

Abstract
Ultraviolet (UV) B causes oxidative stress, which has been implicated in carcinogenesis. We determined if the sensitivity of keratinocytes to UVB-induced oxidative stress is dependent on their differentiation state. In primary cultures of undifferentiated and differentiated mouse keratinocytes, UVB (25 mJ/cm2) stimulated production of reactive oxygen intermediates. This was associated with increased messenger RNA (mRNA) expression of the antioxidant enzymes glutathione peroxidase, heme oxygenase-1 (HO-1) and the glutathione S-transferase (GST), GSTA1-2. The effects of UVB on GSTA1-2 were greater in undifferentiated when compared with differentiated cells. UVB also induced GSTM1, but only in undifferentiated cells. In contrast, UVB reduced expression of manganese superoxide dismutase, metallothionein-2, GSTA3 and microsomal glutathione S-transferase (mGST)3 in both cell types, whereas it had no major effects on catalase, copper–zinc superoxide dismutase, GSTP1, mGST1 or mGST2. Of note, levels of GSTA4 mRNA were 4- to 5-fold greater in differentiated relative to undifferentiated cells. Moreover, whereas GSTA4 was induced by UVB in undifferentiated cells, it was inhibited in differentiated cells. UVB activated p38 and c-jun N-terminal kinase mitogen-activated protein (MAP) kinases in both undifferentiated and differentiated keratinocytes. Whereas inhi bition of these kinases blocked UVB-induced HO-1 in both cell types, GSTA1–2 and GST-4 were only suppressed in undifferentiated cells. In differentiated keratinocytes, p38 inhibition also suppressed GSTA1–2. In contrast, MAP kinase inhibition had no major effects on UVB-induced suppression of GSTA4 in differentiated cells. These data indicate that UVB-induced alterations in antioxidant expression are differentiation dependent. Moreover, MAP kinases are critical regulators of this response. Alterations in antioxidants are likely to be important mechanisms for protecting the skin from UVB-induced oxidative stress.
Introduction
Exposure to ultraviolet (UV) light from sunlight is a major cause of skin cancer (1–3). The higher energy wavelengths of the UVB spectrum (290–320 nm) are considered to be ‘complete carcinogens’, acting both as tumor initiators and tumor promoters (4). In the skin, UVB light is known to induce oxidative stress by generating reactive oxygen intermediates (ROIs) including superoxide anion, hydrogen peroxide and hydroxyl radicals, each of which is important in the pathogenesis of UVB light-induced carcinogenesis (5). These oxidants induce DNA damage leading to mutations (6). ROI can also oxidize proteins and induce lipid peroxidation, resulting in inappropriate or altered cellular signal transduction and aberrant proliferation (7,8).
Key to limiting oxidative stress and preventing UVB-induced damage to the skin is removal or detoxification of ROI. This is accomplished in large part by antioxidant enzymes (9). A number of enzymes have been identified that are involved in this process including the ROI scavengers, superoxide dismutase (SOD), catalase and glutathione peroxidase (GPx), heme oxygenase-1 (HO-1) and metallothionein-2 (MT-2) as well as the glutathione S-transferases (GSTs). UVB-induced oxidative stress has been reported to be associated with transient decreases in SOD, catalase and GPx activity in mouse skin (10). Reduced expression of SOD and catalase activity has also been described in human keratinocytes treated with UVB in vitro (11). In contrast, in human skin, a solar simulator that generates UVB was found to induce messenger RNA (mRNA) for GPx and the GST GSTP1 (12). Although the mouse skin model has been used extensively to investigate mechanisms of UVB-induced oxidative stress, inflammation and carcinogenesis, little is known about the expression of antioxidant enzymes in the tissue and their responses to UVB. In the present studies, we characterized the effects of UVB on expression of antioxidant enzymes in primary cultures of mouse keratinocytes. Since increased oxidative DNA and protein damage as well as lipid peroxidation occurs in suprabasal layers of the epidermis of mouse and human skin in response to UVB (13,14), we used cultures of undifferentiated and differentiated keratinocytes. We found that the keratinocytes were highly responsive to UVB-induced oxidative stress; however, the response was dependent on their differentiation state. Alterations in antioxidant enzyme expression in keratinocytes are likely to be important in controlling oxidative stress in the different layers of the epidermis in response to UVB.
Materials and methods
Chemicals and reagents
Rabbit polyclonal antibody to HO-1 was purchased from Assay Designs (Ann Arbor, MI) and rabbit polyclonal anti-catalase was from Abcam (Cambridge, MA). Rabbit polyclonal antibodies to p38, phospho-p38, c-jun N-terminal kinase (JNK), phospho-JNK, extracellular signal-regulated kinase (ERK) 1/2 and phospho-ERK 1/2 were from Cell Signaling Technology (Beverly, MA), and rabbit anti-keratin 1, keratin 10 and filaggrin from Covance Research Products (Berkley, CA). Goat polyclonal anti-cyclooxygenase-2 (COX-2), rabbit polyclonal anti-copper–zinc superoxide dismutase (Cu, Zn-SOD), rabbit polyclonal anti-α-tubulin and donkey anti-goat secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Horseradish peroxidase-labeled goat anti-rabbit secondary antibodies, detergent-compatible protein assay reagents and the Silver Stain Plus kit were obtained from Bio-Rad Laboratories (Hercules, CA). The Western Lightning enhanced chemiluminescence kit was from PerkinElmer Life Sciences (Boston, MA). M-MLV Reverse Transcriptase was from Promega (Madison, WI). SYBR Green Master Mix and other polymerase chain reaction (PCR) reagents were purchased from Applied Biosystems (Foster City, CA). Cell culture reagents and 2′, 7′-dicholorofluorescein-diacetate were obtained from Invitrogen Corp. (Carlsbad, CA). Collagen IV was purchased from BD Biosciences (San Diego, CA). SP600125 was from Calbiochem (La Jolla, CA) and SB203580, protease inhibitor cocktail and all other chemicals from Sigma (St Louis, MO).
Cells
Primary mouse keratinocytes were isolated from the skin of C57Bl/6J newborn mice and cultured following the procedure described by Hager et al. (15). For experiments, cells grown in six-well collagen IV-coated plates were placed in serum-free low-calcium (0.05 mM) keratinocyte growth medium to maintain the cells in an undifferentiated state. Differentiation was induced by the addition of calcium (0.15 mM) to the medium (16) and was confirmed by the morphological changes and expression of differentiation markers including keratin 1, keratin 10 and filaggrin in control and UVB-treated cells, as analyzed by western blotting (described as follows) (Figure 3A). Expression of these markers was not altered following UVB treatment. In some experiments, primary keratinocytes from the Yale University Cell Culture Facility (New Haven, CT) were used.
Fig 3.
