

Abstract
Ovarian cancer is the fifth leading cause of cancer death for women in the U.S., yet survival rates are over 90% when it is diagnosed at an early stage, highlighting the need for biomarkers for early detection. To enhance the discovery of tumor-specific proteins which could represent novel serum biomarkers for ovarian cancer, we depleted serum of highly abundant proteins which can mask the detection of proteins present in serum at low concentrations. Three commercial immunoaffinity columns were used in parallel to deplete the highly abundant proteins in serum from 60 patients with serous ovarian carcinoma and 60 non-cancer controls. Medium and low abundance serum proteins from each serum pool were then evaluated by the quantitative proteomic technique of Differential-In-Gel-Electrophoresis (DIGE). The number of protein spots that were elevated in ovarian cancer sera by at least 2-fold ranged from 36 to 248, depending upon the depletion and separation methods. From the 33 spots picked for MS analysis, nine different proteins were identified, including the novel candidate ovarian cancer biomarkers leucine-rich alpha-2 glycoprotein-1 and ficolin 3. Western blotting validated the relative increases in serum protein levels for three of the proteins identified, demonstrating the utility of this approach for the identification of novel serum biomarkers for ovarian cancer.
Keywords: Biomarker, Differential In Gel Electrophoresis, Immunodepletion, Ovarian cancer, Proteomics, Serum
Introduction
Ovarian cancer is the fifth leading cause of cancer-related deaths for women in the United States [1], a statistic that could be reduced with improved methods for early detection. Current screening techniques for ovarian cancer are neither adequately sensitive nor specific [2]. CA125 is an antigen present in the sera of the majority of patients diagnosed with ovarian cancer [3, 4]. However, CA125 is less commonly elevated in the sera of women with early stage ovarian cancer [5] and can be detected in other cancers as well as non-malignant conditions [4], making it unsuitable as a screening test for the general population.
Researchers have used a variety of techniques to discover novel protein biomarkers to replace or be used in conjunction with CA125. Gene expression and proteomic profiling of ovarian tumor tissues and cell lines have identified a large number of proteins with increased expression in ovarian cancer [6]. Although proteomic techniques have been used to analyze lysates from ovarian cancer and normal ovary epithelial cells [7-9] and microdissected ovarian tumors [10], they have not been widely used on serum.
In the past decade, the development of quantitative MS-based proteomic techniques has allowed the direct comparison of protein levels present in control and diseased samples. Using Differential-In-Gel-Electrophoresis (DIGE), cancer and control samples are labeled with different fluorescent dyes, then the samples are combined and proteins are separated by 2-DE. Bengtsson et al. [11] used this technique to identify proteins differentially expressed in malignant ovarian cancer tissues compared to normal, benign, or borderline ovarian tissues. It is not known whether any of the proteins identified in these studies are secreted or released ectopically into the sera of patients.
Although MS identification of tissue-derived proteins in plasma is feasible [12], the dynamic range and complexity of proteins present in serum or plasma requires additional fractionation in order to detect low abundance proteins. One approach is to deplete the most highly abundant proteins, comprising ∼ 95% of serum total protein, using immunoaffinity columns [13]. Available depletion strategies have demonstrated effective removal of high abundance proteins and improvement in the detection of less abundant serum proteins [14, 15]. Immunodepletion in combination with DIGE analysis of serum has been used to identify potential biomarkers in lung [16, 17], pancreatic [18, 19], and prostate cancers [20].
In our study, pooled serum samples from 60 patients with serous ovarian carcinoma and 60 non-cancer controls were depleted of high abundance proteins using immunoaffinity depletion columns. The remaining medium and low abundance proteins were then subjected to analyses by DIGE in order to identify proteins with increased abundance in ovarian cancer sera relative to control sera. To the authors' knowledge, this is the first study of serum immunodepletion in combination with DIGE as a means to measure relative protein expression in ovarian cancer patients for the pursuit of serum biomarkers, enabling the discovery of new and potentially valuable biomarkers of ovarian cancer.
Materials and Methods
Patient Samples
De-identified serum samples from 60 patients with serous ovarian carcinoma and 60 female non-cancer controls were obtained from the Gynecologic Oncology Group (GOG) Tissue Bank. The majority of the ovarian cancer serum samples were from patients with stage III serous tumors (44 samples), seven had stage I and II, and nine had stage IV tumors. The average CA125 value was 2,289 units/ml (range 12 – 15,000 units/ml) for the 26 ovarian cancer patients for whom this information was available. The age of ovarian cancer patients ranged from 35-85 years compared to 19-58 years for the non-cancer controls. Cancer and non-cancer control sera were separately pooled into six groups containing serum from ten patients (C1 to C6 for the 60 cancer samples; N1 to N6 for the 60 non-cancer control samples).
Serum depletion by MARS spin column
The multiple affinity removal system (MARS) spin column (Agilent Technologies, Santa Clara, CA) was used to deplete the six most highly abundant proteins present in sera (albumin, alpha-1 antitrypsin, haptoglobin, transferrin, IgA, and IgG) according to the manufacturer's specifications. Briefly, 10 μl of each serum pool (∼ 0.5 mg) was diluted and filtered through a 0.22 μm cellulose acetate spin filter (Agilent Technologies) before applying to the equilibrated MARS column. To maximize recovery, two additional washes were performed and combined with the flow-through fraction. Six 10 μl aliquots of each serum pool (C1 – C6 and N1 – N6) were subjected to depletion and combined. The combined flow-through and wash fractions for each serum pool were designated the low abundance fraction. The high abundance (bound) proteins were eluted from the column and the column was immediately equilibrated for reuse.
