

The hasubanan akaloids are a large collection of natural products isolated from several medicinal herbs that are used in traditional Chinese medicine.[i] Members of this family share a common aza-[4.4.3]-propellane core, but vary substantially in the oxidation patterns of their peripheral structure. The least oxidized hasubanans are 8-demethoxyrunanine (1)[ii] and cepharamine (3);[iii] runanine (2),[iv] aknadinine (4),[v] and hasubanonine (5)[vi] are the result of further oxidation at C8 (Figure 1). These compounds are closely related to an isomeric family of natural products, the cepharatines (7–10), which were isolated in 2011 from S. cepharantha, the same plant from which cepharamine (3) was isolated.[vii] The structural similarities between the hasubanans and the cepharatines have led to the hypothesis that both arise biosynthetically from common precursors. For example, 3, 7, and 8 are proposed to derive from sinoacutine (11),[vii, viii] a compound related, though antipodal, to morphine. Indeed, due to the topographical similarities between compounds 1–5 and morphine, there is speculation that the unnatural enantiomers of the hasubanans may exhibit analgesic properties.[ix]
Figure 1.

Representative members of the hasubanan and cepharatine alkaloids.
The hasubanan alkaloids have been the subject of research by a number of synthetic groups over the past forty years. Although several hasubanans were prepared by total synthesis in racemic form in the early 1970s,[x, xi] the enantioselective chemical synthesis of this family of compounds has proven far more challenging. The first enantioselective total synthesis of a hasubanan alkaloid was the 21-step synthesis of cepharamine (3) reported by Schultz and Wang in 1998.[xii, xiii] As part of a program targeting the total syntheses of several alkaloid natural products, we sought to develop a unified strategy for the enantioselective preparation of the hasubanan and cepharatine alkaloids. From the outset, the objective was to develop a synthetic approach that could provide access to any member of the hasubanan alkaloids, starting with the least oxidized members 1 and 3. Following a plan inspired by nature,[xiv] we envisioned preparing the appropriate aza-propellane skeleton, and then systematically introducing peripheral oxidation as dictated by the target compound. Ideally, the proposed aza-propellane intermediates would also be suited for conversion to the corresponding cepharatine natural products. In this communication, we report our preliminary results that establish the viability of this approach through short and enantioselective total syntheses of the natural products 8-demethoxyrunanine (1) and cepharatines A (7), C (8) and D (10).
In accord with our plan, both the hasubanans and cepharatines were anticipated to arise from an aza-propellane intermediate of the general structure 12 (Figure 2). This aza-propellane intermediate was foreseen to derive from dihydroindolone 13 by an intramolecular Friedel-Crafts-type alkylation. In a following step, oxidation and rearrangement of 12 could then give rise to 17, bearing the cepharatine framework. As an important part of our strategy, it was anticipated that the arene oxidation patterns of either runanine/cepharatine D or cepharamine/cepharatine A could be generated from 13 by simply controlling the site of electrophilic aromatic substitution in the Friedel-Crafts reaction. Literature precedent suggested that the intrinsic selectivity of the dimethoxy substrate 13a (R1 = H, R2 = OMe, R3 = H) would favor reaction at the less sterically encumbered para-position, to provide the product with the runanine oxidation pattern (12a, R1 = H, R2 = OMe, R3 = H).[xv] Alternatively, we anticipated generating the aza-propellane bearing the cepharamine oxidation pattern found in 12b (R1 = OTMSE, R2 = H, R3 = H) by installing an appropriate blocking group in the cyclization substrate (e.g. 13b, R1 = OTMSE, R2 = H, R3 = Br).
Figure 2.

Synthetic plan.
To implement the synthetic plan detailed above, the enantioselective preparation of dihydroindolones 13a/13b would be required. We recently reported the preparation of benzoquinone monoketal-derived N-tert-butanesulfinimine 14, which undergoes highly diastereoselective 1,2-addition with a variety of organometallic reagents.[xvi] Based on this report, we expected dihydroindolones 13a/13b to be accessible from 14 by a short sequence involving Grignard addition, N-methylation, and pyrrolidine formation.