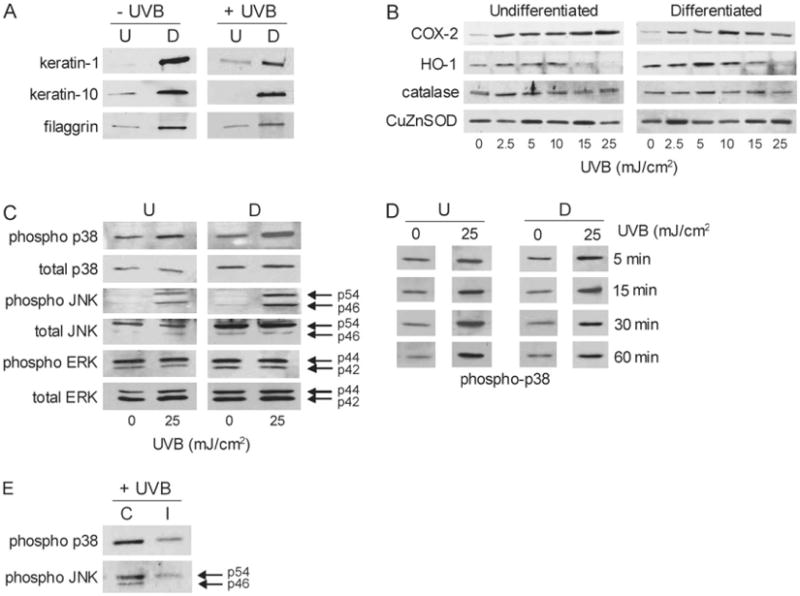
Effects of UVB on differentiation markers and markers of oxidative stress in mouse keratinocytes. Panel (A) western blot showing that differentiation resulted in increased expression of keratin 1, keratin 10 and filaggrin. Control and UVB-treated (25 mJ/cm2) cells were analyzed 24 h after calcium-induced differentiation. Panel (B) effects of UVB on expression of COX-2 and antioxidant enzymes. Cells were analyzed by western blotting 24 h after control or UVB treatment. Panel (C) effects of UVB on MAP kinase expression in undifferentiated and differentiated keratinocytes. Total and phosphorylated p38, JNK and ERK 1/2 MAP kinases were analyzed in the cells by western blotting 15 min after UVB (25 mJ/cm2) or control. Panel (D) MAP kinases were activated from 5 to 60 min following UVB in undifferentiated and differentiated keratinocytes. Total and phosphorylated p38 MAP kinase expression was analyzed 5, 15, 30 and 60 min after UVB (25 mJ/cm2) or control. Expression of p38 MAP kinase was representative of kinase activation. Panel (E) effects of inhibitors on keratinocyte p38 and JNK activation. Undifferentiated cells were treated with a p38 kinase inhibitor (SB203580, 10 μM) or JNK inhibitor (SP600125, 20 μM) and analyzed for p38 or JNK activity by western blotting. Note that the MAP kinase inhibitors effectively suppressed enzyme phosphorylation. The figure shows phosphorylated p38 and JNK expression in control (C) or inhibitor-treated (I) cells.
UVB and inhibitor treatments
UVB was generated from a bank of two Westinghouse FS40BL bulbs calibrated using an International IL-1700 UV-radiometer. As previously reported, the spectrum of the bulbs was described by the manufacturer as ~85% UVB (290–320 nm), 1% UVA (320–400 nm) and, <1% UVC (<290 nm) with the remainder composed of visible light (17). Cells were grown to 90% confluence and exposed to UVB (2.5–25 mJ/cm2) in phosphate-buffered saline as described previously (18). Following UVB treatment, the phosphate-buffered saline was removed and the cells were refed with keratinocyte growth medium containing low (0.05 mM) or high (0.15 mM) calcium. In some experiments, the p38 mitogen-activated protein (MAP) kinase inhibitor SB203580 (10 μM) (19), the JNK inhibitor SP600125 (20 μM) (20) or dimethyl sulfoxide control were added to the medium and the cells incubated at 37°C for 3 h prior to UVB treatment. We have found that higher doses of UVB (>50 mJ/cm2) are cytotoxic to the keratinocytes. Intact skin or other cell types (e.g. human keratino cytes) can be exposed to higher doses of UVB, possibly due to a greater content of antioxidants (21).
Western blotting
Western blotting was performed as described previously (22). Briefly, cell lysates were prepared using sodium dodecyl sulfate lysis buffer (10 mM Tris base and 1% sodium dodecyl sulfate, pH 7.6, supplemented with a protease inhibitor cocktail consisting of 4-(2-aminoethyl)benzenesulfonyl fluoride, aprotinin, bestatin hydrochloride, N-(trans-epoxysuccinyl)-l-leucine 4-guanidinobutylamide, ethylenediaminetetraacetic acid and leupeptin). Proteins (20 μg) from lysates were separated on 10% sodium dodecyl sulfate–polyacrylamide gels and then transferred to nitrocellulose membranes. After incubating the membranes in blocking buffer (5% dry milk Tris-buffered saline with 0.1% Tween 20), the membranes were incubated for 1–2 h at room temperature or overnight at 4°C with primary antibodies followed by horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Protein expression was visualized using enhanced chemiluminescence reagents. Loading of equal amounts of protein on the gels was confirmed by silver staining.
Oxidative metabolism
Intracellular hydrogen peroxide production was measured using 2′, 7′-dicholorofluorescein-diacetate and flow cytometry as described previously (23). Briefly, the cells were incubated with 2′, 7′-dicholorofluorescein-diacetate (5 μM) in phosphate-buffered saline for 15 min, exposed to UVB (25 mJ/cm2) and then immediately analyzed on a Beckman Coulter Cytomics FC 500 flow cytometer equipped with a 488 nm argon laser and CXP software.