Serum depletion by ProteomeLab IgY-12 spin column
The ProteomeLab IgY-12 spin column (Beckman Coulter, Fullerton, CA) was used to deplete twelve highly abundant serum proteins [albumin, total IgG, alpha-1 antitrypsin, IgA, IgM, transferrin, haptoglobin, α1-acid glycoprotein (orosomucoid), α2-macroglobulin, HDL (apolipoproteins A-I and A-II), and fibrinogen] according to the manufacturer's specifications, except that Dulbecco's PBS without calcium chloride or magnesium chloride (Invitrogen, Carlsbad, CA) was substituted for the dilution buffer. Ten μl (∼ 0.5 mg) of each serum pool was diluted and applied to the IgY-12 column, and the medium and low abundance proteins were collected in the flow-through. Two additional washes with PBS were conducted and pooled with flow-through to maximize recovery. The high abundance (bound) proteins were eluted from the column, and complete removal of bound proteins was ensured by monitoring for absorbance at 280 nm. The column was immediately regenerated prior to reuse.
Serum depletion by ProteomeLab IgY-12 liquid chromatography
The same 12 high abundance proteins described above were depleted from pooled serum with the ProteomeLab IgY-12 High Capacity LC10 affinity column (Beckman Coulter) according to the manufacturer's instructions. For each serum pool, 150 μl (∼ 7.5 mg) was diluted into loading buffer and loaded onto the column using an Agilent 1100 Series quaternary pump HPLC (Agilent Technologies). Elution of proteins not bound to the column (medium and low abundance proteins) was monitored by A280. Bound proteins (high abundance proteins) were eluted at low pH and then neutralized. The column was regenerated by washing until the A280 returned to baseline to ensure complete removal of the high abundance proteins and avoid sample carryover between column runs.
Serum concentration and protein determination
Fractions from the three affinity columns were concentrated and the buffer exchanged to PBS using a 5000 MW cut off PES membrane concentrator (VivaScience, Hanover, Germany). Protein concentration was determined using the BCA method (Pierce Protein Research Products, Rockford, IL).
2-DE
Fifty μg of unlabeled serum proteins were separated by isoelectric point (pI range 3.0 to 10.0) in the first dimension and SDS-PAGE in the second dimension, as described below for DIGE. Proteins were visualized by silver staining [21].
Differential In-Gel Electrophoresis (DIGE)
50 μg of medium and low abundance proteins from each serum pool were minimally labeled with Amersham CyDye™, per the manufacturer's protocol (GE Healthcare, Piscataway, NJ). The initial DIGE experiment used sera depleted of high abundance proteins with the MARS column. Ovarian cancer serum pool (C1) and non-cancer serum pool (N1) were labeled with Cy5 and Cy3, respectively. IgY-12 spin column depleted cancer serum pools (C1-C6) were labeled with Cy5 and non-cancer control serum pools (N1-N6) were labeled with Cy3. Paired C and N samples were run on six DIGE gels. For sera depleted using the IgY-12 HPLC method, cancer serum pools C1-C3 were labeled with Cy5, and non-cancer serum pools N1-N3 were labeled with Cy3; cancer serum pools C4-C6 were labeled with Cy3, and non-cancer serum pools N4-N6 were labeled with Cy5. For the IgY-12 HPLC experiments, Cy2 was designated for labeling a pooled internal standard composed of equal quantities from all serum pools. The Cy2 labeled internal control was used to determine the “standardized abundance” (the ratio of Cy3/Cy2 and Cy5/Cy2 channels) for more accurate spot statistics and quantitation, and better spot matching between gels. Six DIGE gels were run with paired ovarian cancer and non-cancer samples, and the internal control.
Equal quantities of the labeled samples were combined to a final volume of 250 μl for 13 cm Immobline™ DryStrip (GE Healthcare), or 200 ul for 11 cm ReadyStrip™ (BioRad, Hercules, CA) with immobilized pH gradient (IPG) running buffer [7 M urea, 2 M thiourea, bromophenol blue, 0.5% v/v ampholyte buffer (GE Healthcare or BioRad)] and 12 mM dithiothreitol (DTT). Samples were rehydrated into Immobline™ DryStrips, pH 3-10 or 4-7, or ReadyStrip™, pH 3.9-5.1 overnight under low current. Immobline™ DryStrip samples were resolved in an Ettan™ IPGphor ™ IEF apparatus (GE Healthcare), whereas the ReadyStrip™ samples were resolved on a BioRad Protean® IEF Cell per manufacturer's protocol. Strips were equilibrated for 0.5 hr in SDS equilibration buffer (50 mM Tris, pH 8.8, 8 M urea, 30% glycerol, 4% SDS, 1% DTT) followed by separation on 8-16% Tris-HCl SDS-PAGE Criterion™ gels (BioRad).
Gel spots were visualized with either the Typhoon 8610 scanner (GE Healthcare) or the Fuji Film FLA-5000 digital scanner (Cypress, CA). Fuji Film ImageGauge v4.1 software was used to export tagged image format (tiff) files for analysis, and composite images consisting of all CyDyes were made with FluorSep v2.2 software (Molecular Dynamics, Sunnyvale, CA). Images were cropped for importing into the DeCyder software with Image Quant v5.2 software (Molecular Dynamics). DeCyder v5.02 (GE Healthcare) software was used to evaluate the differential expression between samples. Spots were considered differentially expressed if they had an average ratio change greater than 2.0 and a student's t-test p-value of ≤ 0.05. Differentially expressed spots were picked for MS identification if they had a volume of at least 5 × 10−6, and were resolved adequately from other spots as determined by visual inspection.
Protein staining, spot excision and protease digestion
Gels were stained with Deep Purple total protein stain (GE Healthcare), and visualized with the Typhoon 8610 scanner. Images were imported into the Genomic Solutions® Investigator ProPic™ instrument (Genomic Solutions Inc, Ann Arbor, MI) for robotic excision. Excised spots were trypsin digested [22] using a Genomic Solutions® ProPrep™ kit and lyophilized.
