

Abstract
Glycogenotic hepatocellular carcinoma (HCC) with glycogen-ground-glass hepatocytes has recently been described as an allegedly “novel variant” of HCC, but neither the historical background nor the heuristic relevance of this observation were put in perspective. In the present contribution, the most important findings in animal models and human beings related to the emergence and further evolution of excessively glycogen storing (glycogenotic) hepatocytes with and without ground glass features during neoplastic development have been summarized. Glycogenotic HCCs with glycogen-ground-glass hepatocytes represent highly differentiated neoplasms which contain subpopulations of cells phenotypically resembling those of certain types of preneoplastic hepatic foci and benign hepatocellular neoplasms. It is questionable whether the occurrence of glycogen-ground-glass hepatocytes in a glycogenotic HCC justifies its classification as a specific entity. The typical appearance of ground-glass hepatocytes is due to a hypertrophy of the smooth endoplasmic reticulum, which is usually associated with an excessive storage of glycogen and frequently also with an expression of the hepatitis B surface antigen. Sequential studies in animal models and observations in humans indicate that glycogen-ground-glass hepatocytes are a facultative, integral part of a characteristic cellular sequence commencing with focal hepatic glycogenosis potentially progressing to benign and malignant neoplasms. During this process highly differentiated glycogenotic cells including ground-glass hepatocytes are gradually transformed via various intermediate stages into poorly differentiated glycogen-poor, basophilic (ribosome-rich) cancer cells. Histochemical, microbiochemical, and molecular biochemical studies on focal hepatic glycogenosis and advanced preneoplastic and neoplastic lesions in tissue sections and laser-dissected specimens in rat and mouse models have provided compelling evidence for an early insulinomimetic effect of oncogenic agents, which is followed by a fundamental metabolic switch from gluconeogenesis towards the pentose-phosphate pathway and the Warburg type of glycolysis during progression from preneoplastic hepatic glycogenosis to the highly proliferative malignant phenotype.
Keywords: Acquired focal hepatic glycogenosis, Inborn hepatic glycogenosis, Hepatic preneoplasia, Hepatic neoplasia, Early metabolic aberrations, Progression-linked metabolic switch
INVITED COMMENTARY ON HOT ARTICLES
The glycogenotic hepatocellular carcinoma (glycogenotic HCC) with glycogen-ground-glass hepatocytes has recently been described as a “novel variant” of HCC by Callea et al[1]. Relating the excessive storage of glycogen (glycogenosis) to a complete absence of glucose-6-phosphatase activity as measured by a microbiochemical approach, the authors referred to the long known finding of a focal hepatic glycogenosis (FHG) as an early event in experimental hepatocarcinogenesis[2], continuing “however, ground-glass cells have not been reported in FHG”. In contrast to this statement, the characteristic hepatocellular phenotype addressed was not only explicitly described for the first time in preneoplastic and neoplastic lesions in rodents[3,4] and in humans[5,6] (Figure 1) decades ago but has also been proven to be heuristically highly relevant in numerous publications since then. Glycogenotic ground-glass hepatocytes (GGH) predominate in subpopulations of many focal precancerous hepatocellular lesions, particularly in preneoplastic FHG and benign hepatocellular neoplasms such as hepatocellular adenomas and focal nodular hyperplasia, but may also occur in more or less extended subpopulations of HCC as observed in various species, including non-human primates and human beings[7,8]. However, glycogen-GGH hardly ever account for whole neoplasms. This also applies to the glycogenotic HCC depicted by Callea et al[1] in which the GGH are mixed with “clear” (glycogenotic) cells without ground-glass features. It is, hence, questionable whether glycogenotic HCC with glycogen-GGH should be considered a specific entity as proposed by Callea et al[1]. Extensive investigations in models of chemical, viral, and hormonal hepatocarcinogenesis and some observations in humans suggest that FHG with and without GGH indicates a critical early metabolic aberration in the pathogenesis of benign and malignant hepatocellular neoplasms[7,8].
Figure 1.
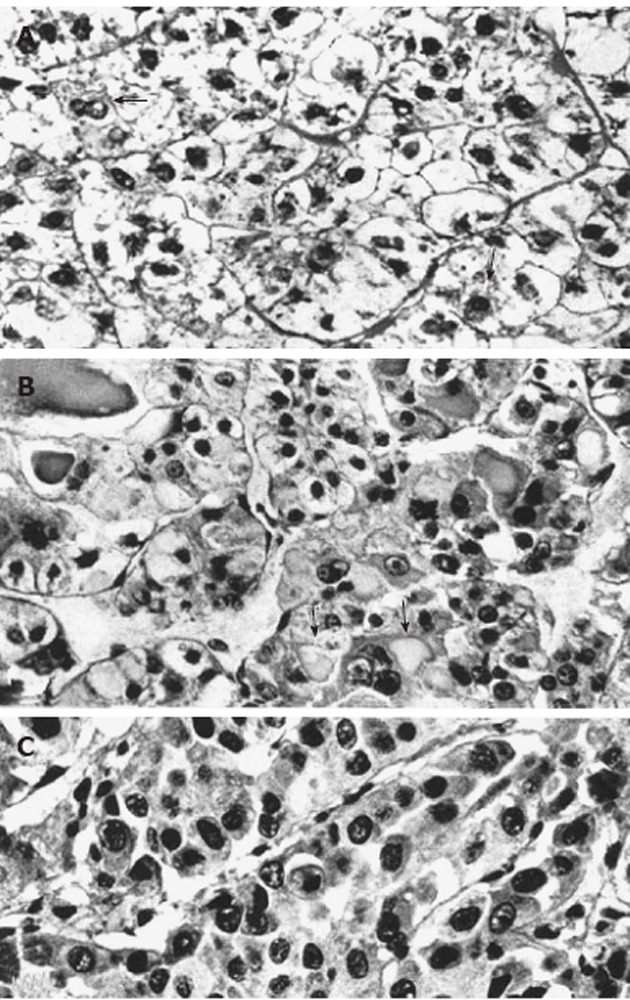
Light micrographs of portions from human hepatocellular neoplasms with and without glycogenosis. A: Clear-cell hepatocellular adenoma consisting predominantly of glycogenotic cells. In some cells (arrows) there is a reduction of glycogen and focal increase in cytoplasmic basophilia; B: Highly differentiated hepatocellular carcinoma (HCC) composed of a mixed population of clear (glycogenotic) cells, acidophilic cells (ground-glass hepatocytes, arrows), and some glycogen-poor, basophilic cells; C: Poorly differentiated, glycogen-free, basophilic HCC. All: Hematoxylin and eosin stain, x 460, from Bannasch et al[6].