Real-time PCR
RNA was isolated from the cells using TRI Reagent (Sigma) following the protocol provided by the manufacturer. RNA was converted to cDNA using M-MLV Reverse Transcriptase. The cDNA was diluted 1:10 in RNase–DNase-free water for PCR analysis. For each gene, a standard curve was generated from serial dilutions of cDNA from the samples. All values were normalized to β-actin (n = 3−6, ±SE). The undifferentiated control was assigned a value of one and all other samples were calculated relative to this control. Real-time PCR was performed on an ABI Prism 7900 Sequence Detection System using 96-well optical reaction plates. SYBR Green was used for detection of the fluorescent signal and the standard curve method was used for relative quantitative analysis. The primer sequences for the genes were generated using Primer Express software (Applied Biosystems) and the oligonucleotides were synthesized by Integrated DNATechnologies (Coralville, IA). The forward and reverse (5′ and 3′) primer sequences were as follows: β-actin, TCACCCA-CACTGTGCCCATCTACGA and GGATGCCACAGGATTCCATACCCA; catalase, ACCAGGGCATCAAAAACTTG and GCCCTGAAGCTTTTT-GTCAG; COX-2, CATTCTTTGCCCAGCACTTCAC and GACCAGGC-ACCAGACCAAAGAC; GSTA1–2, CAGAGTCCGGAAGATTTGGA and CAAGGCAGTCTTGGCTTCTC; GSTA3, GCAAGCCTTGCCAAGATCAA and GGCAGGGAAGTAACGGTTCC; GSTA4, CCCTTGGTTGAAATC-GATGG and GAGGATGGCCCTGGTCTGT; GSTM1, CCTACATGAAGAG-TAGCCGCTACAT and TAGTGAGTGCCCGTGTAGCAA; GSTP1, CCTTG-GCCGCTCTTTGG and GGCCTTCACGTAGTCATTCTTACC; GPx-1, GGTTCGAGCCCAATTTTACA and TCGATGTCGATGGTACGAAA; HO-1, CCTCACTGGCAGGAAATCATC and CCTCGTGGAGACGCTTTACA-TA; interleukin (IL)-1β, CCAAAAGATGAAGGGCTGCT and TCATCTG-GACAGCCCAGGTC; IL-6, ATGAAGTTCCTCTCTGCAAGAGACT and CACTAGGTTTGCCGAGTAGATCTC; IL-10, GTTGCCAAGCCTTATCG-GAA and CCAGGGAATTCAAATGCTCCT; MT-2, TGCAGGAAGTA-CATTTGCATTGTT and TTTTCTTGCAGGAAGTACATTTGC; microsomal glutathione S-transferase (mGST)1, GCTTTGGCAAGGGAGAGAATG and CCTTCTCGTCAGTGCGAACA; mGST2, TGCAGCCTGTCTGGGTCTC and CAGAAATACTTGTGACGGGCG; mGST3, GGAGGTGTACCCTCCC-TTCC and TGGTAAACACCTCCCACCGT; Cu,Zn-SOD, ACCAGTGCAG-GACCTCATTTTAA and TCTCCAACATGCCTCTCTTCATC; manganese superoxide dismutase (Mn-SOD), CACATTAACGCGCAGATCATG and CCA-GAGCCTCGTGGTACTTCTC and tumor necrosis factor-α (TNF-α), AAATT-CGAGTGACAAGCCGTA and CCCTTGAAGAGAACCTGGGAGTAG.
Statistical analysis
Data were evaluated using the Student's t-test and differences were considered statistically significant at P < 0.05.
Results
UVB light induces oxidative stress and upregulates inflammatory markers in keratinocytes
In initial studies, we compared the effects of UVB light on intracellular hydrogen peroxide production in undifferentiated and differentiated keratinocytes. Both cell types were found to constitutively generate low levels of hydrogen peroxide (Figure 1). UVB (25 mJ/cm2) treatment resulted in a 10- to 20-fold increase in hydrogen peroxide production in both cell types. No major differences were observed between undifferentiated and differentiated cells. Previous studies have shown that COX-2, the rate-limiting enzyme in prostaglandin biosynthesis, and inflammatory cytokines such as TNF-α, IL-1β, IL-6 and IL-10 are upregulated by UVB in keratinocytes (24–27). This is considered part of the UVB response of skin (28). Similarly, in mouse keratinocytes, UVB was found to markedly increase expression (25- to 35-fold) of COX-2 mRNA (Figure 2) and protein (Figure 3B). In both undifferentiated and differentiated keratinocytes, this effect was dose dependent (2.5–25 mJ/cm2). Differentiated cells were more responsive to UVB at the higher doses (15 and 25 mJ/cm2) with respect to expression of COX-2 than undifferentiated cells. High expression of COX-2 protein was evident at low doses of UVB (2.5 mJ/cm2), possibly due to efficient mRNA translation. UVB caused 2- to 3-fold increases in TNF-α and IL-10 expression in both undifferentiated and differentiated cells (Figure 2). No differences were evident in TNF-α levels between the cell types, whereas at low UVB doses (2.5–5 mJ/cm2), IL-10 expression was significantly greater in differentiated when compared with undifferentiated cells. UVB also resulted in 3- to 4-fold increases in IL-1β and IL-6 expression in both cell types (Figure 2). However, differentiation modulated UVB-induced expression of these two cytokines at the higher doses (15–25 mJ/cm2); undifferentiated cells were more responsive with respect to IL-1β expression, whereas IL-6 levels were greater in differentiated cells.
Fig 1.

UVB stimulates intracellular hydrogen peroxide production. Undifferentiated and differentiated keratinocytes were incubated with 2′, 7′-dicholorofluorescein-diacetate (5 μM) for 15 min. The cells were then exposed to UVB (25 mJ/cm2) or control and analyzed by flow cytometry. Data are presented on a four-decade log scale.
Fig 2.

Effects of UVB on COX-2 and cytokine mRNA expression. Undifferentiated and differentiated keratinocytes were exposed to UVB (2.5–25 mJ/cm2) or control. After 24 h, RNAwas extracted from the cells and analyzed for gene expression by real-time PCR (n= 3−6, ±SE). Data are presented as fold increase in gene expression relative to unexposed undifferentiated cells. Differences were considered statistically significant at P < 0.05: aundifferentiated fold increase as compared with control; bdifferentiated fold increase versus control; cundifferentiated versus differentiated.
Effects of UVB on expression of antioxidant enzymes
We next compared the effects of UVB on expression of antioxidant enzymes in undifferentiated and differentiated keratinocytes. UVB treatment was associated with increased expression of HO-1 mRNA (Figure 4A) in both cell types which was maximal at 15 mJ/cm2. Differentiated cells were more responsive to UVB than undifferentiated cells at the lower doses (2.5–10 mJ/cm2). Both cell types also expressed HO-1 protein after UVB treatment, reaching maximal levels at 5–10 mJ/cm2 (Figure 3B). As observed with HO-1 mRNA, expression of HO-1 protein was greater in differentiated when compared with undifferentiated cells. Interestingly, in both cell types expression of HO-1 protein was inhibited at the highest dose of UVB (25 mJ/cm2).
Fig 4.

Effects of UVB on antioxidant gene expression. Undifferentiated and differentiated keratinocytes were exposed to UVB (2.5–25 mJ/cm2) or control. After 24 h, RNA was extracted from the cells and analyzed for expression of antioxidant enzymes by real-time PCR (n = 3–6, ±SE). Data are presented as fold change in gene expression relative to unexposed undifferentiated cells. Differences were considered statistically significant at P < 0.05: aundifferentiated fold increase as compared with control; bdifferentiated fold increase versus control; cundifferentiated versus differentiated.