MS analysis of protein spots
Lyophilized tryptic peptides were reconstituted in water/ACN/formic acid (95:5:0.1) and injected into a Michrom Bioresources Paradigm 2D capillary LC system (Auburn, CA) online with a linear ion trap (LTQ, Thermo Scientific, Waltham, MA). Peptides were desalted and concentrated on a Paradigm Platinum Peptide Nanotrap (Michrom Bioresources, Inc.) precolumn and eluted onto a Magic C18 AQ reversed-phase column (Michrom Bioresources, Inc.), at a flow rate of ∼250 nl/min. The peptides were separated during a 60 min (10-40% ACN) linear gradient and ionized with a voltage of 2.0 kV applied distally on the column. The LTQ was set to positive polarity and scans were acquired using a data-dependent acquisition method: one survey (MS) scan followed by MS/MS (relative collision energy of 35%) on the four most abundant ions detected in the survey scan. Dynamic exclusion was employed for 30 sec time intervals.
Database searching and protein identification
MS/MS data from the LTQ were analyzed with Sequest embedded in BioWorks Browser (v 3.3, Thermo Scientific) and searched against a database of human, mouse, and rat [NCBI (http://www.ncbi.nlm.nih.gov)] non-redundant protein sequences (extracted on 122409) in addition to 179 common contaminant proteins (Thermo Scientific), for a total of 206,318 proteins. Searches were conducted with a fragment ion mass tolerance of 1.00 Da and a parent ion tolerance of 1.00 Da. The search parameters were: trypsin digestion, fixed carbamidomethyl modification of cysteine and variable oxidation of methionine, with three trypsin missed cleavage sites allowed. The dta/out files generated by Bioworks were analyzed in Scaffold (v_2_00_03, Proteome Software Inc., Portland, OR) to validate MS/MS based peptide and protein identifications. Peptide identifications were accepted if they could be established at >95.0% probability as specified by the Peptide Prophet algorithm [23]. Protein identifications were accepted if they could be established at >99.0% probability by the Protein Prophet algorithm [24], and contained at least 3 identified peptides. Spots that could not be identified using the above criteria were assigned to the next highest scoring protein if, upon manual inspection of the spectra, all peaks could be assigned and greater than 3 consecutive product ions were present.
Western Blots
Fifty μg of IgY-12 HPLC column depleted sera in Laemmli loading buffer (BioRad) containing 2% β-mercaptoethanol (v/v), was separated on a 4-20% gradient Tris-HCl Criterion™ gel (BioRad) in Tris-glycine buffer [0.1% SDS (w/v), 25 mM Tris, 192 mM glycine, pH 8.3]. Separated proteins were electroblotted using a Criterion™ Blotter (BioRad) onto PVDF membrane (Pall Corporation, Pensacola, FL) in transfer buffer (20% methanol, 25 mM Tris base, 192 mM glycine, pH 8.0). The blots were blocked with 5% non-fat dried milk, in TBS with 0.05% Tween-20, then incubated in antibodies against human alpha-1 antichymotrypsin (mAb 213907; R&D Systems, Minneapolis, MN); ficolin 3 (mAb 2A6; R&D Systems); and leucine-rich alpha-2 glycoprotein-1 (LRG1) (mAb 2E3; Abnova, Taiwan). After washing, blots were incubated with a horseradish peroxidase conjugated secondary antibody (Pierce Protein Research Products). Proteins were visualized with ECL using SuperSignal West Femto Maximum Sensitivity substrate (Pierce Protein Research Products). Images were collected by exposure to Kodak x500 film (Midwest Scientific, Valley Park, MO). A duplicate gel was run and stained with CBB as a loading control.
Results
Removal of high abundance proteins from serum
Pooled sera were depleted of the high abundance proteins using one of three commercial immunoaffinity depletion columns: MARS, IgY-12 spin, or the IgY-12 HPLC.
To demonstrate the efficacy of immunodepletion by the MARS spin column, serum proteins from all twelve serum pools were separated by 2-DE before and after fractionation. Overall, the immunodepletion was quite reproducible between serum pools. A representative example of the protein spot patterns from whole sera for one cancer and one non-cancer serum pool were visually compared to protein patterns from the different fractions for each pool (Fig. 1A-C). Spot patterns for the high abundance fractions (Fig. 1B, B′) were similar between the cancer and non-cancer serum pools. In the low abundance fractions (Fig. 1C, C′), the number and resolution of spots was superior to that seen for whole sera. In addition, the medium and low abundance fractions (Fig. 1C, C′; region within white rectangle) contained numerous protein spots not visible in the unfractionated sera (Fig. 1A, A′), demonstrating the enrichment of proteins in the low abundance fraction, particularly in the pI range of 4 to 7. Furthermore, many proteins <60 kDa in size appeared to be present in higher amounts in cancer sera (Fig. 1C; white rectangle) compared to control sera (Fig. 1C′; white rectangle); demonstrating the potential of the depletion strategy to enhance the identification of candidate serum biomarkers for ovarian cancer.
Figure 1. Depletion of highly abundant proteins from sera by MARS column.

A) 2-DE of whole sera. Fifty μg of undepleted sera from pooled cancer (A) and non-cancer (A′) samples were separated by isoelectric point and then by molecular size. Proteins were visualized by silver stain.
B) 2-DE of sera fractionated by the MARS column. Fifty μg of the high abundance proteins from cancer (B) and non-cancer (B′) serum pools were separated by 2-DE as described above.
C) Fifty μg of the medium and low abundance proteins recovered from the MARS column for one cancer serum pool (C) and non-cancer serum pool (C′) were separated by 2-DE. Box indicates a region of the gel with protein spots of increased intensity in the ovarian cancer sera. Arrows show haptoglobin subunits.
D) DIGE analysis of medium and low abundance proteins from sera depleted by the MARS column. Fifty μg of medium and low abundance proteins from ovarian cancer serum pool C1 was labeled with Cy5 (red) and 50 μg of medium and low abundance proteins from non-cancer serum pool N1 was labeled with Cy3 (green). Proteins present equally in both serum pools appear as yellow spots.