Animal models of hepatocarcinogenesis
The observations in humans were preceded by several seminal findings in animal models of hepatocarcinogenesis as repeatedly reviewed[2,9]. In their pioneering electron microscopic investigations in rats continuously exposed to 3-methyl-dimethylaminoazobenzene, Porter et al[10] detected a hypertrophy of the smooth endoplasmic reticulum in many hepatocytes, and addressed its light microscopic counterpart as “hyaline degeneration”[11]. The authors related this characteristic subcellular change to a decreased rather than an increased storage of glycogen and felt “that only cells which, through mutation, loose the normal tendency to differentiate for glycogenesis will survive and so will be selected out for continued growth and differentiation”[10]. However, investigations in other models of rat hepatocarcinogenesis, employing both continuous or limited (stop model) exposure to N-nitrosomorpholine at various dose levels and time schedules[3,4], or ethionine for up to approximately 20 wk[12] revealed that hypertrophy of the smooth endoplasmic reticulum is often associated with an excessive storage of glycogen, the smooth membranes forming either a typical network or peculiar lamellar complexes which often show a close spatial relationship with glycogen particles (Figure 2) but may also be free of glycogen forming “fingerprints”[4]. Steiner et al[12] designated the complexes of the smooth endoplasmic reticulum associated with glycogen as “glycogen-bodies”, and speculated that these formations indicate a “resistance” to the carcinogen, reflecting a reactivation of the glycogen-storing ability after an early loss of glycogen in response to toxicity. In contrast to both of these considerations, many studies on experimental hepatocarcinogenesis in different species revealed that FHG composed of glycogenotic clear and/or acidophilic cells, the latter showing a pronounced hypertrophy of the smooth endoplasmic reticulum (corresponding to glycogenotic GGH), regularly occur in a multi-centric fashion in early stages of neoplastic development induced in small rodents by a variety of chemicals[3,4,9,13]. More recently, typical glycogenotic GGH were also found after chronic infection of woodchucks with hepadnaviridae[14-16], in hepatitis B virus (HBV)-transgenic mice[17], and during hormonal hepatocarcinogenesis in diabetic animals after intrahepatic transplantation of pancreatic islets in rats and mice[18,19].
Figure 2.
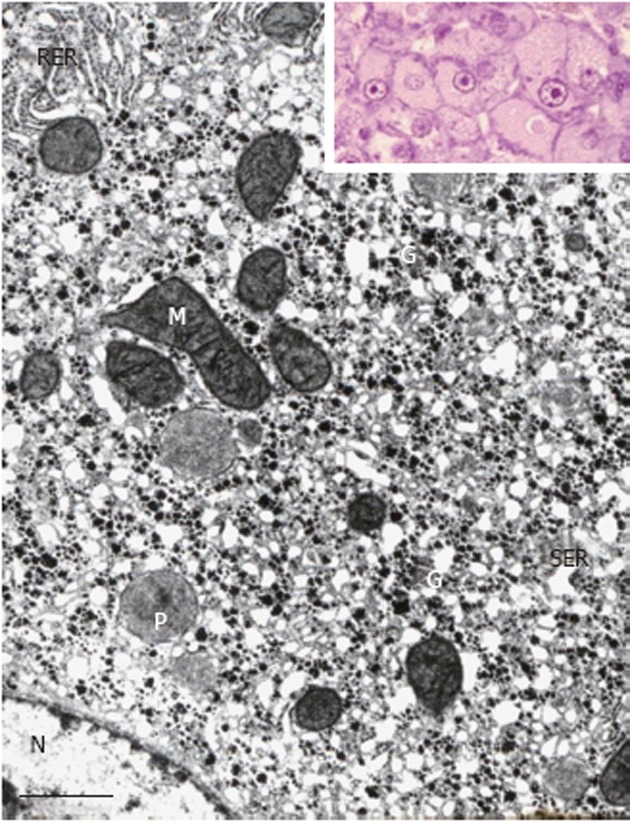
Portion of a glycogenotic acidophilic hepatocyte (corresponding to glycogen-ground-glass hepatocyte) induced in rat liver by N-nitrosomorpholine. Note abundant α- and β-glycogen particles (G) in close spatial relationship with large network complexes of proliferated smooth endoplasmic reticulum (SER). Rough endoplasmic reticulum (RER), mitochondria (M), peroxisomes (P), and nucleus (N). Inset: Group of acidophilic hepatocytes as seen under the light microscope. Transmission electron microscopy, lead citrate. Bar: 1 μm.
As demonstrated particularly in stop models of hepatocarcinogenesis in rats exposed to N-nitrosomorpholine or thioacetamide, glycogenotic hepatocytes with and without hypertrophy of the smooth endoplasmic reticulum may persist for weeks and months after withdrawal of the carcinogen, and may persist as almost entire populations of hepatocellular adenomas (“hyperplastic nodules”, “neoplastic nodules”) and in subpopulations of highly differentiated HCCs[4,20,21]. These observations are neither compatible with the idea of a preferential cellular survival by loss of differentiation for glycogenesis[10] nor with the notion of a “resistance” of these cells to carcinogen[12,22,23]. The functional significance of the persistent hypertrophy of the smooth endoplasmic reticulum has remained obscure, but many findings suggest that the organelle represents a metabolic compartment whose increase is a facultative consequence of the disturbed carbohydrate metabolism characterizing the acquired hepatocellular glycogenosis[4,24].
Independent of the observation of an acquired FHG produced in rat liver by N-nitrosomorpholine[5], Gössner et al[25] described a focal reduction in the activity of glucose-6-phosphatase in rats exposed to N-nitrosodiethylamine. A causal relationship between this enzyme deficiency and the accumulation of glycogen in preneoplastic FHG and “hyperplastic liver nodules” has been suggested by a number of authors[3,4,26,27]. This conclusion appears to be supported by the well known high risk of children suffering from an inborn hepatic glycogenosis type I, especially type Ia (von Gierke), due to a genetically fixed deficiency of the glucose-6-phosphatase, to develop hepatocellular adenomas and carcinomas when passing through adolescence[2,9,28]. This interpretation is in line with the recent finding that the targeted deletion of liver glucose-6-phosphatase in a knock-out mouse model results in hepatic glycogenosis and steatosis, and eventually also in multiple hepatocellular adenomas in all animals beyond 18 mo of age[29].
It is important to realize, however, that correlative cytochemical studies in rodent models of hepatocarcinogenesis have shown that the focal decrease of glucose-6-phosphatase activity in FHG is regularly combined with decrease or increase in the activity of many other enzymes[7,8,30], especially enzymes of the carbohydrate metabolism[2,24,31]. In addition, over-expression of the key enzyme of de novo fatty acid synthase has been described in FHG in the N-nitrosomorpholine stop-model[32]. In rats exposed to N-2-fluorenylacetamide, Williams et al[33] demonstrated that the preneoplastic glycogenotic clear cell foci are resistant to the storage of iron.
For a long time the cause of these complex metabolic alterations in FHG remained elusive. More recently, however, histochemical, microbiochemical and molecular biochemical studies on FHG and advanced preneoplastic and neoplastic liver lesions in tissue sections and laser-dissected specimens obtained from small rodents exposed to chemical carcinogens or oncogenic viruses provided evidence for an early insulin-like (insulinomimetic) effect of these agents[7,24,34-36]. This notion has been corroborated by a number of studies on hormonal hepatocarcinogenesis induced in diabetic rats and mice by local hyperinsulinemia[18,19,32,37-39].
The phenotype of preneoplastic FHG is not stable, but undergoes dramatic changes during progression to the benign and/or malignant neoplastic phenotype[3,4]. Collectively, all types of specific focal hepatocellular lesions appearing during the preneoplastic phase in rodents have been termed foci of altered hepatocytes, and have been widely used as early indicators of neoplastic development in toxicologic pathology[40-42]. The characteristic sequence of cellular changes starting with FHG follows an ordered pattern, passing through intermediate or mixed cell foci composed of glycogenotic, intermediate, and glycogen-poor basophilic (ribosome-rich) cell types, the latter corresponding to the typical cell type in poorly differentiated HCCs[2,4,24]. Detailed morphological analysis of intermediate cell types at the light- and electron microscopic level has suggested that the hypertrophied smooth endoplasmic reticulum is usually transformed into rough endoplasmic reticulum by the addition of ribosomes during this phenotypic conversion[4,9,20]. Frequently, the intermediate cells show an accumulation of neutral fat, often leading to a combination of glycogenosis and steatosis[2,20]. Evidence of this sequence of cellular changes was originally provided by light- and electron-microscopic studies in rats exposed for the lifetime or for limited time periods to N-nitrosomorpholine[3,4]. And it has since been substantiated by a series of morphometric studies[43-45] and by similar observations in other rodent models of hepatocarcinogenesis elicited by several “genotoxic” and “nongenotoxic” chemicals[7,41], by local hyperinsulinemia[18], by hepadnaviridae[14,15], and by oncogenic transgenes[17,46].Most recently, multiple FHG, more advanced types of foci of altered hepatocytes, hepatocellular adenomas and HCC indicative of such a sequence, including an intermediate steatosis, were also observed in a knock-out mouse model with a reduced expression of the mitochondrial protein frataxin, which is responsible for the inherited neurodegenerative disease Friedreich´s ataxia in humans[47].