In both undifferentiated and differentiated keratinocytes, UVB treatment (2.5–25 mJ/cm2) also resulted in increased expression of GPx-1 and GSTA1–2 mRNA (Figures 4 and 5). At doses >10 mJ/cm2, undifferentiated cells were more sensitive than differentiated cells with respect to GSTA1–2 expression (Figure 5). GSTM1 mRNA expression was also induced by UVB, but only in undifferentiated cells (Figure 5). In contrast, UVB significantly suppressed the expression of Mn-SOD, MT-2, GSTA3 and mGST3 in both cell types, whereas no major changes were observed in catalase, Cu,Zn-SOD, GSTP1, mGST1 or mGST2 (Figures 4 and 5). Similarly, no alterations in catalase or Cu,Zn-SOD protein expression were evident (Figure 3B). Interestingly, constitutive levels of GSTA4 mRNA were 4- to 5-fold greater in differentiated when compared with undifferentiated cells. Moreover, whereas GSTA4 expression increased in undifferentiated cells after UVB treatment, it decreased in differentiated cells (Figure 5).
Fig 5.

Effects of UVB on GST expression. Undifferentiated and differentiated keratinocytes were exposed to UVB (2.5–25 mJ/cm2) or control. After 24 h, RNA was extracted from the cells and analyzed for expression of cytosolic and mGSTs by real-time PCR (n = 3−6, ±SE). Data are presented as fold change in gene expression relative to unexposed undifferentiated cells. Differences were considered statistically significant at P < 0.05: aundifferentiated fold increase as compared with control; bdifferentiated fold increase versus control; cundifferentiated versus differentiated.
Effects of UVB on MAP kinase activation in keratinocytes
MAP kinases are important regulators of gene expression (29). In further studies, the effects of UVB on these signaling molecules and their role in antioxidant gene expression was examined. Both undifferentiated and differentiated keratinocytes constitutively expressed total p38, JNK and ERK 1/2 MAP kinases (Figure 3C). p38 kinase and JNK expression levels were greater in differentiated relative to undifferentiated cells. Keratinocytes also constitutively expressed the activated phosphorylated forms of p38 and ERK 1/2 kinases, but not JNK. In both undifferentiated and differentiated cells, UVB (25 mJ/cm2) enhanced the expression of activated phospho-p38 and JNK, but not ERK 1/2 MAP kinase (Figure 3C). Kinase activation was both rapid and prolonged with increased phosphorylation evident from 5 min to 1 h after UVB (Figure 3D). Greater amounts of activated p38 and JNK were evident in differentiated cells, following UVB treatment when compared with undifferentiated cells. To determine if these MAP kinases play a role in regulating UVB-induced alterations in expression of antioxidants, we used SB203580, a p38 kinase inhibitor, and SP600125, a JNK inhibitor. As shown in Figure 3E, these inhibitors effectively suppressed UVB-induced phosphorylation of the p38 kinase and JNK. In these studies, expressions of HO-1,GSTA1–2andGSTA4,whicharehighlyresponsivetoUVB,were analyzed. Both p38 and JNK inhibitors were found to suppress UVB-induced expression of HO-1, GSTA1–2 and GSTA4 in undifferentiated cells (Figure 6). In these cells, the p38 inhibitor was more effective in suppressing HO-1 and GSTA4 expression, whereas the JNK inhibitor was more effective in blocking GSTA1–2. In differentiated cells, UVB-induced HO-1 expression was blocked by both p38 and JNK inhibition, whereas GST1–2 was only suppressed by the p38 inhibitor. Inhibiting p38 was more effective in suppressing HO-1 in these cells than inhibiting JNK. The p38 and JNK inhibitors were unable to reverse the inhibitory effects of UVB on GSTA4 in differentiated cells and, in fact, further suppressed expression of this enzyme (Figure 6).
Fig 6.

Effects of p38 and JNK MAP kinase inhibitors on UVB-induced expression of antioxidant enzymes. Undifferentiated and differentiated keratinocytes were incubated with SB203580 (10 μM), a p38 MAP kinase inhibitor, or SP600125 (20 μM), a JNK MAP kinase inhibitor, for 3 h and then exposed to UVB or control at the indicated doses. After 24 h, RNA was extracted from the cells and analyzed for mRNA expression by real-time PCR (n = 3−6, ±SE). Data are presented as fold change in gene expression relative to unexposed undifferentiated cells. Differences were considered statistically significant at P < 0.05: ap38 kinase inhibitor treatment versus control and bJNK inhibitor treatment versus control.
Discussion
Oxidative stress is recognized as an important factor contributing to the development of many cutaneous diseases including cancer (30,31). This is thought to be due to the production of ROI, which induce lipid peroxidation and generate oxidized DNA (6). Exposure to UVB and production of ROI are associated with oxidative stress in both intact skin and isolated keratinocytes (32). The present study demonstrates that UVB treatment of mouse keratinocytes stimulates the production of hydrogen peroxide. However, no major differences were evident in the response of undifferentiated and differentiated cells, suggesting that the two cell types possess similar antioxidant capacity. This is supported by our findings that both undifferentiated and differentiated keratinocytes express generally similar levels of SOD, catalase and GPx-1 mRNA. In the skin, UVB-induced oxidative stress is associated with an inflammatory response (28). Consistent with this, we found that in mouse keratinocytes, UVB induces expression of COX-2, a key enzyme regulating the production of prostaglandins, lipid-derived mediators known to contribute to the carcinogenic process (33). Expression for COX-2 was greater in differentiated when compared with undifferentiated cells treated with UVB. These findings are in accord with studies demonstrating suprabasal expression of COX-2 in UVB-treated human skin (25). Increased responsiveness of differentiated keratinocytes to UVB-induced COX-2 may be due to increased signaling regulating COX-2 expression in these cells. In this regard, the present study shows that differentiated keratinocytes express increased levels of phospho-p38 and phospho-JNK, two MAP kinases known to regulate COX-2 in human keratinocytes (29). TNF-α, IL-lβ, IL-6 and IL-10 were also induced by UVB in both undifferentiated and differentiated keratinocytes, although no major differences were noted in their expression between the two cell types. Thus, regulation of cytokine and COX-2 expression is distinct in undifferentiated and differentiated keratinocytes. TNF-α, IL-lβ, IL-6 and IL-10 are inflammatory cytokines known to regulate cellular responses to oxidative stress, in part, through upregulation of antioxidants (34–36). Expression of these cytokines in keratinocytes may be important in protecting the cells from UVB-induced oxidative damage.