The ProteomeLab™ IgY-12 spin and HPLC affinity columns were also used for serum depletion. Fractionated sera were then subjected to 1-DE and 2-DE (Supplemental Figs. 1 and 2). As expected, the high abundance proteins were depleted from the whole sera, and the low abundance proteins enriched. Similar to the results from the MARS depleted sera (Fig. 1A-C), a visual comparison of the numbers and intensities of the medium and low abundant protein spots observed by 2-DE in cancer and non-cancer sera suggested the differential expression of various serum proteins in ovarian cancer relative to control sera (Supplemental Fig. 2).
Identification of candidate biomarkers by DIGE
DIGE analysis was performed for a single pair of pooled samples (C1 and N1) immunodepleted by MARS. In order to determine the optimal pI and molecular weight separations for effective DIGE analysis of serum, we started with the pI range 3 – 10. Image analysis using DeCyder software revealed hundreds of proteins with differential expression in ovarian cancer sera compared to control sera (Fig. 1D). A total of 1256 protein spots were detected, of which 145 (11.5%) were 2-fold or more abundant in the cancer vs. control sera; 63 (5%) were 2-fold or less abundant in the cancer vs. control sample; and 1048 (83.4%) were nearly equal in abundance in both samples (Table 1). In this experiment, spots were not excised for protein identification since the objective was to optimize the DIGE conditions; and in many cases the spots were inadequately resolved, or did not contain a sufficient amount of protein for successful identification by MS.
Table 1. Summary of DIGE Experiments.
| Depletion Method | Serum Pool | DIGE Label | pH Range | Total Number of Spots | Number of Spots 2-Fold Up* | Number of Spots 2-Fold Down† | Number of Spots Chosen for MS | Number of Proteins Identified |
|---|---|---|---|---|---|---|---|---|
| MARS | C1/N1 | Cy5-red/Cy3-green | 3.0 - 10.0 | 1256 | 145 | 63 | ∼ | ∼ |
| IgY Spin | C1-C6/N1-N6 | Cy5-red/Cy3-green | 4.0 - 7.0 | 876 | 248 | 102 | 16 | 6 |
| IgY HPLC | C1-C3/N1-N3 | Cy5-red/Cy3-green | 3.9 -5.1 | 961 | 36 | 49 | 17 | 4 |
| C4-C6/N4-N6 | Cy3-green/Cy5-red |
2-fold up indicates the number of protein spots that were 2-fold or more upregulated in the ovarian cancer serum pools compared to the non-cancer control sera as determined by the DeCyder software.
2-fold down indicates the number of protein spots that were 2-fold or more downregulated in the ovarian cancer serum pools compared to the non-cancer control sera as determined by the DeCyder software.
Six biological replicates of medium and low abundance proteins from pooled IgY-12 spin column depleted sera were co-resolved in six DIGE gels at pI 4.0 – 7.0 (Fig. 2A). More than 870 protein spots were detected using the Decyder Biological Variance Analysis software, which compared and analyzed all six gels collectively. Of the total spots detected, 248 spots (28%) were >2-fold more abundant in the cancer vs. control serum pools (p-value <0.05; Table 1). Sixteen spots (labeled on Fig. 2B) with a significant increase in abundance in all the ovarian cancer sample sets were excised for identification by MS. Three subunits of the serum protein haptoglobin were identified in 8 distinct spots (Table 2). Serum amyloid-P component, alpha-1 antichymotrypsin, and ficolin 3 were also identified (Table 2). Identification of ficolin 3 in spot 620 required reducing the search criteria to two peptides; the spectra for these peptides were verified by manual inspection (Supplemental Fig. 3). No proteins were identified for five of the excised spots. Peptides used for the identification of each protein are listed in Supplemental Table 1.
Figure 2. DIGE analysis of medium and low abundance serum proteins depleted by the IgY-12 spin column.
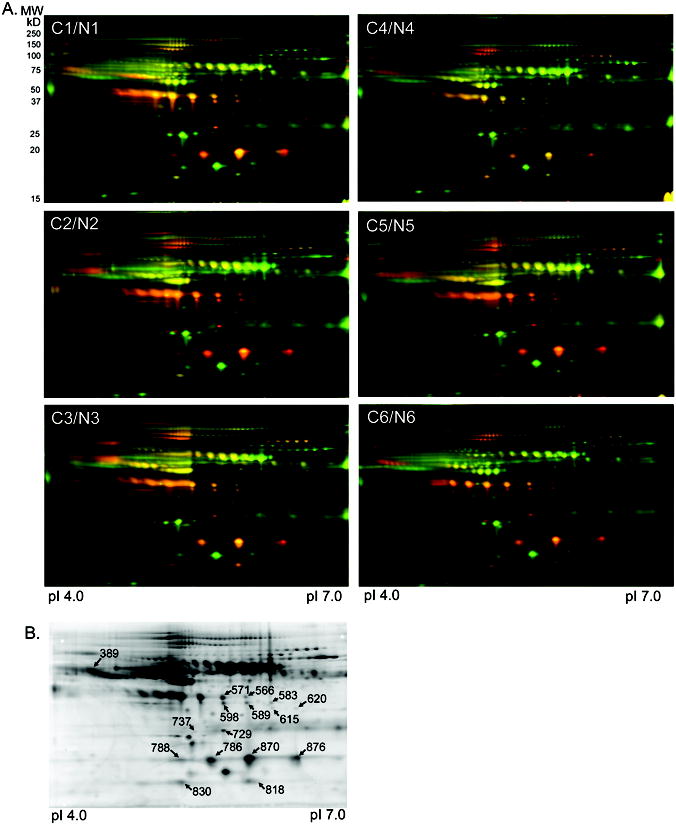
A) DIGE gels for all 12 serum pools depleted of high abundance proteins by the IgY-12 spin column. Fifty μg of the medium and low abundance proteins recovered from the IgY-12 spin column were labeled with Cy5 (red; cancer serum pools C1-C6) or Cy3 (green; non-cancer serum pools N1-N6). Proteins present equally in both serum pools appear as yellow spots. Proteins were separated by isoelectric point and then by molecular size.