The conversion of the highly differentiated glycogenotic clear or acidophilic to the de-differentiated glycogen-poor, basophilic (ribosome-rich) phenotype is associated with a fundamental metabolic switch characterized by a reduction in gluconeogenesis, an activation of the pentose phosphate pathway and the Warburg type of glycolysis as detailed elsewhere[7,8,24], and by an ever increasing cell proliferation which is inversely related to the gradual reduction of the glycogen initially stored in excess[48]. Based on these observations, Kopp-Schneider et al[49] developed the so-called color-shift model of hepatocarcinogenesis considering epigenetic changes in parenchymal colonies rather than multiple successive genomic mutations in single cells as the main cause of neoplastic cell conversion induced in the liver by exogenous oncogenic agents. The importance of epigenetic events in chemical hepatocarcinogenesis has been discussed by several authors previously[7,50], and has been emphasized in recent years by Pogribny et al[51,52].
Human hepatocarcinogenesis
In human pathology, the predominance of clear (glycogenotic) cells (Figure 1A and B) in a minor proportion of HCCs[6,53], comprising about 8% in 150 cases studied by Buchanan et al[54], and in many hepatocellular adenomas[6,53-56] is well known. A favorable prognosis of the clear-cell variant of HCC has been reported[57]. Sasaki et al[58] described two cases of clear-cell HCC associated with hypoglycemia and hypercholesterolemia, and postulated a disturbed glucose metabolism of the tumor tissue, directed to lipogenesis and/or glucogenesis. Acquired FHG has been considered a preneoplastic condition in humans[6]. This idea was supported by the fortuitous observation of FHG (clear-cell foci) in HCC-bearing livers of children suffering from different disorders[59,60], in women after long-term use of oral contraceptives[61], in about 12% of 95 males studied in a consecutive autopsy series in Finland[62], in patients with Crohn’s disease treated over years with azathioprine which is apparently also responsible for associated HCC development[63-67], and in a variety of other chronic liver diseases prone to develop HCC[8,24,53,68,69]. Special cases are patients with genetic hemochromatosis endowed with a high risk of developing HCC, which frequently show FHG excluding iron similar to the iron-resistant FHG observed in experimental hepatocarcinogenesis in rodents[8,53,69-73].
Particularly relevant are systematic histochemical and histological investigations on the phenotype and proliferation kinetics of foci and nodules of altered hepatocytes in more than 150 explanted and resected human livers with and without HCC[68,74,75]. The results suggest that foci of altered hepatocytes are proliferative preneoplastic lesions, mixed cell foci (frequently with “small cell change”) being more advanced than FHG (Figure 3), potentially transforming into nodules of altered hepatocytes, highly differentiated HCC containing glycogenotic clear and ground-glass hepatocytes (Figure 4), and eventually also glycogen-poor, basophilic HCC (Figure 1C)[68]. In keeping with these findings, clear-cell change, steatosis and small cell change have been considered histological features predicting malignant transformation in non-malignant hepatocellular nodules[76]. Analysis of clonality and chromosomal aberrations in nodules of altered hepatocytes microdissected from cirrhotic livers revealed a loss of chromosomal inactivation mosaicism in three large “regenerative” nodules and in all (12) nodules of altered hepatocytes with small cell change, indicating their neoplastic nature[77]. Even among 60 nodules of altered hepatocytes without small cell change, almost 50% (29) were shown to be monoclonal, whereas FHG and 14 “regenerative” nodules were found to be polyclonal. Interestingly, Cai et al[78] using a similar approach to analyze focal nodular hyperplasia, the pathogenesis and neoplastic nature of which has been debated for decades[53], found that this lesion, as a whole, is polyclonal, but represents a cluster of nodules of altered hepatocytes, some of which are monoclonal harboring chromosomal aberrations as in hepatocellular adenomas. The lack of genomic alterations in fatty and clear-cell changes in HCC and precursor nodular lesions in cirrhotic livers emphasized by some authors[79] is in line with the polyclonal nature of many of these lesions[77], but in view of the increasing evidence for a decisive role of epigenetic events in the development and progression of human HCC[80] the findings by Laurent et al[79] do not argue against a preneoplastic nature for these cellular changes.
Figure 3.
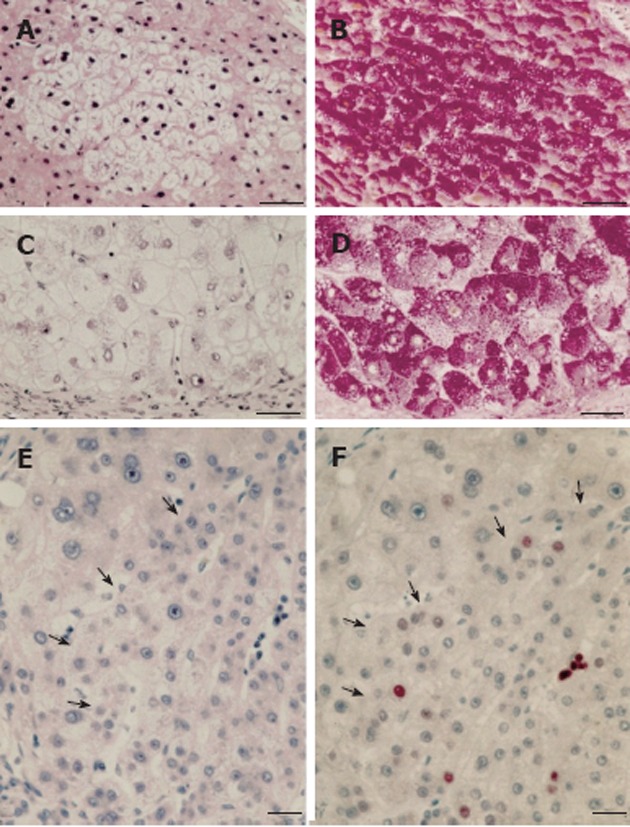
Human focal (A, B) and nodular (C, D) hepatic glycogenosis, and more advanced mixed cell populations (E, F) with intrafocal small cell change. A, B: Hepatic vein. Perivenular glycogenolytic (clear cell) focus in an hepatocellular carcinoma (HCC)-bearing liver with hepatitis B virus (HBV)-associated cirrhosis, demonstrated in serial sections with hematoxylin and eosin (A) and periodic acid schiff (PAS) (B)-reaction counterstained with orange G and iron hematoxilin (Tri-PAS). Bar: 100 μm; C, D: Glycogenotic (clear cell) nodule in an HCC-free liver with HBV-associated cirrhosis demonstrated in serial sections with hematoxylin-eosin (C) and Tri-PAS (D). Bar: 100 μm; E, F: Hematoxylin and eosin (E), proliferating cell nuclear antigen (F) immunostaining. Increased cell proliferation (arrows) in a mixed cell focus with less pronounced glycogen storage (not shown) and with intrafocal small cell change in liver with cryptogenic cirrhosis, demonstrated in serial sections. Bar: 50 μm, from Su et al[68].