Following UVB treatment, HO-1 and GPx-1 increased in undifferentiated and differentiated keratinocytes. Lower doses of UVB were more effective in inducing HO-1 in differentiated cells. HO-1 expression is regulated by MAP kinases; increased activity of these kinases in differentiated cells may account for greater HO-1 expression. HO-1 is a stress protein and increases in its expression at lower doses of UVB may indicate that differentiated cells are more sensitive to UVB-induced oxidative stress (37). Earlier studies have showed that HO-1 is induced by UVA with little or no effects of UVB except at very high doses (50–550 mJ/cm2) (38,39). In contrast, our studies show that HO-1 mRNA and protein expression in keratinocytes is inhibited at higher doses of UVB. GPx-1 is an antioxidant that functions to reduce hydroperoxides to their corresponding alcohols utilizing glutathione. In contrast to HO-1, similar increases in expression of GPx-1 were noted in differentiated and undifferentiated cells after UVB. This may be due to distinct pathways regulating the expression of these antioxidants in the two cell types.
Interestingly, following UVB, MT-2 and Mn-SOD significantly decreased in both differentiated and undifferentiated keratinocytes. MT-2 functions to sequester metals such as zinc, important in its antioxidant activity (40). Zinc and other heavy metals regulate the expression of MT-2, and it is possible that UVB interferes with this protein in keratinocytes. Yamada et al. (41) demonstrated that UV light blocks transcriptional activation of MT-2 in human skin-derived fibroblasts and a similar mechanism may also function in mouse keratinocytes. We also found that UVB had no effect on the expression of Cu,Zn-SOD in the cells. Cu,Zn-SOD and Mn-SOD are distinct gene products; Cu,Zn-SOD is localized in cytosolic and lysosomal cell fractions and in mitochondrial intermembraneous space, whereas Mn-SOD is concentrated in mitochondrial matrix and inner membrane (42). Multiple SODs are important in protecting different intracellular compartments from superoxide, a radical that does not readily cross membranes. UVB damages mitochondria and this may trigger signals that reduce expression of Mn-SOD (43).
The GST enzymes are comprised of a large superfamily of ubiquitous cytosolic and microsomal phase II detoxification enzymes that conjugate reduced glutathione to electrophilic compounds, decreasing their potential cytotoxicity. Cytosolic GSTs are divided into three major gene families, alpha (GSTA), mu (GSTM) and pi (GSTP), each with preferred substrates (44). Although total GST activity increases significantly in skin following UVB exposure (45,46), there has been limited investigation on the involvement of GST isozymes in this cellular response. We found that GSTA1–2 and GSTA4 were upregulated by UVB in undifferentiated keratinocytes. These findings are in accord with previous studies, showing that GSTA4 expression is upregulated in response to oxidative stress induced by UVB (47,48) and that overexpression of GSTA1 protects against hydrogen peroxide-induced oxidative stress (49). GSTA and, to a lesser extent, GSTP, remove lipid peroxidation products, thereby breaking radical-forming chain reactions (50). This activity is probably important in the antioxidant response of undifferentiated mouse keratinocytes to UVB. Of interest was our observation that the response of differentiated keratinocytes to UVB with respect to GSTA1–2 and GSTA4 expression was distinct. Thus, differentiated cells expressed greater constitutive levels of these cytosolic GSTs. This is consistent with the earlier work, showing that differentiated cells are more resistant to oxidant-induced stress (51), and that the differentiation process itself results in an increase in GST activity (52). GSTA1–2 also increased in differentiated keratinocytes in response to UVB, although the response was attenuated. In contrast, GSTA4 expression decreased following UVB treatment. The mechanism for differential responsiveness of these enzymes to UVB in undifferentiated and differentiated cells is not known. Previous studies have shown that GSTA4 regulates proliferation and protects against apoptosis (53) and it is possible that UVB-induced decreases in its expression promotes turnover of differentiated keratinocytes in the skin during oxidative stress. Also of note was our observation that expression of GSTM1, which protects against protein oxidation, initially declined in response to UVB and then increased. Greater increases inGSTM1 were observed in undifferentiated cells. GSTM1 catalyzes the conjugation of electrophiles to reduced glutathione (GSH) and may be important in protecting basal cells from cellular oxidative damage (44). GSTA3, which is involved in steroid biosynthesis, as well as GSTP1, which removes DNA and protein oxidation products (50), were unaffected by UVB. Although expressed in keratinocytes, it appears that these enzymes are not involved in the UVB response to oxidative stress.
mGSTs are enzymes that play a role in protection against microsomal lipid peroxidation (54). Although expression of mGST1 and mGST2 were not significantly altered by UVB, expression of mGST3 was markedly reduced in both undifferentiated and differentiated keratinocytes. mGST3 is involved in synthesizing leukotriene A4, a proinflammatory lipid mediator, and UVB-induced decreases in its expression may regulate the resolution of inflammation (50).
It is well recognized that ROI can activate a number of signaling pathways controlling gene expression (32,42,55). This is thought to occur by direct or indirect activation of growth factors or growth factor receptors via a process referred to as ‘the UV response’ (56). This response was initially characterized with respect to activation of the immediate early genes, c-fos and c-jun and the transcription factors nuclear factor-kappa B and AP-1 (57,58) and is regulated by members of the MAP kinase family. In mouse keratinocytes, only p38 and JNK MAP kinases were activated by UVB. This is in accord with responses observed in PAM212 mouse keratinocytes (18) and human keratinocytes (59). Interestingly, greater amounts of constitutive expression of total and activated forms of JNK and p38 MAP kinase were noted in differentiated cells. Increases in activated JNK have been described previously in suprabasal layers of human skin (60). This may be due to increased sensitivity of the differentiated cells to autocrine growth factor activation or a requirement for JNK in the regulation of gene transcription in differentiating keratinocytes (61). A variety of additional growth factors including activators of MAP kinase signaling have been identified in primary mouse keratinocytes as well as in mouse skin (62,63). Activation of MAP kinases promotes differentiation (61), although the mechanisms mediating this effect remain to be determined.
Studies using MAP kinase inhibitors demonstrated that JNK and p38 regulate keratinocyte expression of HO-1, GSTA1–2 and GSTA4. Moreover, with respect to expression of the GST's, differentiation appears to regulate cellular responsiveness to the kinase inhibitors. Previous studies have shown that both the p38 and JNK MAP kinases regulate expression of HO-1 (64). The fact that inhibition of p38 was more effective than JNK in suppressing UVB-induced HO-1 suggests that this kinase plays a more prominent role in regulating expression of this antioxidant. A similar pattern of inhibition was noted in the regulation of GSTA4 in undifferentiated cells. In this regard, p38 is known to regulate GSTA4 in murine hepatocytes stimulated with epidermal growth factor (65). In contrast, JNK appeared to play a more prominent role in regulating GSTA1–2 expression in undifferentiated cells. These data indicate that regulation of expression of these antioxidants in the cells occurs via distinct mechanisms. Although p38 and JNK inhibitors did not reverse the suppressive effects of UVB on GSTA4 in differentiated keratinocytes, only p38 inhibition was able to inhibit UVB-induced GSTA1–2 in these cells. Thus, it appears that the differentiation process alters the mechanisms by which UVB modulates expression of the GSTs. These data are consistent with studies showing that keratinocyte differentiation is associated with alterations in expression of a variety of transcription factors including AP-1, SP-1 and peroxisome proliferator-activated receptor (PPAR-γ) (66,67).
