.75-84.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li L, Shen G, Zhang ZG, et al. Canonical heterotrimeric G proteins regulating mating and virulence of Cryptococcus neoformans. Mol Biol Cell. 2007;18:4201–4209. doi: 10.1091/mbc.E07-02-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang P, Perfect JR, Heitman J. The G-protein beta subunit GPB1 is required for mating and haploid fruiting in Cryptococcus neoformans. Mol Cell Biol. 2000;20:352–362. doi: 10.1128/mcb.20.1.352-362.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chasse SA, Flanary P, Parnell SC, et al. Genome-scale analysis reveals Sst2 as the principal regulator of mating pheromone signaling in the yeast Saccharomyces cerevisiae. Eukaryot Cell. 2006;5:330–346. doi: 10.1128/EC.5.2.330-346.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang P, Cutler J, King J, Palmer D. Mutation of the regulator of G protein signaling Crg1 increases virulence in Cryptococcus neoformans. Eukaryot Cell. 2004;3:1028–1035. doi: 10.1128/EC.3.4.1028-1035.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Segers GC, Regier JC, Nuss DL. Evidence for a role of the regulator of G-protein signaling protein CPRGS-1 in Galpha subunit CPG-1-mediated regulation of fungal virulence, conidiation, and hydrophobin synthesis in the chestnut blight fungus Cryphonectria parasitica. Eukaryot Cell. 2004;3:1454–1463. doi: 10.1128/EC.3.6.1454-1463.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu H, Suresh A, Willard FS, et al. Rgs1 regulates multiple Galpha subunits in Magnaporthe pathogenesis, asexual growth and thigmotropism. EMBO J. 2007;26:690–700. doi: 10.1038/sj.emboj.7601536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen G, Wang Y, Whittington A, Li L, Wang P. The RGS protein Crg2 regulates pheromone and cyclic AMP signaling in Cryptococcus neoformans. Eukaryot Cell. 2008;7:1540–1548. doi: 10.1128/EC.00154-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xue C, Hsueh Y, Chen L, Heitman J. The RGS protein Crg2 regulates both pheromone and cAMP signalling in Cryptococcus neoformans. Mol Microbiol. 2008;70:379–395. doi: 10.1111/j.1365-2958.2008.06417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nielsen K, Cox GM, Wang P, et al. Sexual cycle of Cryptococcus neoformans var grubii and virulence of congenic a and alpha isolates. Infect Immun. 2003;71:4831–4841. doi: 10.1128/IAI.71.9.4831-4841.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alvarez M, Casadevall A. Phagosome extrusion and host-cell survival after Cryptococcus neoformans phagocytosis by macrophages. Curr Biol. 2006;16:2161–2165. doi: 10.1016/j.cub.2006.09.061. [DOI] [PubMed] [Google Scholar]
- 25.Fan W, Kraus PR, Boily MJ, Heitman J. Cryptococcus neoformans gene expression during murine macrophage infection. Eukaryot Cell. 2005;4:1420–1433. doi: 10.1128/EC.4.8.1420-1433.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feldmesser M, Kress Y, Novikoff P, Casadevall A. Cryptococcus neoformans is a facultative intracellular pathogen in murine pulmonary infection. Infect Immun. 2000;68:4225–4237. doi: 10.1128/iai.68.7.4225-4237.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma H, Croudace JE, Lammas DA, May RC. Expulsion of live pathogenic yeast by macrophages. Curr Biol. 2006;16:2156–2160. doi: 10.1016/j.cub.2006.09.032. [DOI] [PubMed] [Google Scholar]
- 28.Macura N, Zhang T, Casadevall A. Dependence of macrophage phagocytic efficacy on antibody concentration. Infect Immun. 2007;75:1904–1915. doi: 10.1128/IAI.01258-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perfect JR. Management of cryptococcosis: how are we doing? PLoS Med. 2007;4:e47. doi: 10.1371/journal.pmed.0040047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aberg JA. Pulmonary cryptococcosis in normal hosts: treat or observe? Chest. 2003;124:2049–2051. doi: 10.1378/chest.124.6.2049. [DOI] [PubMed] [Google Scholar]