Figure 4.
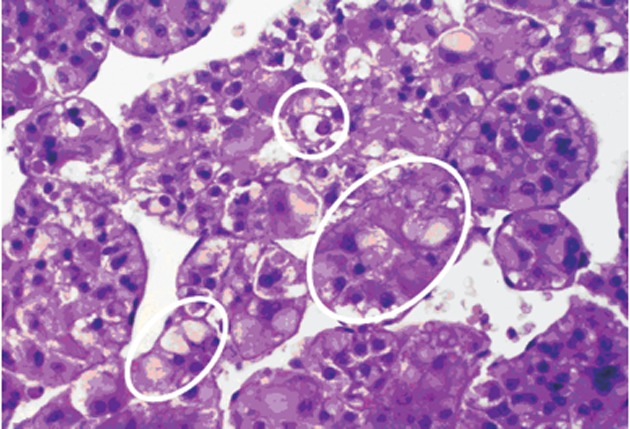
Portion of a human trabecular hepatocellular carcinoma with mixed populations, including clear (glycogenotic) cells (small upper white circle), acidophilic (glycogenotic) ground-glass cells (somewhat larger white circle at the bottom, left), transitions from ground-glass hepatocytes into basophilic cell populations (largest white circle in the middle, right), and basophilic cell populations in the middle and at the bottom right (not marked by circles). Hematoxylin and eosin.
A pronounced hypertrophy of the smooth endoplasmic reticulum was discovered in biopsies from cirrhotic livers and liver cell carcinomas at the light and electron microscopic level (Figure 1B) almost half a century ago, and related to aberrations of glycogen metabolism including glycogenosis from the very beginning[5,6]. The altered hepatocytes (Figures 1B and 4) were designated as “acidophilic” (or “eosinophilic”). Later on, Popper et al[81,82] found frequent association of this phenomenon with the expression of the hepatitis B surface antigen (HBsAg) localized in the lumen of hypertrophied smooth endoplasmic reticulum (Figure 5), and coined the term “ground glass hepatocyte” which has become an important diagnostic entity in chronic liver diseases elicited by HBV and has dominated the literature since then[83]. Wills[84] described HBsAg-free “ground glasslike hepatocytes” exhibiting “glycogen bodies” in liver biopsies from an immunosuppressed (azathioprine, prednisone, clonidine, frusemide) renal transplant patient, similar to those observed in experimental chemical hepatocarcinogenesis[4,12].
Figure 5.
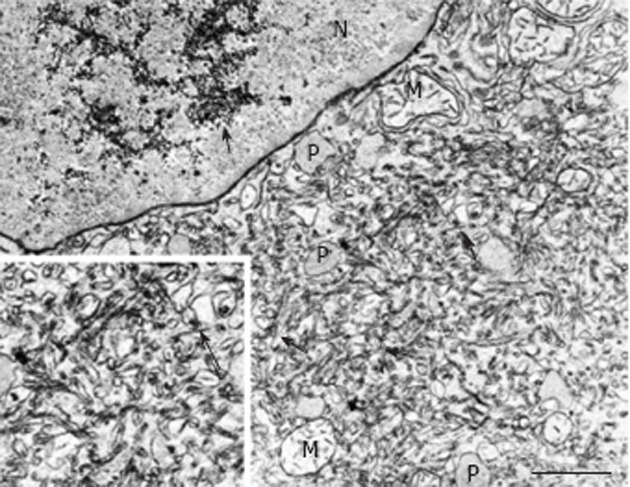
Portion of a ground-glass hepatocyte from a human liver with hepatitis B virus-associated cirrhosis, showing an accumulation of glycogen granules (long arrow) in the nucleus (N) and abundant smooth endoplasmic reticulum containing filamentous hepatitis B surface antigen (short arrows). M: Mitochondria: P: Peroxisome. Transmission electron microscopy, lead citrate. Bar: 1 μm.
Irrespective of the expression of HBsAg, glycogenotic hepatocytes showing hypertrophy of the smooth endoplasmic reticulum (corresponding to GGH) have often been observed in human FHG, hepatocellular adenomas, and HCC and considered preneoplastic or highly differentiated neoplastic phenotypes[6,53,68]. In 30 specimens of HBV-associated cirrhosis, GGH were identified in 17 of 25 showing HBsAg expression[85] (Figure 5). Without mentioning any particular relationship to glycogen, others described GGH containing pre-S mutants in chronic HBV infection, postulating that they represent preneoplastic lesions[83]. Wisell et al[86] depicted partially persisting “glycogen pseudoground glass hepatocytes” in 12 patients immunosuppressed for numerous indications, but detected neither viral particles nor hypertrophy of the smooth endoplasmic reticulum under the electron microscope. Similar observations were reported by Bejarano et al[87] but no efforts were made in either of these studies to directly compare GGH in serial sections of defined focal lesions at the light and electron microscopic level. Different types of altered hepatocytes, which superficially resemble glycogenotic GGH appearing during hepatocarcinogensis, have also been observed in several other diseases but will not be further discussed in this context[83,86,87].
An intriguing form of acquired hepatic glycogenosis was discovered by Mauriac et al[88] in a child with poorly controlled insulin-dependent diabetes type 1. The excessive storage of glycogen resulted in hepatomegaly and was associated with growth retardation, delayed puberty, and a cuchingoid face (named after Harvey Williams Cushing). Many additional cases resembling Mauriac’s syndrome, especially with respect to hepatic glycogenosis, have subsequently been described[89-91]. Torbenson et al[91] emphasized that this “glycogenic hepatopathy” is an underrecognized complication of diabetes mellitus. In addition to children with insulin-dependent diabetes, hepatomegaly due to glycogen storage has also been recognized in adults afflicted by non-insulin-dependent diabetes type 2 with poor glycemic control[91-93]. To the best of my knowledge neither GGH nor a relationship of glycogenic hepatopathy to the evolution of HCC in patients suffering from diabetes mellitus has hitherto been described, but it might be timely to take a closer look into this possibility.
The high risk of diffuse glycogenosis characterizing inborn hepatic glycogen storage diseases, particularly glycogen storage disease type I due to glucose-6-phosphatase deficiency, developing into hepatocellular adenomas potentially progressing to HCC has been well established[2,9,28,94] since the first description of a case by Mason et al[95]. In the meantime, hepatocellular neoplasms are now known to also be occasionally found in other types of glycogen storage disease, namely glycogenoses type III (amylo-1,6-glucosidase deficiency), type IV (α-1,4-glucan:α-1,4-glucan-6-glycosyl transferase deficiency) and type VI (phosphorylase deficiency, Hers disease)[94,96-99]. I am not aware of any explicit report of GGH in inborn glycogenoses, but from the findings outlined it is obvious that a more detailed comparison of the molecular, metabolic, and morphological aspects of hepatocarcinogenesis in inborn and acquired (focal) hepatic glycogenosis should help to further elucidate the pathogenesis of hepatocellular neoplasms, and facilitate development of appropriate measures for the prevention and therapy of this frequently fatal disease. From a diagnostic point of view it appears to be of great advantage to use the characteristic changes in hepatocellular glycogen content during hepatocarcinogenesis as simple “superficial” histochemical markers of complex basic aberrations at the molecular and metabolic level.
ACKNOWLEDGMENTS
I am indebted to Qin Su, MD, PhD, Professor, Shanghai, China, and Malcolm Moore, PhD, Nagoya, Japan, for critical reading of the manuscript.