B) A representative deep purple stained DIGE gel of sera depleted with the IgY-12 spin column. Spots picked for identification by MS are labeled with the master spot number assigned by the Decyder software. Identity of the proteins for each spot is listed in Table 2. The peptides identified for each protein are listed in Supplemental Table 1.
Table 2.
MS identification of proteins from IgY-12 spin column depleted sera.
| Spot No.* | Accession Number | Protein Name† | p-value | Fold Change | pI‡ | MW kDa pre (obs)¶ | % Coverage | Unique Peptides |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| 389 | gi|50659080 | alpha-1 antichymotrypsin | 0.0002 | 4.93 | 4.28 | 48 (100) | 22 | 9 |
| 566 | NI | 0.00067 | 2.61 | |||||
| 571 | gi|4826762 | haptoglobin beta subunit§ | 0.045 | 2.49 | 5.2 | 45 (40) | 15 | 4 |
| 583 | NI | 0.0004 | 2.78 | |||||
| 589 | gi|4826762 | haptoglobin beta subunit§ | 3.80E-05 | 3.31 | 5.88 | 45 (40) | 18 | 6 |
| 598 | gi|4826762 | haptoglobin beta subunit§ | 5.40E-06 | 4.11 | 5.65 | 45 (40) | 26 | 7 |
| gi|4502133 | serum amyloid P component | 25 (40) | 14 | 3 | ||||
| 615 | NI | 0.0004 | 3.45 | |||||
| ║620 | gi27759776 | ficolin 3 | 3.20E-05 | 3.77 | 6.34 | 32 (28) | 3 | 2 |
| 729 | gi|4502133 | serum amyloid P component | 4.20E-06 | 4.38 | 5.66 | 25 (26) | 29 | 8 |
| 737 | NI | 3.10E-06 | 2.77 | |||||
| 786 | gi|4826762 | haptoglobin alpha-2 subunit§ | 3.60E-07 | 6.78 | 5.54 | 45 (18) | 7 | 6 |
| 788 | gi|4826762 | haptoglobin alpha-2 subunit§ | 1.90E-09 | 7.30 | 5.25 | 45 (18) | 7 | 6 |
| 818 | NI | 0.0032 | 2.81 | |||||
| 830 | gi|4826762 | haptoglobin alpha-1 subunit§ | 0.0014 | 2.13 | 5.27 | 45 (13) | 7 | 3 |
| 870 | gi|4826762 | haptoglobin alpha-2 subunit§ | 0.01 | 3.07 | 5.91 | 45 (19) | 9 | 7 |
| 876 | gi|4826762 | haptoglobin alpha-2 subunit§ | 1.00E-03 | 9.10 | 6.33 | 45 (19) | 7 | 5 |
Spot number corresponds to DIGE gel in Fig. 2B
Proteins identified with a Protein Prophet probablility score of 100% are listed for each spot, unless noted. Peptides identified for each protein are listed in Supplemental Table 1.
pI = observed pI on 2D gel in Fig. 2B.
Predicted, unprocessed molecular weight (observed molecular weight, Fig. 2B).
The haptoglobin subunits are derived from a common processed preprotein. Individual subunit assignments were estimated from the location of the spot in the 2D gel, compared to the Swiss 2D PAGE reference map for human plasma (ExPASy Proteomics Server:http://ca.expasy.org).
Ficolin 3 was identified in spot 620 with 91% probability. Spectra were manually verified (Supplemental Figure 1).
NI = No Protein Identified.
DIGE analysis of the 12 serum pools depleted by the IgY-12 HPLC was conducted using pI from 3.9 to 5.1 in order to better separate the proteins within this pI range (Fig. 3A). A total of 961 protein spots were detected collectively among the six DIGE gels (Table 1), of which 36 spots (4%) showed significantly increased levels in all six ovarian cancer samples compared to control samples. Seventeen spots were excised from these gels, and MS for 16 of the spots identified at least one protein (Table 3 and Fig. 3B). Inter-alpha (globulin) inhibitor H4 was identified in three spots with the most peptides of all proteins that met our criteria. Similarly, alpha-1 antichymotrypsin (AACT) was identified in seven spots, while complement component 3 and leucine rich alpha-2 glycoprotein 1 (LRG1) were each identified in three spots (Table 3). Relative to the control serum pools, ten of the proteins identified were increased by >2-fold in the ovarian cancer serum pools, and three proteins were increased by 1.5- to 1.8-fold (Table 3). The proteins from each spot that met our threshold for protein identification are listed in Table 3. Due to comigration of proteins in the gels, several spots contained more than one protein identified with high probability (99% probability, three peptide minimum). As a result, the distinct protein(s) that contributed to the differential expression detected by the DeCyder software were ambiguous. For example, the proteins inter-alpha (globulin) inhibitor H4, protein S alpha, and complement component 1s were all detected in spot 217. Peptides used in the identification of each protein are listed in Supplemental Table 1.
Figure 3. DIGE analysis of medium and low abundance serum proteins depleted by the IgY-12 HPLC column.