- 31.Shirley RM, Baddley JW. Cryptococcal lung disease. Curr Opin Pulm Med. 2009;15:254–260. doi: 10.1097/MCP.0b013e328329268d. [DOI] [PubMed] [Google Scholar]
- 32.Aberg JA, Mundy LM, Powderly WG. Pulmonary cryptococcosis in patients without HIV infection. Chest. 1999;115:734–740. doi: 10.1378/chest.115.3.734. [DOI] [PubMed] [Google Scholar]
- 33.Shen G, Wang YL, Whittington A, Li L, Wang P. The RGS Protein Crg2 Regulates Pheromone and cAMP Signaling in Cryptococcus neoformans. Eukaryotic Cell. 2008;7:1540–1548. doi: 10.1128/EC.00154-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 35.Skaletsky SRAHJ. Bioinformatics Methods and Protocols: Methods in Molecular Biology. Totowa, NJ: Humana Press; 2000. Primer3 on the WWW for general users and for biologist programmers; pp. 365–386. [DOI] [PubMed] [Google Scholar]
- 36.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kligman AM, Mescon H. The periodic acid-Schiff stain for the demonstration of fungi in animal tissue. J Bacteriol. 1950;60:415–421. doi: 10.1128/jb.60.4.415-421.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shao X, Mednick A, Alvarez M, et al. An innate immune system cell is a major determinant of species-related susceptibility differences to fungal pneumonia. J Immunol. 2005;175:3244–3251. doi: 10.4049/jimmunol.175.5.3244. [DOI] [PubMed] [Google Scholar]
- 39.Ma H, Hagen F, Stekel DJ, et al. The fatal fungal outbreak on Vancouver Island is characterized by enhanced intracellular parasitism driven by mitochondrial regulation. Proc Natl Acad Sci USA. 2009;106:12980–12985. doi: 10.1073/pnas.0902963106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cox GM, McDade HC, Chen SC, et al. Extracellular phospholipase activity is a virulence factor for Cryptococcus neoformans. Mol Microbiol. 2001;39:166–175. doi: 10.1046/j.1365-2958.2001.02236.x. [DOI] [PubMed] [Google Scholar]
- 41.Cruickshank JG, Cavill R, Jelbert M. Cryptococcus neoformans of unusual morphology. Appl Microbiol. 1973;25:309–312. doi: 10.1128/am.25.2.309-312.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rivera J, Casadevall A. Mouse genetic background is a major determinant of isotype-related differences for antibody-mediated protective efficacy against Cryptococcus neoformans. J Immunol. 2005;174:8017–8026. doi: 10.4049/jimmunol.174.12.8017. [DOI] [PubMed] [Google Scholar]
- 43.Osterholzer JJ, Milam JE, Chen G, et al. Role of dendritic cells and alveolar macrophages in regulating early host defense against pulmonary infection with Cryptococcus neoformans. Infect Immun. 2009;77:3749–3758. doi: 10.1128/IAI.00454-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Noverr MC, Williamson PR, Fajardo RS, Huffnagle GB. CNLAC1 is required for extrapulmonary dissemination of Cryptococcus neoformans but not pulmonary persistence. Infect Immun. 2004;72:1693–1699. doi: 10.1128/IAI.72.3.1693-1699.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.D’Souza CA, Alspaugh JA, Yue C, et al. Cyclic AMP-dependent protein kinase controls virulence of the fungal pathogen Cryptococcus neoformans. Mol Cell Biol. 2001;21:3179–3191. doi: 10.1128/MCB.21.9.3179-3191.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Okagaki LH, Strain AK, Nielsen JN, et al. Cryptococcal cell morphology affects host cell interactions and pathogenicity. PLoS Pathog. 2010;6:e1000953. doi: 10.1371/journal.ppat.1000953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zaragoza O, García-Rodas R, Nosanchuk JD, et al. Fungal cell gigantism during mammalian infection. PLoS Pathog. 2010;6:e1000945. doi: 10.1371/journal.ppat.1000945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin XR, Jackson JC, Feretzaki M, Xue CY, et al. Transcription factors Mat2 and Znf2 operate cellular circulauits orchestrating opposite- and same-sex mating in Cryptococcus neoformans. PloS Pathog. 2010;6:e1000953. doi: 10.1371/journal.pgen.1000953. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.