Footnotes
Peer reviewers: Markus Peck-Radosavljevic, Professor, Department of Internal Medicine III, Division of Gastroenterology and Hepatology, University of Vienna, Waehringer Guertel 18-20, A-1090 Vienna, Austria; Dr. Maria Cristina Carrillo, Instituto de Fisiologia Experimental, Departamento de Ciencias Fisiologicas, Suipacha 570, Rosario 2000, Argentina
S- Editor Xiong L L- Editor A E- Editor Xiong L
References
- 1.Callea F, Giovannoni I, Stefanelli M, Villanacci V, Lorini G, Francalanci P. Glycogenotic hepatocellular carcinoma with glycogen-ground-glass hepatocytes: histological, histochemical and microbiochemical characterization of the novel variant. Histopathology. 2012;60:1010–1012. doi: 10.1111/j.1365-2559.2011.04168.x. [DOI] [PubMed] [Google Scholar]
- 2.Bannasch P, Hacker HJ, Klimek F, Mayer D. Hepatocellular glycogenosis and related pattern of enzymatic changes during hepatocarcinogenesis. Adv Enzyme Regul. 1984;22:97–121. doi: 10.1016/0065-2571(84)90010-4. [DOI] [PubMed] [Google Scholar]
- 3.Bannasch P, Mueller HA. [Light microscopic studies on the effects of n-nitrosomorpholine on the liver of rats and mice] Arzneimittelforschung. 1964;14:805–814. [PubMed] [Google Scholar]
- 4.Bannasch P. The cytoplasm of hepatocytes during carcinogenesis. Electron and light microscopical investigations of the nitrosomorpholine-intoxicated rat liver. In: Rentchnick P, editor. Recent Results in Cancer Research. Vol. 19. Heidelberg: Springer; 1968. pp. 1–100. [Google Scholar]
- 5.Klinge O, Bannasch P. [The increase of smooth endoplasmatic reticulum in hepatocytes of human liver punctates] Verh Dtsch Ges Pathol. 1968;52:568–573. [PubMed] [Google Scholar]
- 6.Bannasch P, Klinge O. [Hepatocellular glycogenosis and hepatoma development in man] Virchows Arch A Pathol Pathol Anat. 1971;352:157–164. [PubMed] [Google Scholar]
- 7.Bannasch P. Pathogenesis of hepatocellular carcinoma: sequential cellular, molecular, and metabolic changes. Prog Liver Dis. 1996;14:161–197. [PubMed] [Google Scholar]
- 8.Bannasch P, Schröder CH. Tumors and tumor-like lesions of the liver and biliary tract: pathogenesis of primary liver tumors. In: MacSween RNM, Burt AD, Portman BC, Ishak KG, Scheuer PJ, et al., editors. Pathology of the Liver. 4th ed. London: Churchill Livingstone; 2002. pp. 777–825. [Google Scholar]
- 9.Bannasch P, Mayer D, Hacker HJ. Hepatocellular glycogenosis and hepatocarcinogenesis. Biochim Biophys Acta. 1980;605:217–245. doi: 10.1016/0304-419x(80)90005-0. [DOI] [PubMed] [Google Scholar]
- 10.Porter KR, Bruni C. An electron microscope study of the early effects of 3’-Me-DAB on rat liver cells. Cancer Res. 1959;19:997–1009. [PubMed] [Google Scholar]
- 11.Bruni C. Hyaline degeneration of rat liver cells studied with the electron microscope. Lab Invest. 1960;9:209–215. [PubMed] [Google Scholar]
- 12.Steiner JW, Miyai K, Phillips MJ. Electron microscopy of membrane-particle arrays in liver cells of ethionine-intoxicated rats. Am J Pathol. 1964;44:169–214. [PMC free article] [PubMed] [Google Scholar]
- 13.Carter JH, Carter HW, Deddens JA, Hurst BM, George MH, DeAngelo AB. A 2-year dose-response study of lesion sequences during hepatocellular carcinogenesis in the male B6C3F(1) mouse given the drinking water chemical dichloroacetic acid. Environ Health Perspect. 2003;111:53–64. doi: 10.1289/ehp.5442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bannasch P, Khoshkhou NI, Hacker HJ, Radaeva S, Mrozek M, Zillmann U, Kopp-Schneider A, Haberkorn U, Elgas M, Tolle T. Synergistic hepatocarcinogenic effect of hepadnaviral infection and dietary aflatoxin B1 in woodchucks. Cancer Res. 1995;55:3318–3330. [PubMed] [Google Scholar]
- 15.Radaeva S, Li Y, Hacker HJ, Burger V, Kopp-Schneider A, Bannasch P. Hepadnaviral hepatocarcinogenesis: in situ visualization of viral antigens, cytoplasmic compartmentation, enzymic patterns, and cellular proliferation in preneoplastic hepatocellular lineages in woodchucks. J Hepatol. 2000;33:580–600. doi: 10.1034/j.1600-0641.2000.033004580.x. [DOI] [PubMed] [Google Scholar]
- 16.Xu C, Yamamoto T, Zhou T, Aldrich CE, Frank K, Cullen JM, Jilbert AR, Mason WS. The liver of woodchucks chronically infected with the woodchuck hepatitis virus contains foci of virus core antigen-negative hepatocytes with both altered and normal morphology. Virology. 2007;359:283–294. doi: 10.1016/j.virol.2006.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toshkov I, Chisari FV, Bannasch P. Hepatic preneoplasia in hepatitis B virus transgenic mice. Hepatology. 1994;20:1162–1172. [PubMed] [Google Scholar]
- 18.Dombrowski F, Bannasch P, Pfeifer U. Hepatocellular neoplasms induced by low-number pancreatic islet transplants in streptozotocin diabetic rats. Am J Pathol. 1997;150:1071–1087. [PMC free article] [PubMed] [Google Scholar]
- 19.Dombrowski F, Mathieu C, Evert M. Hepatocellular neoplasms induced by low-number pancreatic islet transplants in autoimmune diabetic BB/Pfd rats. Cancer Res. 2006;66:1833–1843. doi: 10.1158/0008-5472.CAN-05-2787. [DOI] [PubMed] [Google Scholar]
- 20.Bannasch P. Cytology and cytogenesis of neoplastic (hyperplastic) hepatic nodules. Cancer Res. 1976;36:2555–2562. [PubMed] [Google Scholar]
- 21.Bannasch P, Hesse J, Angerer H. [Hepatocellular glycogenosis and the genesis of so-called hyperplastic liver nodules in thioacetamide intoxicated rats (author’s transl)] Virchows Arch B Cell Pathol. 1974;17:29–50. [PubMed] [Google Scholar]
- 22.Farber E. Hyperplastic liver nodules. In: Busch H, et al., editors. Methods Cancer Research. Vol. 7. New York: Academic Press; 1973. pp. 345–379. [Google Scholar]
- 23.Merkow LP, Epstein SM, Slifkin M, Farber E, Pardo M. Ultrastructural alterations within hyperplastic liver nodules induced by ethionine. Cancer Res. 1971;31:174–178. [PubMed] [Google Scholar]
- 24.Bannasch P, Klimek F, Mayer D. Early bioenergetic changes in hepatocarcinogenesis: preneoplastic phenotypes mimic responses to insulin and thyroid hormone. J Bioenerg Biomembr. 1997;29:303–313. doi: 10.1023/a:1022438528634. [DOI] [PubMed] [Google Scholar]
- 25.Gössner W, Friedrich-Freska H. [Histochemical studies on glucose-6-phosphatase in the rat liver during cancerization by nitrosamine] Z Naturforsch B. 1964;19:862–863. [PubMed] [Google Scholar]