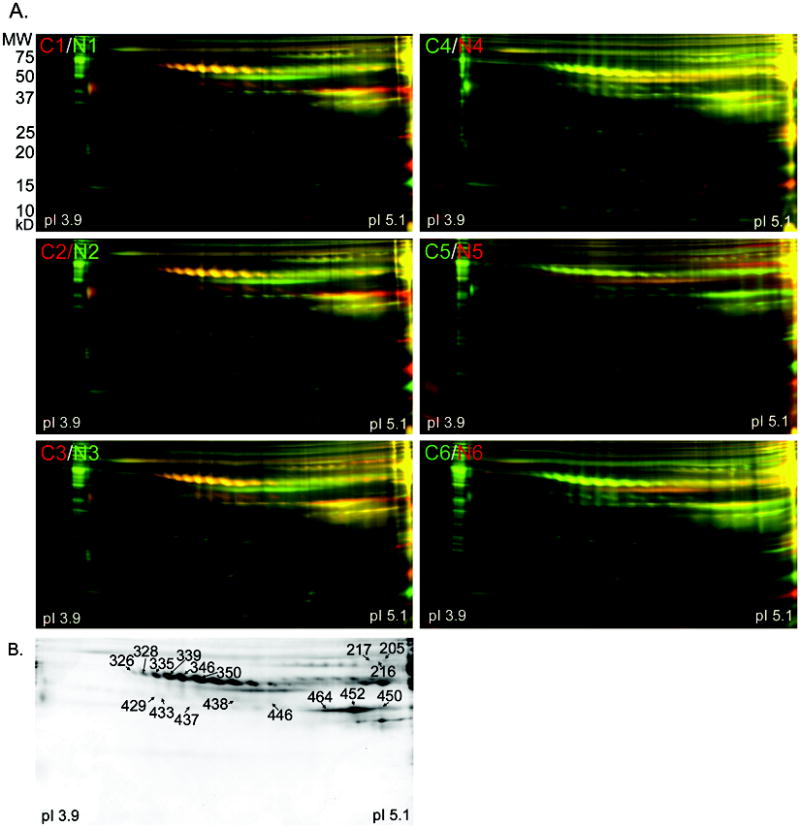
A) DIGE gels for all 12 serum pools depleted of high abundance proteins by the IgY-12 HPLC column. Fifty μg of the medium and low abundance proteins recovered from the IgY-12 HPLC column were label for each serum pool. Cancer serum pools C1-C3 were labeled with Cy5 (red), and C4-C6 were labeled with Cy3 (green). Non-cancer serum pools N1-N3 were labeled with Cy3 (green), and N4-N6 were labeled with Cy5 (red). Proteins present equally in both serum pools appear as yellow spots. Proteins were separated by isoelectric point and then by molecular size.
B) A representative deep purple stained DIGE gel of sera depleted with the IgY-12 HPLC column. Spots picked for identification by MS are labeled with the master spot number assigned by the Decyder software. The identity of the proteins for each spot is listed in Table 3. The peptides identified for each protein are listed in Supplemental Table 1.
Table 3.
MS identification of proteins from IgY-12 HPLC depleted sera.
| Spot No.* | Accession Number | Protein Name† | p-value | Fold Change | pI‡ | MW kDa pre (obs) ║ | % Coverage | Unique Peptides |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| 205 | gi|31542984 | inter-alpha (globulin) inhibitor H4 | 0.006283 | 2.31 | 5.04 | 103 (78) | 18 | 16 |
| gi|115298678 | complement component 3 precursor | 187 (78) | 12 | 15 | ||||
| gi|4501987 | afamin precursor | 69 (78) | 21 | 11 | ||||
| gi|7385856 | complement component 1 inhibitor | 55 (78) | 19 | 10 | ||||
| gi|192447438 | protein S, alpha | 75 (78) | 16 | 9 | ||||
| gi|21071030 | alpha 1B-glycoprotein precursor | 54 (78) | 26 | 9 | ||||
| gi|4503635 | coagulation factor II preproprotein | 70 (78) | 8 | 4 | ||||
| gi|178557739 | complement component 4B preproprotein | 192 (78) | 2 | 3 | ||||
| gi|50659080 | alpha-1 antichymotrypsin | 48 (78) | 6 | 3 | ||||
| 216 | gi|31542984 | inter-alpha (globulin) inhibitor H4 | 0.01218 | 2.60 | 5.11 | 103 (78) | 2 | 2 |
| 217 | gi|31542984 | inter-alpha (globulin) inhibitor H4 | 0.00874 | 2.30 | 4.98 | 103 (78) | 18 | 16 |
| gi|192447438 | protein S, alpha | 75 (78) | 13 | 9 | ||||
| gi|115298678 | complement component 3 precursor | 187 (78) | 7 | 8 | ||||
| gi|7385856 | complement component 1 inhibitor | 55 (78) | 13 | 6 | ||||
| gi|41393602 | complement component 1, s subcomponent | 77 (78) | 12 | 6 | ||||
| gi|21071030 | alpha 1B-glycoprotein precursor | 54 (78) | 9 | 4 | ||||
| gi|4501987 | afamin precursor | 69 (78) | 6 | 3 | ||||
| gi|4503635 | coagulation factor II preproprotein | 70 (78) | 5 | 3 | ||||
| gi|178557739 | complement component 4B preproprotein | 192 (78) | 4 | 3 | ||||
| gi|4505047 | lumican precursor | 38 (78) | 9 | 3 | ||||
| 326 | gi|50659080 | alpha-1 antichymotrypsin | 3.46E-06 | 2.60 | 4.17 | 48 (60) | 9 | 4 |
| 328 | gi|50659080 | alpha-1 antichymotrypsin | 3.02E-06 | 2.90 | 4.22 | 48 (60) | 27 | 8 |
| 335 | gi|50659080 | alpha-1 antichymotrypsin | 1.69E-06 | 2.80 | 4.25 | 48 (60) | 29 | 9 |
| 339 | gi|50659080 | alpha-1 antichymotrypsin | 1.31E-06 | 2.65 | 4.3 | 48 (60) | 5 | 2 |
| 346 | gi|50659080 | alpha-1 antichymotrypsin | 2.60E-06 | 2.45 | 4.3 | 48 (55) | 39 | 17 |
| 350 | gi|50659080 | alpha-1 antichymotrypsin | 1.44E-05 | 2.25 | 4.4 | 48 (55) | 40 | 17 |
| gi|156231037 | kininogen 1 isoform 1 | 72 (55) | 7 | 4 | ||||
| 429 | gi|16418467 | leucine-rich alpha-2-glycoprotein 1 | 0.01237 | 1.50 | 4.26 | 38 (45) | 13 | 5 |
| 433 | gi|16418467 | leucine-rich alpha-2-glycoprotein 1 | 0.06842 | 1.40 | 4.28 | 38 (45) | 13 | 5 |
| 437 | gi|16418467 | leucine-rich alpha-2-glycoprotein 1 | 0.2296 | 1.20 | 4.37 | 38 (45) | 13 | 5 |
| 438 | gi|50659080 | alpha-1 antichymotrypsin | 0.09471 | 1.42 | 4.5 | 48 (60) | 23 | 7 |