In summary, our data demonstrate that UVB light induces oxidative stress in primary cultures of mouse keratinocytes by increasing the production of ROI and inflammatory mediators and enhancing the activity of p38 and JNK MAP kinases. This leads to increased expression of critical antioxidant enzymes including HO-1, GPx-1, GSTA1–2, and, in undifferentiated cells, GSTA4. In contrast, UVB also reduced expression of Mn-SOD, MT-2, GSTA3 and mGST3. GSTA4 also decreased in differentiated cells. Whether or not these responses protect the skin from UVB will depend on the levels of expression of functional proteins for these enzymes in the different cell types and their precise roles in regulating oxidative stress and/or repairing cellular damage. Recent studies have shown that UVB-induced DNA photoproducts are also critical mediators of skin cell damage (68). Further studies are needed to determine the precise role of DNA photoproducts versus oxidative stress in initiating the carcinogenic process.
Acknowledgments
The contents are solely the responsibility of the authors and do not necessarily represent the official views of the federal government.
Funding: National Institutes of Health (AR055073, CA100994, CA093798, ES004738, GM034310 and ES005022); New Jersey State Commission on Cancer Research fellowship (05-2413-CCR-E0); National Institutes of Health CounterACT Program through the National Institute of Arthritis and Musculoskeletal and Skin Diseases (U54AR055073).
Abbreviations
- COX-2
cyclooxygenase-2
- Cu,Zn-SOD
copper-zinc super oxide dismutase
- ERK
extracellular signal-regulated kinase
- GPx
glutathione peroxidase
- GST
glutathione S-transferase
- HO-1
heme oxygenase-1
- IL
interleukin
- JNK
c-jun N-terminal kinase
- MAP
mitogen-activated protein
- mGST
microsomal glutathione S-transferase
- mRNA
messenger RNA
- Mn-SOD
manganese superoxide dismutase
- MT-2
metallothionein-2
- PCR
polymerase chain reaction
- ROI
reactive oxygen intermediate
- SOD
superox ide dismutase
- TNF-α
tumor necrosis factor-α
- UV
ultraviolet
Footnotes
Conflict of Interest Statement: None declared.
References
- 1.Wikonkai NM, et al. Ultraviolet radiation induced signature mutations in photocarcinogenesis. J Invest Dermatol Symp Proc. 1999;4:6–10. doi: 10.1038/sj.jidsp.5640173. [DOI] [PubMed] [Google Scholar]
- 2.Kraemer KH. Sunlight and skin cancer: another linked revealed. Proc Natl Acad Sci USA. 1997;94:11–14. doi: 10.1073/pnas.94.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang W, et al. UVB-induced apoptosis drives clonal expansion during skin tumor development. Carcinogenesis. 2005;26:249–257. doi: 10.1093/carcin/bgh300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cadet J, et al. Effects of UVand visible radiations on cellular DNA. Curr Probl Dermatol. 2001;29:62–73. doi: 10.1159/000060654. [DOI] [PubMed] [Google Scholar]
- 5.Ichihashi M, et al. UV-induced skin damage. Toxicology. 2003;189:21–39. doi: 10.1016/s0300-483x(03)00150-1. [DOI] [PubMed] [Google Scholar]
- 6.Reid TM, et al. Tandem double CC→TT mutations are produced by reactive oxygen species. Proc Natl Acad Sci USA. 1993;90:3904–3907. doi: 10.1073/pnas.90.9.3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bender K, et al. UV-induced signal transduction. J Photochem Photobiol B. 1997;37:1–17. doi: 10.1016/s1011-1344(96)07459-3. [DOI] [PubMed] [Google Scholar]
- 8.Darr D, et al. Free radicals in cutaneous biology. J Invest Dermatol. 1994;102:671–675. doi: 10.1111/1523-1747.ep12374036. [DOI] [PubMed] [Google Scholar]
- 9.Afaq F, et al. Effects of solar radiation on cutaneous detoxification pathways. J Photochem Photobiol B. 2001;63:61–69. doi: 10.1016/s1011-1344(01)00217-2. [DOI] [PubMed] [Google Scholar]
- 10.Shindo Y, et al. Dose-response effects of acute ultraviolet irradiation on antioxidants and molecular markers of oxidation in murine epidermis and dermis. J Invest Dermatol. 1994;102:470–475. doi: 10.1111/1523-1747.ep12373027. [DOI] [PubMed] [Google Scholar]
- 11.Sasaki H, et al. Effects of a single exposure to UVB radiation on the activities and protein levels of copper-zinc and manganese superoxide dis-mutase in cultured human keratinocytes. Photochem Photobiol. 1997;65:707–713. doi: 10.1111/j.1751-1097.1997.tb01914.x. [DOI] [PubMed] [Google Scholar]
- 12.Smith G, et al. Quantitative real-time reverse transcription-polymerase chain reaction analysis of drug metabolizing and cytoprotective genes in psoriasis and regulation by ultraviolet radiation. J Invest Dermatol. 2003;121:390–398. doi: 10.1046/j.1523-1747.2003.12354.x. [DOI] [PubMed] [Google Scholar]
- 13.Theile JJ, et al. Protein oxidation in human stratum corneum: susceptibility of keratins to oxidation in vitro and presence of a keratin oxidation gradient in vivo. J Invest Dermatol. 1999;113:335–339. doi: 10.1046/j.1523-1747.1999.00693.x. [DOI] [PubMed] [Google Scholar]
- 14.Sander CS, et al. Photoaging is associated with protein oxidation in human skin in vivo. J Invest Dermatol. 2002;118:618–625. doi: 10.1046/j.1523-1747.2002.01708.x. [DOI] [PubMed] [Google Scholar]
- 15.Hager B, et al. Long-term culture of murine epidermal keratinocytes. J Invest Dermatol. 1999;112:971–976. doi: 10.1046/j.1523-1747.1999.00605.x. [DOI] [PubMed] [Google Scholar]
- 16.Yuspa SH, et al. Signal transduction for proliferation and differentiation in keratinocytes. Ann N Y Acad Sci. 1988;548:191–196. doi: 10.1111/j.1749-6632.1988.tb18806.x. [DOI] [PubMed] [Google Scholar]
- 17.Kramer-Stickland K, et al. Effect of UVB on hydrolysis of α-tocopherol acetate to α-tocopherol in mouse skin. J Invest Dermatol. 1998;111:302–307. doi: 10.1046/j.1523-1747.1998.00273.x. [DOI] [PubMed] [Google Scholar]