- 26.Epstein S, Ito N, Merkow L, Farber E. Cellular analysis of liver carcinogenesis: the induction of large hyperplastic nodules in the liver with 2-fluorenylacetamide or ethionine and some aspects of their morphology and glycogen metabolism. Cancer Res. 1967;27:1702–1711. [PubMed] [Google Scholar]
- 27.Friedrich-Freksa H, Gössner W, Börner P. [Histochemical investigations of carcinogenesis in rat liver after continuous application of diethylnitrosamine] Z Krebsforsch. 1969;72:226–239. [PubMed] [Google Scholar]
- 28.Bianchi L. Glycogen storage disease I and hepatocellular tumors. Eur J Pediatr. 1993;152 Suppl 1:S63–S70. doi: 10.1007/BF02072092. [DOI] [PubMed] [Google Scholar]
- 29.Mutel E, Abdul-Wahed A, Ramamonjisoa N, Stefanutti A, Houberdon I, Cavassila S, Pilleul F, Beuf O, Gautier-Stein A, Penhoat A, et al. Targeted deletion of liver glucose-6 phosphatase mimics glycogen storage disease type 1a including development of multiple adenomas. J Hepatol. 2011;54:529–537. doi: 10.1016/j.jhep.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 30.Moore MA, Tatematsu M. Are the Phenotypes of Preneoplastic Lesions of Significance for Cancer Prevention? 1. Liver. Asian Pac J Cancer Prev. 2001;2:27–42. [PubMed] [Google Scholar]
- 31.Hacker HJ, Moore MA, Mayer D, Bannasch P. Correlative histochemistry of some enzymes of carbohydrate metabolism in preneoplastic and neoplastic lesions in the rat liver. Carcinogenesis. 1982;3:1265–1272. doi: 10.1093/carcin/3.11.1265. [DOI] [PubMed] [Google Scholar]
- 32.Evert M, Schneider-Stock R, Dombrowski F. Overexpression of fatty acid synthase in chemically and hormonally induced hepatocarcinogenesis of the rat. Lab Invest. 2005;85:99–108. doi: 10.1038/labinvest.3700206. [DOI] [PubMed] [Google Scholar]
- 33.Williams GM, Klaiber M, Parker SE, Farber E. Nature of early appearing, carcinogen-induced liver lesions to iron accumulation. J Natl Cancer Inst. 1976;57:157–165. doi: 10.1093/jnci/57.1.157. [DOI] [PubMed] [Google Scholar]
- 34.Klimek F, Bannasch P. Isoenzyme shift from glucokinase to hexokinase is not an early but a late event in hepatocarcinogenesis. Carcinogenesis. 1993;14:1857–1861. doi: 10.1093/carcin/14.9.1857. [DOI] [PubMed] [Google Scholar]
- 35.Nehrbass D, Klimek F, Bannasch P. Overexpression of insulin receptor substrate-1 emerges early in hepatocarcinogenesis and elicits preneoplastic hepatic glycogenosis. Am J Pathol. 1998;152:341–345. [PMC free article] [PubMed] [Google Scholar]
- 36.Aleem E, Nehrbass D, Klimek F, Mayer D, Bannasch P. Upregulation of the insulin receptor and type I insulin-like growth factor receptor are early events in hepatocarcinogenesis. Toxicol Pathol. 2011;39:524–543. doi: 10.1177/0192623310396905. [DOI] [PubMed] [Google Scholar]
- 37.Scharf JG, Ramadori G, Dombrowski F. Analysis of the IGF axis in preneoplastic hepatic foci and hepatocellular neoplasms developing after low-number pancreatic islet transplantation into the livers of streptozotocin diabetic rats. Lab Invest. 2000;80:1399–1411. doi: 10.1038/labinvest.3780147. [DOI] [PubMed] [Google Scholar]
- 38.Evert M, Sun J, Pichler S, Slavova N, Schneider-Stock R, Dombrowski F. Insulin receptor, insulin receptor substrate-1, Raf-1, and Mek-1 during hormonal hepatocarcinogenesis by intrahepatic pancreatic islet transplantation in diabetic rats. Cancer Res. 2004;64:8093–8100. doi: 10.1158/0008-5472.CAN-04-2040. [DOI] [PubMed] [Google Scholar]
- 39.Evert M, Calvisi DF, Evert K, De Murtas V, Gasparetti G, Mattu S, Destefanis G, Ladu S, Zimmermann A, Delogu S, et al. V-AKT murine thymoma viral oncogene homolog/mammalian target of rapamycin activation induces a module of metabolic changes contributing to growth in insulin-induced hepatocarcinogenesis. Hepatology. 2012;55:1473–1484. doi: 10.1002/hep.25600. [DOI] [PubMed] [Google Scholar]
- 40.Histologic typing of liver tumors of the rat. Institute of Laboratory Animal Resources, National Research Council, National Academy of Sciences, Washington, D.C. J Natl Cancer Inst. 1980;64:177–206. [PubMed] [Google Scholar]
- 41.Bannasch P. Preneoplastic lesions as end points in carcinogenicity testing. I. Hepatic preneoplasia. Carcinogenesis. 1986;7:689–695. doi: 10.1093/carcin/7.5.689. [DOI] [PubMed] [Google Scholar]
- 42.Bannasch P, Haertel T, Su Q. Significance of hepatic preneoplasia in risk identification and early detection of neoplasia. Toxicol Pathol. 2003;31:134–139. doi: 10.1080/01926230390173923. [DOI] [PubMed] [Google Scholar]
- 43.Moore MA, Mayer D, Bannasch P. The dose dependence and sequential appearance of putative preneoplastic populations induced in the rat liver by stop experiments with N-nitrosomorpholine. Carcinogenesis. 1982;3:1429–1436. doi: 10.1093/carcin/3.12.1429. [DOI] [PubMed] [Google Scholar]
- 44.Enzmann H, Bannasch P. Potential significance of phenotypic heterogeneity of focal lesions at different stages in hepatocarcinogenesis. Carcinogenesis. 1987;8:1607–1612. doi: 10.1093/carcin/8.11.1607. [DOI] [PubMed] [Google Scholar]
- 45.Weber E, Bannasch P. Dose and time dependence of the cellular phenotype in rat hepatic preneoplasia and neoplasia induced by continuous oral exposure to N-nitrosomorpholine. Carcinogenesis. 1994;15:1235–1242. doi: 10.1093/carcin/15.6.1235. [DOI] [PubMed] [Google Scholar]
- 46.Kim CM, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature. 1991;351:317–320. doi: 10.1038/351317a0. [DOI] [PubMed] [Google Scholar]
- 47.Thierbach R, Florian S, Wolfrum K, Voigt A, Drewes G, Blume U, Bannasch P, Ristow M, Steinberg P. Specific alterations of carbohydrate metabolism are associated with hepatocarcinogenesis in mitochondrially impaired mice. Hum Mol Genet. 2012;21:656–663. doi: 10.1093/hmg/ddr499. [DOI] [PubMed] [Google Scholar]
- 48.Zerban H, Radig S, Kopp-Schneider A, Bannasch P. Cell proliferation and cell death (apoptosis) in hepatic preneoplasia and neoplasia are closely related to phenotypic cellular diversity and instability. Carcinogenesis. 1994;15:2467–2473. doi: 10.1093/carcin/15.11.2467. [DOI] [PubMed] [Google Scholar]