| gi|16418467 | leucine-rich alpha-2-glycoprotein 1 | 38 (45) | 16 | 6 | ||||
| 446 | NI | 0.01822 | 1.82 | |||||
| 450 | gi|115298678 | complement component 3 precursor | 0.08128 | 2.12 | 5.00 | 187 (40) | 14 | 22 |
| gi|4826762 | haptoglobin beta subunit§ | 45 (40) | 24 | 8 | ||||
| gi|178557739 | complement component 4B preproprotein | 192 (40) | 2 | 4 | ||||
| 452 | gi|115298678 | complement component 3 precursor | 0.01713 | 1.79 | 4.93 | 187 (40) | 16 | 27 |
| gi|4826762 | haptoglobin beta subunit§ | 45 (40) | 26 | 9 | ||||
| gi|178557739 | complement component 4B preproprotein | 193 (40) | 5 | 9 | ||||
| 464 | gi|115298678 | complement component 3 precursor | 0.0352 | 1.82 | 4.81 | 187 (40) | 13 | 19 |
| gi|178557739 | complement component 4B preproprotein | 192 (40) | 10 | 13 | ||||
| gi|4826762 | haptoglobin beta subunit§ | 45 (40) | 14 | 5 | ||||
Spot number corresponds to DIGE gel in Fig. 3B.
Proteins identified with a Protein Prophet probablility score of 100% are listed for each spot, unless noted. Peptides identified for each protein are listed in Supplemental Table 1.
pI = observed pI on 2D gels in Fig. 3B.
Predicted, unprocessed molecular weight (observed molecular weight, Fig. 3B).
The haptoglobin subunits are derived from a common processed preprotein. Individual subunit assignments were estimated from the location of the spot in the 2D gel, compared to the Swiss 2D PAGE reference map for human plasma (ExPASy Proteomics Server: http://ca.expasy.org).
NI = No Protein Identified.
Validation of protein expression by immunoblotting
Results of the DIGE experiments were validated by Western blot. We used immunodepleted serum for the initial validation, as the presence of the highly abundant serum proteins interferes with detection of low abundance proteins by Western blot. Monoclonal antibodies to AACT, LRG1, and ficolin 3 were used to probe Western blots of medium and low abundance proteins from serum pools depleted by the IgY-12 HPLC column. We chose proteins for validation that showed robust levels of expression in ovarian cancer sera by DIGE or were identified in multiple spots, and have not been previously reported to be upregulated in the sera of ovarian cancer patients. We were particularly interested in validating ficolin 3 and LRG1, as the peptide identification and/or quantitation values were weak and these are potentially tumor-specific proteins. For example, ficolin 3 was identified with only two peptides. The mAb against ficolin 3 recognized two protein bands of ∼32 and ∼65 kDa; both bands were more intense in the ovarian cancer serum pools compared to the control sera (Fig. 4A), supporting the DIGE results. The ficolin 3 monomer is predicted to be 32 kDa, while the native form is a polymer [25]. Although the levels of LRG1 in ovarian cancer sera were only modestly increased, it was identified in five spots, and Western blots probed with a mAb to LRG1 validated the increased levels of LRG1 in ovarian cancer sera compared to non-cancer sera. AACT was detected as a protein of ∼ 56-65 kDa that was visibly increased in the cancer serum pools by Western blotting (Fig. 4A). In the DIGE experiments, six spots were identified as AACT (Tables 2 and 3), with an average 3-fold increase in the cancer serum pools; this was corroborated by the Western blotting results. Fig. 4B shows a duplicate gel stained with CBB as a loading control.
Figure 4. Western blot validation of proteins overexpressed in cancer sera.
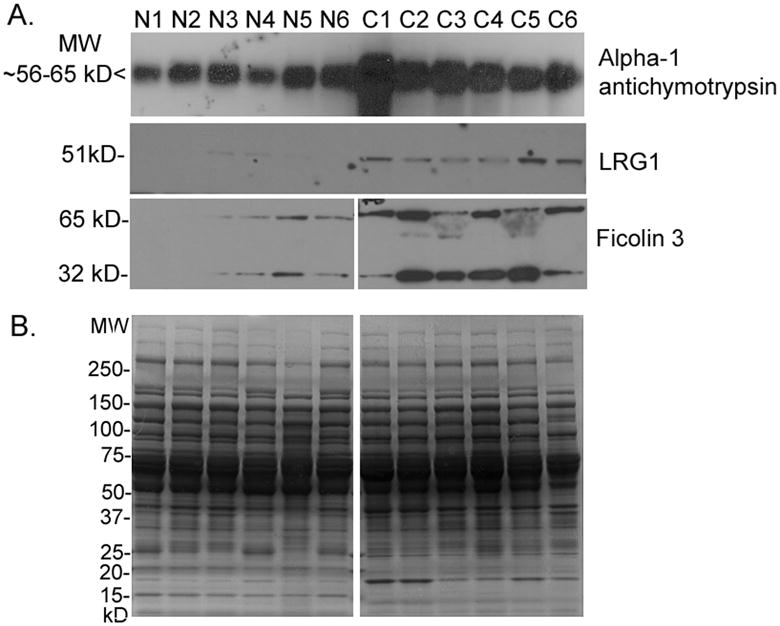
A) Western blots of 50 μg of medium and low abundance proteins from each non-cancer (N1-N6) and cancer (C1-C6) serum pool depleted by the IgY-12 HPLC column were probed with the antibodies to AACT, LRG1, and ficolin 3.