- 18.Sur R, et al. UVB light suppresses nitric oxide production by murine keratinocytes and macrophages. Biochem Pharmacol. 2002;64:1469–1481. doi: 10.1016/s0006-2952(02)01419-3. [DOI] [PubMed] [Google Scholar]
- 19.Chouinard N, et al. UVB-mediated activation of p38 mitogen-activated protein kinase enhances resistance of normal keratinocytes to apoptosis by stabilizing cytoplasmic p53. Biochem J. 2002;365:133–145. doi: 10.1042/BJ20020072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mizuno H, et al. Effects of MAP kinase inhibitors on epidermal growth factor-induced neoplastic transformation of human keratinocytes. Mol Carcinog. 2006;45:1–9. doi: 10.1002/mc.20160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Black HS. The defensive role of antioxidants in skin carcinogenesis. In: Fuchs J, Packer L, editors. Oxidative Stress in Dermatology. Marcel Decker, Inc.; New York: 1993. pp. 243–270. [Google Scholar]
- 22.Vetrano AM, et al. Characterization of the oxidase activity in mam malian catalase. J Biol Chem. 2005;280:35372–35381. doi: 10.1074/jbc.M503991200. [DOI] [PubMed] [Google Scholar]
- 23.Heck DE, et al. Epidermal growth factor suppresses nitric oxide and hydrogen peroxide production by keratinocytes. J Biol Chem. 1992;267:21277–21280. [PubMed] [Google Scholar]
- 24.Isoherranen K, et al. Ultraviolet irradiation induces cyclooxyge-nase-2 expression in keratinocytes. Br J Dermatol. 1999;140:1017–1022. doi: 10.1046/j.1365-2133.1999.02897.x. [DOI] [PubMed] [Google Scholar]
- 25.Buckman SY, et al. COX-2 expression is induced by UVB exposure in human skin: implications for the development of skin cancer. Carcinogenesis. 1998;19:723–729. doi: 10.1093/carcin/19.5.723. [DOI] [PubMed] [Google Scholar]
- 26.Yarosh D, et al. Regulation of TNFα production and release in human and mouse keratinocytes and mouse skin after UV-B irradiation. Photodermatol Photoimmunol Photomed. 2000;16:263–270. doi: 10.1034/j.1600-0781.2000.160606.x. [DOI] [PubMed] [Google Scholar]
- 27.Enk CD, et al. Induction of IL-10 gene expression in human kerati-nocytes by UVB exposure in vivo and in vitro. J Immunol. 1995;154:4851–4856. [PubMed] [Google Scholar]
- 28.Halliday GM. Inflammation, gene mutation and photoimmunoesup-pression in response to UVR-induced oxidative damage contributes to pho-tocarcinogenesis. Mutat Res. 2005;571:107–120. doi: 10.1016/j.mrfmmm.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 29.Bode AM, et al. Mitogen-activated protein kinase activation in UV-induced signal transduction. Sci STKE. 2003;2003:RE2. doi: 10.1126/stke.2003.167.re2. [DOI] [PubMed] [Google Scholar]
- 30.Trouba KJ, et al. Oxidative stress and its role in skin disease. Antioxid Redox Signal. 2002;4:665–673. doi: 10.1089/15230860260220175. [DOI] [PubMed] [Google Scholar]
- 31.Black HS. Potential involvement of free radical reactions in ultraviolet light-mediated cutaneous damage. Photochem Photobiol. 1987;46:213–221. doi: 10.1111/j.1751-1097.1987.tb04759.x. [DOI] [PubMed] [Google Scholar]
- 32.Sander CS, et al. Role of oxidative stress and the antioxidant net work in cutaneous carcinogenesis. Int J Dermatol. 2004;43:326–335. doi: 10.1111/j.1365-4632.2004.02222.x. [DOI] [PubMed] [Google Scholar]
- 33.Marks F, et al. A causal relationship between unscheduled eicosa-noid signaling and tumor development: cancer chemoprevention by inhibitors of arachidonic acid metabolism. Toxicology. 2000;153:11–26. doi: 10.1016/s0300-483x(00)00301-2. [DOI] [PubMed] [Google Scholar]
- 34.Feng L, et al. Involvement of reactive oxygen intermediates in cyclooxygenase-2 expression induced by interleukin-1, tumor necrosis factor-alpha, and lipopolysaccharide. J Clin Invest. 1995;95:1669–1675. doi: 10.1172/JCI117842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feldmeyer L, et al. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Curr Biol. 2007;17:1140–1145. doi: 10.1016/j.cub.2007.05.074. [DOI] [PubMed] [Google Scholar]
- 36.Staniforth V, et al. Caffeic acid suppresses UVB radiation-induced expression of interleukin-10 and activation of mitogen-activated protein kinases in mouse. Carcinogenesis. 2006;27:1803–1811. doi: 10.1093/carcin/bgl006. [DOI] [PubMed] [Google Scholar]
- 37.Applegate LA, et al. Induction of heme oxygenase: a general response to oxidant stress in cultured mammalian cells. Cancer Res. 1991;51:974–978. [PubMed] [Google Scholar]
- 38.Allanson M, et al. Immunoprotective UVA (320–400 nm) irradiation upregulates heme oxygenase-1 in the dermis and epidermis of hairless mouse skin. J Invest Dermatol. 2004;122:1030–1036. doi: 10.1111/j.0022-202X.2004.22421.x. [DOI] [PubMed] [Google Scholar]
- 39.Obermuller-Jevic UC, et al. The effect of beta-carotene on the expression of interleukin-6 and heme oxygenase-1 in UV-irradiated human skin and fibroblasts in vitro. FEBS Lett. 2001;509:186–190. doi: 10.1016/s0014-5793(01)03169-6. [DOI] [PubMed] [Google Scholar]
- 40.Coyle P, et al. Metallothionein: the multipurpose protein. Cell Mol Life Sci. 2002;59:627–647. doi: 10.1007/s00018-002-8454-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamada H, et al. Ultraviolet irradiation increases the sensitivity of cultured human skin cells to cadmium probably through the inhibition of metallothionein gene expression. Toxicol Appl Pharmacol. 2004;200:251–257. doi: 10.1016/j.taap.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 42.Fridovich I. The biology of oxygen radicals. Science. 1978;201:875–880. doi: 10.1126/science.210504. [DOI] [PubMed] [Google Scholar]
- 43.Denning MF, et al. Caspase activation and disruption of mitochon-drial membrane potential during UV radiation-induced apoptosis of human keratinocytes requires activation of protein kinase C. Cell Death Differ. 2002;9:40–52. doi: 10.1038/sj.cdd.4400929. [DOI] [PubMed] [Google Scholar]