- 49.Kopp-Schneider A, Portier C, Bannasch P. A model for hepatocarcinogenesis treating phenotypical changes in focal hepatocellular lesions as epigenetic events. Math Biosci. 1998;148:181–204. doi: 10.1016/s0025-5564(97)10007-4. [DOI] [PubMed] [Google Scholar]
- 50.Ghoshal AK, Farber E. The induction of liver cancer by dietary deficiency of choline and methionine without added carcinogens. Carcinogenesis. 1984;5:1367–1370. doi: 10.1093/carcin/5.10.1367. [DOI] [PubMed] [Google Scholar]
- 51.Pogribny IP, Ross SA, Wise C, Pogribna M, Jones EA, Tryndyak VP, James SJ, Dragan YP, Poirier LA. Irreversible global DNA hypomethylation as a key step in hepatocarcinogenesis induced by dietary methyl deficiency. Mutat Res. 2006;593:80–87. doi: 10.1016/j.mrfmmm.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 52.Pogribny IP, Shpyleva SI, Muskhelishvili L, Bagnyukova TV, James SJ, Beland FA. Role of DNA damage and alterations in cytosine DNA methylation in rat liver carcinogenesis induced by a methyl-deficient diet. Mutat Res. 2009;669:56–62. doi: 10.1016/j.mrfmmm.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 53.Altmann HW. Hepatic neoformations. Pathol Res Pract. 1994;190:513–577. doi: 10.1016/S0344-0338(11)80394-8. [DOI] [PubMed] [Google Scholar]
- 54.Buchanan TF, Huvos AG. Clear-cell carcinoma of the liver. A clinicopathologic study of 13 patients. Am J Clin Pathol. 1974;61:529–539. doi: 10.1093/ajcp/61.4.529. [DOI] [PubMed] [Google Scholar]
- 55.Garancis JC, Tang T, Panares R, Jurevics I. Hepatic adenoma. Biochemical and electron microscopic study. Cancer. 1969;24:560–568. doi: 10.1002/1097-0142(196909)24:3<560::aid-cncr2820240320>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 56.Hamperl H. “Adrenal rest-tumors” (hypernephromas) of the liver. Z Krebsforsch. 1970;74:310–317. doi: 10.1007/BF00531211. [DOI] [PubMed] [Google Scholar]
- 57.Wu PC, Lai CL, Lam KC, Lok AS, Lin HJ. Clear cell carcinoma of liver. An ultrastructural study. Cancer. 1983;52:504–507. doi: 10.1002/1097-0142(19830801)52:3<504::aid-cncr2820520321>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 58.Sasaki K, Okuda S, Takahashi M, Sasaki M. Hepatic clear cell carcinoma associated with hypoglycemia and hypercholesterolemia. Cancer. 1981;47:820–822. doi: 10.1002/1097-0142(19810215)47:4<820::aid-cncr2820470432>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 59.Balázs M. [Light and electron microscopie examination of a case of primary liver carcinoma in an infant (author’s transl)] Zentralbl Allg Pathol. 1976;120:3–13. [PubMed] [Google Scholar]
- 60.Cain H, Kraus B. [Developmental anomalies of the liver and carcinoma of the liver in infants and children (author’s transl)] Dtsch Med Wochenschr. 1977;102:505–509. doi: 10.1055/s-0028-1104920. [DOI] [PubMed] [Google Scholar]
- 61.Fischer G, Hartmann H, Droese M, Schauer A, Bock KW. Histochemical and immunohistochemical detection of putative preneoplastic liver foci in women after long-term use of oral contraceptives. Virchows Arch B Cell Pathol Incl Mol Pathol. 1986;50:321–337. doi: 10.1007/BF02889911. [DOI] [PubMed] [Google Scholar]
- 62.Karhunen PJ, Penttilä A. Preneoplastic lesions of human liver. Hepatogastroenterology. 1987;34:10–15. [PubMed] [Google Scholar]
- 63.Cattan S, Wendum D, Chazouilleres O, Schmitz J, Gendre JP. Hepatocellular carcinoma and focal hepatic glycogenosis after prolonged azathioprine therapy. Hum Pathol. 2000;31:874–876. doi: 10.1053/hupa.2000.7629. [DOI] [PubMed] [Google Scholar]
- 64.Klein WM, Molmenti EP, Colombani PM, Grover DS, Schwarz KB, Boitnott J, Torbenson MS. Primary liver carcinoma arising in people younger than 30 years. Am J Clin Pathol. 2005;124:512–518. doi: 10.1309/TT0R7KAL32228E99. [DOI] [PubMed] [Google Scholar]
- 65.Chen SC, Cummings OW, Hartley MP, Filomena CA, Cho WK. Hepatocellular carcinoma occurring in a patient with Crohn’s disease treated with both azathioprine and infliximab. Dig Dis Sci. 2006;51:952–955. doi: 10.1007/s10620-005-9009-9. [DOI] [PubMed] [Google Scholar]
- 66.Murakami A, Tanaka Y, Ueda M, Nagano Y, Kunisaki R, Morimoto M, Enaka M, Tanabe M, Kawachi K, Sasaki T, et al. Hepatocellular carcinoma occurring in a young Crohn’s disease patient. Pathol Int. 2009;59:492–496. doi: 10.1111/j.1440-1827.2009.02399.x. [DOI] [PubMed] [Google Scholar]
- 67.Ishida M, Naka S, Shiomi H, Tsujikawa T, Andoh A, Nakahara T, Saito Y, Kurumi Y, Takikita-Suzuki M, Kojima F, et al. Hepatocellular carcinoma occurring in a Crohn’s disease patient. World J Gastroenterol. 2010;16:3215–3218. doi: 10.3748/wjg.v16.i25.3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Su Q, Benner A, Hofmann WJ, Otto G, Pichlmayr R, Bannasch P. Human hepatic preneoplasia: phenotypes and proliferation kinetics of foci and nodules of altered hepatocytes and their relationship to liver cell dysplasia. Virchows Arch. 1997;431:391–406. doi: 10.1007/s004280050116. [DOI] [PubMed] [Google Scholar]
- 69.Evert M, Dombrowski F. [Hepatocellular carcinoma in the non-cirrhotic liver] Pathologe. 2008;29:47–52. doi: 10.1007/s00292-007-0953-3. [DOI] [PubMed] [Google Scholar]
- 70.Hirota N, Hamazaki M, Williams GM. Resistance to iron accumulation and presence of hepatitis B surface antigen in preneoplastic and neoplastic lesions in human hemochromatotic livers. Hepatogastroenterology. 1982;29:49–51. [PubMed] [Google Scholar]
- 71.Mori H, Tanaka T, Sugie S, Takahashi M, Williams GM. DNA content of liver cell nuclei of N-2-fluorenylacetamide-induced altered foci and neoplasms in rats and human hyperplastic foci. J Natl Cancer Inst. 1982;69:1277–1282. [PubMed] [Google Scholar]
- 72.Deugnier YM, Charalambous P, Le Quilleuc D, Turlin B, Searle J, Brissot P, Powell LW, Halliday JW. Preneoplastic significance of hepatic iron-free foci in genetic hemochromatosis: a study of 185 patients. Hepatology. 1993;18:1363–1369. [PubMed] [Google Scholar]
- 73.Sá Cunha A, Blanc JF, Trillaud H, De Ledinghen V, Balabaud C, Bioulac-Sage P. Hypervascular nodule in a fibrotic liver overloaded with iron: identification of a premalignant area with preserved liver architecture. Comp Hepatol. 2005;4:5. doi: 10.1186/1476-5926-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bannasch P, Jahn U, Hacker H, Su Q, Hoffmann W, Pichlmayr R, Otto G. Focal hepatic glycogenosis. Int J Oncol. 1997;10:261–268. doi: 10.3892/ijo.10.2.261. [DOI] [PubMed] [Google Scholar]