In the forward sense, our synthesis began with N-tert-butanesulfinimine 14,[xvi] easily prepared on multigram scale in two steps from commercially available 2-bromo-4-methoxyphenol (Scheme 1). Addition of Grignard reagent 20a at low temperatures followed by in situ N-methylation provided sulfinamide 21 in 77% yield, which was isolated as a single diastereomer. Analysis of the crude reaction mixture determined that the 1,2-addition proceeded in 96:4 dr. Notably, hydrolysis of the dimethyl acetal occurs during the mildly acidic workup without detectable quantities of undesired dienone-phenol rearrangementxvii products.
Scheme 1.

Synthesis of enantioenriched dihydroindolones 13a and 26.
Construction of the required pyrrolidine ring was accomplished by a three step sequence that began with Pd-catalyzed cross coupling between vinyl bromide 21 and ethoxy vinylstannane 23 to yield enol ether 24 in 90% yield (Scheme 1). After considerable experimentation, it was found that brief exposure of 24 to 1M HCl in THF at 0 °C cleaved the sulfinamide and promoted intramolecular condensation to provide the corresponding indolone.[xviii] Chemoselective mono-reduction of the indolone was achieved using sodium borohydride and acetic acid, furnishing the desired dihydroindolone 13a in 96% yield over two steps. With an eye toward preparing cepharamine (3) or cepharatines A (7) and C (8), dihydroindolone 26, bearing a differentially protected arene, was prepared through an analogous route from 20b.
With access to dihydroindolones 13a and 26, our efforts turned to implementing the key intramolecular Friedel-Crafts reactions (Scheme 2). A small screen of Lewis acids revealed that exposure of dienone 13a to BF3•Et2O promoted cyclization; however, the yield of recovered 12a was moderate.[xix] Thus, we turned to the use of strong Brønsted acids, and were pleased to find that use of excess TfOH in dichloromethane[xx] smoothly promoted cyclization exclusively at C14, delivering the desired propellane 12a in 97% yield. It is proposed that selective addition to the trisubstituted alkene, in preference to the less-hindered disubstituted alkene, results from the formation of a discrete, protonated intermediate that favors the more stable, tertiary carbocation at C14. Given our desire to access the cepharamine oxidation pattern, the TMSE-protected substrate 26 was brominated and exposed to the TfOH cyclization conditions in situ to furnish aza-propellane 30. Monitoring this reaction revealed that cleavage of the TMSE group is rapid, and cyclization of the phenol occurs upon warming the reaction to room temperature.
Scheme 2.

Preparation of aza-propellanes 12a and 30.
Having developed an efficient and unified approach to aza-propellanes bearing either the runanine or cepharamine oxidation patterns, we turned to the remaining challenge of adjusting the oxidation level of C7 to that found in both 1 and 3. To this end, exploratory studies were carried out using 12a (Scheme 3). We were pleased to find that exposure of enone 12a to standard nucleophilic epoxidation conditions[xxi] (H2O2, LiOH, MeOH) followed by heating to 50 °C provided 8-demethoxyrunanine (1), albeit in low yield (10–15% yield). Presuming that 1 was formed via the epoxide, we hoped to optimize the yield of the overall process by isolating this intermediate. Ultimately, it was determined that the combination of t-butylhydroperoxide (TBHP) and Triton B in THF provided clean conversion to epoxide 27. However, attempts to purify the reaction mixture by silica gel chromatography led to isolation of epoxide 27 along with hemiaminal 28, a compound bearing the cepharatine framework.[xxii] One proposed mechanism for the formation of 28 begins with nitrogen-assisted opening of the epoxide followed by β-elimination of the aziridinium to give enol 15 (see Figure 2, R1 = H, R2 = OMe). Enol-facilitated elimination of the amine and intramolecular aminal formation would provide 28. This rearrangement can be suppressed by purification of epoxide 27 using Florisil. Though pleased by our ability to generate the cepharatine framework from the hasubanan core, we continued to explore conditions for the formation of 1. After an extensive survey of reaction conditions,[xxiii] it was discovered that use of tetrabutylammonium methoxide[xxiv] in THF at 50 °C for 12 h provided the natural product 8-demethoxyrunanine (1) in 68% yield. Using this sequence, 1 is prepared in only nine steps and in 19% overall yield from commercially available phenol 18.