- 44.Hayes JD, et al. Glutathione transferases. Annu Rev Pharmacol Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 45.Das M, et al. Altered patterns of cutaneous xenobiotic metabolism in UVB-induced squamous cell carcinoma in SKH-1 hairless mice. J Biol Chem. 1985;84:532–536. doi: 10.1111/1523-1747.ep12273527. [DOI] [PubMed] [Google Scholar]
- 46.Sultana S, et al. Inhibition of benzoyl peroxide and ultraviolet-B radiation induced oxidative stress and tumor promotion markers by cyclo-artenol in murine skin. Redox Rep. 2003;8:105–112. doi: 10.1179/135100003125001422. [DOI] [PubMed] [Google Scholar]
- 47.Hiratsuka A, et al. Marked expression of glutathione S-transferase A4-4 detoxifying 4-hydroxy-2(E)-nonenal in the skin of rats irradiated by ultraviolet B-band light (UVB) Biochem Biophys Res Commun. 1999;260:740–746. doi: 10.1006/bbrc.1999.0971. [DOI] [PubMed] [Google Scholar]
- 48.Desmots F, et al. Immunohistological analysis of glutathione trans-ferase A4 distribution in several human tissues using a specific polyclonal antibody. J Histochem Cytochem. 2001;49:1573–1580. doi: 10.1177/002215540104901211. [DOI] [PubMed] [Google Scholar]
- 49.Liang FQ, et al. Enhanced expression of glutathione-S-transferase A1-1 protects against oxidative stress in human retinal pigment epithelial cells. Exp Eye Res. 2005;80:113–119. doi: 10.1016/j.exer.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 50.Hayes JD, et al. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defense against oxidative stress. Free Radical Res. 1999;31:273–300. doi: 10.1080/10715769900300851. [DOI] [PubMed] [Google Scholar]
- 51.Zuliani T, et al. Hydrogen peroxide-induced cell death in normal human keratinocytes is differentation dependent. Free Rad Biol Med. 2005;38:307–316. doi: 10.1016/j.freeradbiomed.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 52.Vessey DA, et al. Differentiation-induced enhancement of the abil ity of cultured human keratinocytes to suppress oxidative stress. J Invest Dermatol. 1995;104:355–358. doi: 10.1111/1523-1747.ep12665382. [DOI] [PubMed] [Google Scholar]
- 53.Sharma R, et al. Antioxidant role of glutathione S-transferases: pro tection against oxidant toxicity and regulation of stress-mediated apoptosis. Antioxid Redox Signal. 2004;6:289–300. doi: 10.1089/152308604322899350. [DOI] [PubMed] [Google Scholar]
- 54.Mosialou E, et al. Microsomal glutathione transferase: lipid-derived substrates and lipid dependence. Arch Biochem Biophys. 1995;320:210–216. doi: 10.1016/0003-9861(95)90002-0. [DOI] [PubMed] [Google Scholar]
- 55.Yu BP. Cellular defenses against damage from reactive oxygen species. Physiol Rev. 1994;74:139–162. doi: 10.1152/physrev.1994.74.1.139. [DOI] [PubMed] [Google Scholar]
- 56.Rosette C, et al. Ultraviolet light and osmotic stress: activation of the JNK cascade through multiple growth factor and cytokine receptors. Science. 1996;274:1194–1197. doi: 10.1126/science.274.5290.1194. [DOI] [PubMed] [Google Scholar]
- 57.Shah G, et al. Mechanism of induction of c-fos by ultraviolet B (290–320 nm) in mouse JB6 epidermal cells. Cancer Res. 1993;53:38–45. [PubMed] [Google Scholar]
- 58.Devary Y, et al. NF-kappa B activation by ultraviolet light not de pendent on a nuclear signal. Science. 1993;261:1442–1445. doi: 10.1126/science.8367725. [DOI] [PubMed] [Google Scholar]
- 59.Assefa Z, et al. Differential stimulation of ERK and JNK activities by ultraviolet B irradiation and epidermal growth factor in human kerati-nocytes. J Invest Dermatol. 1997;108:886–891. doi: 10.1111/1523-1747.ep12292595. [DOI] [PubMed] [Google Scholar]
- 60.Takahashi H, et al. Expression of human cystatin A by keratinocytes is positively regulated via the Ras/MEKK1/MKK7/JNK signal transduction pathways but negatively regulated via the Ras/Raf-1/MEK1/ERK pathway. J Biol Chem. 2001;276:36632–36638. doi: 10.1074/jbc.M102021200. [DOI] [PubMed] [Google Scholar]
- 61.Gazel A, et al. Inhibition of JNK promotes differentiation of epi dermal keratinocytes. J Biol Chem. 2006;281:20530–20541. doi: 10.1074/jbc.M602712200. [DOI] [PubMed] [Google Scholar]
- 62.Coffey RJ, Jr, et al. Growth modulation of mouse keratinocytes by transforming growth factors. Cancer Res. 1988;48:1596–1602. [PubMed] [Google Scholar]
- 63.Kock A, et al. Human keratinocytes are a source for tumor necrosis factor alpha: evidence for synthesis and release upon stimulation with endotoxin or ultraviolet light. J Exp Med. 1990;172:1609–1614. doi: 10.1084/jem.172.6.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kietzmann T, et al. Transcriptional regulation of heme oxygenase-1 gene expression by MAP kinases of the JNK and p38 pathways in primary cultures of rat hepatocytes. J Biol Chem. 2003;278:17927–17936. doi: 10.1074/jbc.M203929200. [DOI] [PubMed] [Google Scholar]
- 65.Desmots F, et al. Pro-inflammatory cytokines tumor necrosis factor alpha and interleukin-6 and survival factor epidermal growth factor posi tively regulate the murine GSTA4 enzyme in hepatocytes. J Biol Chem. 2002;277:17892–17900. doi: 10.1074/jbc.M112351200. [DOI] [PubMed] [Google Scholar]
- 66.Jang SI, et al. Loricrin expression in cultured human keratinocytes is controlled by a complex interplay between transcription factors of the Sp1, CREB, AP1, and AP2 families. J Biol Chem. 2002;277:42268–42279. doi: 10.1074/jbc.M205593200. [DOI] [PubMed] [Google Scholar]
- 67.Rivier M, et al. Differential expression of peroxisome proliferator-activated receptor subtypes during the differentiation of human keratino-cytes. J Invest Dermatol. 1998;111:1116–1121. doi: 10.1046/j.1523-1747.1998.00439.x. [DOI] [PubMed] [Google Scholar]
- 68.Jans J, et al. Differential role of basal keratinocytes in UV-induced immunosuppression and skin cancer. Mol Cell Biol. 2006;26:8515–8526. doi: 10.1128/MCB.00807-06. [DOI] [PMC free article] [PubMed] [Google Scholar]