- 75.Su Q, Bannasch P. Relevance of hepatic preneoplasia for human hepatocarcinogenesis. Toxicol Pathol. 2003;31:126–133. doi: 10.1080/01926230390173905. [DOI] [PubMed] [Google Scholar]
- 76.Terasaki S, Kaneko S, Kobayashi K, Nonomura A, Nakanuma Y. Histological features predicting malignant transformation of nonmalignant hepatocellular nodules: a prospective study. Gastroenterology. 1998;115:1216–1222. doi: 10.1016/s0016-5085(98)70093-9. [DOI] [PubMed] [Google Scholar]
- 77.Gong L, Li YH, Su Q, Chu X, Zhang W. Clonality of nodular lesions in liver cirrhosis and chromosomal abnormalities in monoclonal nodules of altered hepatocytes. Histopathology. 2010;56:589–599. doi: 10.1111/j.1365-2559.2010.03523.x. [DOI] [PubMed] [Google Scholar]
- 78.Cai YR, Gong L, Teng XY, Zhang HT, Wang CF, Wei GL, Guo L, Ding F, Liu ZH, Pan QJ, et al. Clonality and allelotype analyses of focal nodular hyperplasia compared with hepatocellular adenoma and carcinoma. World J Gastroenterol. 2009;15:4695–4708. doi: 10.3748/wjg.15.4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Laurent C, Quaglia A, Foroni L, Bioulac-Sage P, Balabaud C, Dhillon AP. Genomic alteration is not associated with fatty and clear cell change in hepatocellular carcinomas and its precursor nodular lesions in cirrhotic liver. Hepatol Res. 2006;36:40–47. doi: 10.1016/j.hepres.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 80.Pogribny IP, Rusyn I. Role of epigenetic aberrations in the development and progression of human hepatocellular carcinoma. Cancer Lett. 2012:Feb 2 [Epub ahead of print]. doi: 10.1016/j.canlet.2012.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hadziyannis S, Gerber MA, Vissoulis C, Popper H. Cytoplasmic hepatitis B antigen in “ground-glass” hepatocytes of carriers. Arch Pathol. 1973;96:327–330. [PubMed] [Google Scholar]
- 82.Popper H. The ground glass hepatocyte as a diagnostic hint. Hum Pathol. 1975;6:517–520. doi: 10.1016/s0046-8177(75)80069-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Su IJ, Wang HC, Wu HC, Huang WY. Ground glass hepatocytes contain pre-S mutants and represent preneoplastic lesions in chronic hepatitis B virus infection. J Gastroenterol Hepatol. 2008;23:1169–1174. doi: 10.1111/j.1440-1746.2008.05348.x. [DOI] [PubMed] [Google Scholar]
- 84.Wills EJ. Ground glasslike hepatocytes produced by glycogen-membrane complexes (“glycogen bodies”) Ultrastruct Pathol. 1992;16:491–503. doi: 10.3109/01913129209057834. [DOI] [PubMed] [Google Scholar]
- 85.Su Q, Zerban H, Otto G, Bannasch P. Cytokeratin expression is reduced in glycogenotic clear hepatocytes but increased in ground-glass cells in chronic human and woodchuck hepadnaviral infection. Hepatology. 1998;28:347–359. doi: 10.1002/hep.510280209. [DOI] [PubMed] [Google Scholar]
- 86.Wisell J, Boitnott J, Haas M, Anders RA, Hart J, Lewis JT, Abraham SC, Torbenson M. Glycogen pseudoground glass change in hepatocytes. Am J Surg Pathol. 2006;30:1085–1090. doi: 10.1097/01.pas.0000208896.92988.fc. [DOI] [PubMed] [Google Scholar]
- 87.Bejarano PA, Garcia MT, Rodriguez MM, Ruiz P, Tzakis AG. Liver glycogen bodies: ground-glass hepatocytes in transplanted patients. Virchows Arch. 2006;449:539–545. doi: 10.1007/s00428-006-0286-2. [DOI] [PubMed] [Google Scholar]
- 88.Mauriac P. Gros ventre, hepatomegalie, troubles de las croissance chez les enfants diabetiques traits depuis plusieurs annes par l’insuline. Gaz Hebd Sci Med Bordeaux. 1930;26:402–404. [Google Scholar]
- 89.Evans RW, Littler TR, Pemberton HS. Glycogen storage in the liver in diabetes mellitus. J Clin Pathol. 1955;8:110–113. doi: 10.1136/jcp.8.2.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chatila R, West AB. Hepatomegaly and abnormal liver tests due to glycogenosis in adults with diabetes. Medicine (Baltimore) 1996;75:327–333. doi: 10.1097/00005792-199611000-00003. [DOI] [PubMed] [Google Scholar]
- 91.Torbenson M, Chen YY, Brunt E, Cummings OW, Gottfried M, Jakate S, Liu YC, Yeh MM, Ferrell L. Glycogenic hepatopathy: an underrecognized hepatic complication of diabetes mellitus. Am J Surg Pathol. 2006;30:508–513. doi: 10.1097/00000478-200604000-00012. [DOI] [PubMed] [Google Scholar]
- 92.Vaishnava H, Gulati PD, Damodaran VN. Observations on the structure and function of liver in Indian diabetics. Diabetologia. 1970;6:21–26. doi: 10.1007/BF00425887. [DOI] [PubMed] [Google Scholar]
- 93.Tsujimoto T, Takano M, Nishiofuku M, Yoshiji H, Matsumura Y, Kuriyama S, Uemura M, Okamoto S, Fukui H. Rapid onset of glycogen storage hepatomegaly in a type-2 diabetic patient after a massive dose of long-acting insulin and large doses of glucose. Intern Med. 2006;45:469–473. doi: 10.2169/internalmedicine.45.1548. [DOI] [PubMed] [Google Scholar]
- 94.Wolfsdorf JI, Weinstein DA. Glycogen storage diseases. Rev Endocr Metab Disord. 2003;4:95–102. doi: 10.1023/a:1021831621210. [DOI] [PubMed] [Google Scholar]
- 95.Mason HH, Andersen DH. Glycogen disease of the liver (von Gierke’s disease) with hepatomata; case report with metabolic studies. Pediatrics. 1955;16:785–800. [PubMed] [Google Scholar]
- 96.Alshak NS, Cocjin J, Podesta L, van de Velde R, Makowka L, Rosenthal P, Geller SA. Hepatocellular adenoma in glycogen storage disease type IV. Arch Pathol Lab Med. 1994;118:88–91. [PubMed] [Google Scholar]
- 97.Labrune P, Trioche P, Duvaltier I, Chevalier P, Odièvre M. Hepatocellular adenomas in glycogen storage disease type I and III: a series of 43 patients and review of the literature. J Pediatr Gastroenterol Nutr. 1997;24:276–279. doi: 10.1097/00005176-199703000-00008. [DOI] [PubMed] [Google Scholar]
- 98.Demo E, Frush D, Gottfried M, Koepke J, Boney A, Bali D, Chen YT, Kishnani PS. Glycogen storage disease type III-hepatocellular carcinoma a long-term complication? J Hepatol. 2007;46:492–498. doi: 10.1016/j.jhep.2006.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Manzia TM, Angelico R, Toti L, Cillis A, Ciano P, Orlando G, Anselmo A, Angelico M, Tisone G. Glycogen storage disease type Ia and VI associated with hepatocellular carcinoma: two case reports. Transplant Proc. 2011;43:1181–1183. doi: 10.1016/j.transproceed.2011.01.129. [DOI] [PubMed] [Google Scholar]