Scheme 3.

Enantioselective synthesis of 8-demethoxyrunanine (1) and cepharatine D (10).
Following completion of the synthesis of 8-demethoxyrunanine (1), attention turned to improving the yield of hemiaminal 28 and elaborating it to cepharatine D (10). After screening several reaction parameters, it was determined that epoxidation of 12a followed by prolonged exposure to silica gel provided direct access to aminal 28 in 76% yield from propellane 12a (Scheme 3). Desaturation of aminal 28 was carried out by deprotonation with excess KHMDS at −78 °C followed by addition of N-t-butylbenzenesulfinimidoyl chloride (29),[xxv] providing cepharatine D (10) in 9 steps and 22% overall yield from 18.
Having converted propellane 12a to the natural products 1 and 10, we sought to carry out a similar reaction sequence to convert bromo-propellane 30 to the corresponding compounds cepharamine (3) and cepharatine A (7) (Scheme 4). In contrast to the epoxidation of 12a, epoxidation of enone 30 proceeded sluggishly, and despite considerable attempts at optimization, epoxide 31 was isolated in only 40% yield.
Scheme 4.

Enantioselective synthesis of cepharatines A (7) and C (8)
Efforts to drive the epoxidation reaction to completion were complicated by competitive oxidative rearrangement of the epoxide product, resulting in formation of a lactone byproduct.[xix] Unfortunately, exposure of epoxide 31 to Bu4NOMe in THF (identical conditions to those utilized to convert 27 to 1) provided only trace amounts of enol ether 32, as detected by 1H NMR analysis of the crude reaction mixture. Reasoning that deprotonation of the phenolic O-H might contribute to the poor reactivity, several protected variants of 30 were prepared (e.g. Me-, MOM-, allyl-, and Bn-protected phenols).
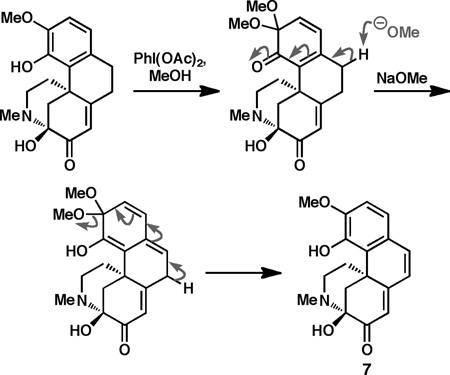
Whereas the epoxidation step proceeded with improved efficiency for these substrates, exposure of the epoxides to a variety of methoxide sources still provided prohibitively low quantities of the desired methyl enol ethers (analogous to 32). These studies illustrate how subtle perturbations in the arene oxidation patterns can strikingly alter the reactivity of the aza-propellane framework. Similarly, treatment of enone 30 under the tandem epoxidation/rearrangement conditions identified for the conversion of 12a to 28 provided lower yields of the corresponding hemiaminal (33) (Scheme 4). However, we were pleased to find that selective hydrodebromination of the aryl bromide followed by treatment with PhI(OAc)2 and base cleanly provided cepharatine A (7) in good yield over two steps.[xxvi] Finally, cepharatine A could be converted to cepharatine C (8) by exposure to methanol under mildly acidic conditions. Using this reaction sequence, 7 and 8 could be prepared in 10 and 11 steps and each in 10% overall yield, respectively, from commercially available starting materials.
In conclusion, a unified synthetic strategy has resulted in the short, enantioselective total syntheses of 8-demethoxyrunanine (1) and cepharatines A (7), C (8), and D (10). Key to this synthetic strategy was the use of benzoquinone monoketal-derived N-tert-butanesulfinimine 14 to prepare 4-aminocyclohexadienones 21 and 22 with excellent stereocontrol. Depending on the reaction sequences, either the runanine or cepharamine arene oxidation patterns could be achieved by way of a regioselective intramolecular Friedel-Crafts-type alkylation. Moreover, it was shown that the hasubanan framework rearranges under mild conditions providing access to the cepharatine natural products. Ongoing studies in our laboratory are focused on the development of oxidation strategies to access cepharamine and the more oxidized members of the hasubanans, as well as the application of this general approach to the synthesis of the related acutumine[xxvii] family of alkaloids.
Supplementary Material
Acknowledgments
We thank Drs. Michael Day and Larry Henling for X-ray crystallographic structural determination, as well as Prof. Brian Stoltz, Dr. Scott Virgil, and the Caltech Center for Catalysis and Chemical Synthesis for access to analytical equipment. Fellowship support was provided by the Gates Millennium Scholars Program (R.N.), the NIH (R.N., FGM098025A) and the Amgen Scholars Program (K.V.C.). HRMS and X-ray crystallographic data were obtained on instruments purchased through awards to the California Institute of Technology by the NSF CRIF program (CHE-0639094, CHE-0541745). Financial support from the California Institute of Technology and the NSF (CAREER-1057143) is gratefully acknowledged.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- i.Matsui M. In: The Alkaloids. Brossi A, editor. Vol. 33. Academic Press, Inc; 1988. pp. 307–347. [Google Scholar]
- ii.Wang X, Jin H, Li Z, Qin G. Fitoterapia. 2007;78:593. doi: 10.1016/j.fitote.2007.03.018. [DOI] [PubMed] [Google Scholar]
- iii.Tomita M, Kozuka M. Tetrahedron Lett. 1966;7:6229. [Google Scholar]
- iv.Zhi-Da M, Ge L, Guang-Xi X, Iinuma M, Tanaka T, Mizuno M. Phytochemistry. 1985;24:3084. [Google Scholar]
- v.Moza BK, Bhaduri B, Basu DK, Kunitomo J, Okamoto Y, Yuge E, Nagai Y. Tetrahedron. 1970;26:427. [Google Scholar]
- vi.Tomita M, Ibuka T, Inubushi Y, Watanabe Y, Matsui M. Tetrahedron Lett. 1964;5:2937. [Google Scholar]
- vii.He L, Zhang Y-H, Guan H-Y, Zhang J-X, Sun Q-Y, Hao X-J. J. Nat. Prod. 2011;74:181. doi: 10.1021/np1005696. [DOI] [PubMed] [Google Scholar]
- viii.Kashiwaba N, Morooka S, Kimura M, Ono M, Toda J, Suzuki H, Sano T. J. Nat. Prod. 1996;59:476. [Google Scholar]
- ix.Schultz and Wang first hypothesized about the analgesic activities of the unnatural antipodes of the hasubanans. See ref 12.
- x.Completed total syntheses: Hasubanonine, Ibuka T, Tanaka K, Inubushi Y. Tetrahedron Lett. 1970;11:4811.; Ibuka T, Tanaka K, Inubushi Y. Chem. Pharm. Bull. 1974;22:782.; Metaphanine, Ibuka T, Tanaka K, Inubushi Y. Tetrahedron Lett. 1972;13:1393.; Ibuka T, Tanaka K, Inubushi Y. Chem. Pharm. Bull. 1974;22:907.; Cepharamine, Inubushi Y, Kitano M, Ibuka T. Chem. Pharm. Bull. 1971;19:1820.; Kametani T, Nemoto H, Kobari T, Shishido K, Fukumoto K. Chem. Ind. 1972;13:538..
- xi.Partial or formal syntheses: Tomita M, Ibuka T, Kitano M. Tetrahedron Lett. 1966;7:6233.; Ibuka T, Kitano M. Chem. Pharm. Bull. 1967;15:1944. doi: 10.1248/cpb.15.1944.; Tomita M, Kitano M, Ibuka T. Tetrahedron Lett. 1968;9:3391.; Evans DA. Tetrahedron Lett. 1969;10:1573.; Evans DA, Bryan CA, Wahl GM. J. Org. Chem. 1970;35:4122.; Keely SL, Jr, Martinez AJ, Tahk FC. Tetrahedron. 1970;26:4729.; Evans DA, Bryan CA, Sims CL. J. Am. Chem. Soc. 1972;94:2891.; Kametani T, Kobari T, Fukumoto K. J. Chem. Soc. Chem. Commun. 1972:288.; Belleau B, Wong H, Monkovic I, Perron YG. J. Chem. Soc. Chem. Commun. 1974:603. doi: 10.1021/ja00804a082.; Kametani T, Kobari T, Shishido K, Fukumoto K. Tetrahedron. 1974;30:1059.; Monkovic I, Wong H, Belleau B, Pachter IJ, Perron YG. Can. J. Chem. 1975;53:2515. doi: 10.1021/ja00804a082.; Monkovic I, Wong H. Can. J. Chem. 1976;54:883.; Shiotani S, Kometani T. Tetrahedron Lett. 1976;17:767.; Bruderer H, Knopp D, Daly JJ. Helv. Chim. Acta. 1977;60:1935.; Schwartz MA, Wallace RA. Tetrahedron Lett. 1979;20:3257.; Trauner D, Porth S, Opatz T, Bats JW, Giester G, Mulzer J. Synthesis. 1998:653.; Jones SB, He L, Castle SL. Org. Lett. 2006;8:3757. doi: 10.1021/ol0613564.; Nguyen TX, Kobayashi Y. J. Org. Chem. 2008;73:5536. doi: 10.1021/jo800793s.; Nielsen DK, Nielsen LL, Jones SB, Toll L, Asplund MC, Castle SL. J. Org. Chem. 2009;74:1187. doi: 10.1021/jo802370v..
- xii.Schultz AG, Wang AH. J. Am. Chem. Soc. 1998;120:8259. [Google Scholar]
- xiii.As we were preparing this manuscript, a synthetic strategy similar to ours was reported by Herzon and coworkers, see: Herzon SB, Calandra NA, King SM. Angew. Chem. Int. Ed. 2011 doi: 10.1002/anie.201102226.
- xiv.For an account on this topic, see: Ishihara Y, Baran PS. Synlett. :1733..
- xv.Selected examples: Gensler WJ, Bluhm AL. J. Org. Chem. 1956;21:336. Padwa A, Danca MD. Org. Lett. 2002;4:715. doi: 10.1021/ol017193v..
- xvi.Chuang KV, Navarro R, Reisman SE. Chem. Sci. 2011;2:1086. [Google Scholar]
- xvii.Wilds AL, Djerassi C. J. Am. Chem. Soc. 1946;68:1715. doi: 10.1021/ja01213a010. [DOI] [PubMed] [Google Scholar]
- xviii.Use of prolonged reaction times or weaker acids for sulfinamide cleavage resulted in diminished yields of 13a due to competitive decomposition of the indolone intermediate. In addition, attempts to utilize the des-N-methyl analogue of 21 provided poor yields in the indolone formation step.
- xix.See Supporting Information.
- xx.Li W-DZ, Wang X-W. Org. Lett. 2007;9:1211. doi: 10.1021/ol070024b. [DOI] [PubMed] [Google Scholar]
- xxi.Antony P, Sigueiro R, Huet T, Sato Y, Ramalanjaona N, Rodrigues LC, Mourino A, Moras D, Rochel N. J. Med. Chem. 2010;53:1159. doi: 10.1021/jm9014636. [DOI] [PubMed] [Google Scholar]
- xxii.Castle and coworkers have isolated a similar intermediate during the course of synthetic studies toward the hasubanans. See ref 11s.
- xxiii.All other conditions screened provided hemiaminal 28 as the major product or led to significant decomposition.
- xxiv.Di Bussolo V, Caselli M, Romano MR, Pineschi M, Crotti P. J. Org. Chem. 2004;69:8702. doi: 10.1021/jo048981b. [DOI] [PubMed] [Google Scholar]
- 25.(a) Mukaiyama T, Matsuo J, Kitagawa H. Chem. Lett. 2000;29:1250. [Google Scholar]; (b) Matsuo J, Aizawa Y. Tetrahedron Lett. 2005;46:407. [Google Scholar]
-
xxvi.In this transformation, desaturation presumably occurs by phenolic oxidation to the ortho-quinone monoketal, followed by base-mediated elimination of methanol and rearomatization.
- 27.Tomita M, Okamoto Y, Kikuchi T, Osaki K, Nishikawa M, Kamiya K, Sasaki Y, Matoba K, Goto K. Tetrahedron Lett. 1967;8:2421. [Google Scholar]

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.