B) A duplicate gel was stained with CBB as a loading control.
Discussion
In this study, we provide evidence that the use of immunodepletion to remove highly abundant proteins from serum followed by DIGE analysis can be used to quantitate protein levels in serum from ovarian cancer patients and non-cancer controls for the discovery of candidate biomarkers. By these techniques, we identified nine proteins present at higher levels in ovarian cancer sera than in control sera.
Several of the proteins identified in this study, for example, haptoglobin [26-30] and inter-alpha globulin inhibitor H4 [31], have previously been shown to be increased in the sera of ovarian cancer patients by proteomic and other techniques, validating our screen for potential biomarkers in serum. Haptoglobin was first shown to be elevated in the sera of women with ovarian cancer almost 40 years ago [28]. This finding has subsequently been confirmed numerous times, most recently by 2-DE [26], SELDI, and ELISA [29]. Haptoglobin is one of the highly abundant proteins targeted by the immunodepletion columns, and was visibly depleted from the non-cancer serum (Fig. 1C′, arrow) by the MARS column. Visual examination of the MARS-fractionated sera by silver stained 2-DE gels revealed increased levels of haptoglobin in the cancer serum pool in both the high abundance (Fig. 1B, arrow) and low abundance (Fig. 1C, arrow) fractions. We conclude that the MARS column effectively depleted haptoglobin from serum except when present at elevated levels, such as in sera from ovarian cancer patients, in which case the column depletion capacity is insufficient for complete removal of the protein.
We also identified several less well-studied proteins that were elevated in ovarian cancer sera and have the potential to serve as biomarkers. For example, LRG1 was expressed at increased levels in ovarian cancer sera by DIGE, and subsequently was validated by Western blot (Fig. 4A), and by ELISA in individual, undepleted serum samples (manuscript in preparation). Although LRG1 has been classified as an “acute phase” protein [32, 33], we have evidence that LRG1 is also produced by ovarian cancer cells and may contribute to the increased levels of LRG1 found in patient sera (manuscript in preparation). Others have found by 2-DE, that specific isoforms of LRG1 have increased expression in the proliferative endometrium of women undergoing fertility treatment who subsequently became pregnant independent of treatment [34], and in the peritoneal fluid of women with uterine leiomyomas [35]. Interestingly, Ferrero and colleagues also saw a significant correlation between leiomyoma size and LRG1 expression [35]. We identified LRG1 peptides in DIGE spots representing four different isoforms, although only one spot was significantly increased in ovarian cancer sera, and only a single protein band of ∼51 kDa was detected by Western blot. Taken together, these data indicate a potential role for at least one isoform of LRG1 in the physiology of the female reproductive organs, and a potential biomarker for ovarian cancer.
We also found that ficolin 3 was more than 3-fold elevated in ovarian cancer sera by DIGE analysis, and its relative expression was validated by Western blot. Although ficolin 3 is thought to play a role in immune activation [36, 37], it has been identified by MS in ovarian cancer ascites cells [38, 39] suggesting a potential role in ovarian cancer, as well. Ficolin 3 has also been identified in a DIGE analysis of serum in prostate cancer progression [20]. Thus, ficolin 3 and LRG1 both warrant further investigation into their expression in ovarian cancer cells and their specificity as potential biomarkers for ovarian cancer.
We also validated the level of serum expression for alpha-1 antichymotrypsin, an acute phase protein that may play a role in ovarian cancer by binding and regulating members of the kallikrein family of serine proteases [40]. Alpha-1 antichymotrypsin was detected as a protein of ∼ 56-65 kDa that was visibly increased in the cancer serum pools by Western blotting (Fig. 4). In the DIGE experiments, six spots were identified as AACT (Tables 2 and 3), with an average 3-fold increase in the cancer serum pools (ranging from a 2.25 to 4.9-fold increase in the cancer sera), which is corroborated by the Western blotting results (Fig. 4).
Several of the proteins found to be increased in ovarian cancer serum relative to control samples were acute phase proteins that can be elevated in response to infection or injury [32], and as such may not be specific to ovarian cancer. Nevertheless, in a recent analysis of 204 serum markers on a large cohort of sera from women with ovarian cancer as well as benign gynecological disease, several acute phase and inflammatory proteins were among the best discriminators of malignancy [41]. These findings support the notion that inflammation plays a role in cancer initiation and progression [42] and indicate that acute phase proteins may prove to be critical in the development of a robust “multi-analyte” panel of ovarian cancer biomarkers.
Others have used post-translational modifications as a method to detect and enrich for biomarkers in serum of ovarian cancer patients. Ogata and colleagues used immunodepletion and 2-DE to analyze post-translationally modified proteins by means of specific gel staining methods [43]. They identified a phosphorylated isoform of fibrinogen alpha that was upregulated in ovarian cancer patients. Furthermore, Jackson et al. [44] used DIGE and 2-D lectin profiling for the discovery of glycoprotein biomarkers. They found the albumin-related protein, afamin, was present at decreased levels in ovarian cancer compared to control sera.
Immunodepletion of serum followed by DIGE analysis has been used for biomarker discovery in a number of other cancer types including prostate [20], pancreatic [18, 19], and lung cancers [16, 17]. Similar to our results in ovarian cancer, serum levels of haptoglobin, inter-alpha globulin inhibitor H4, and LRG1 were elevated in the medium and low abundance serum proteins of lung and pancreatic cancers [16, 18, 19]. Taken together, these results suggest that these proteins may be indicative of a general response to cancer, rather than tumor-specific proteins; although in the cases of haptoglobin and LRG1, there is evidence that these proteins are synthesized by ovarian cancer cells [30].
For biomarker discovery, we used 60 serum samples from patients with primarily stage III and stage IV serous ovarian carcinoma and 60 serum samples from female non-cancer “controls” that were randomly pooled into six groups of ten patient samples. The samples were pooled to reduce variation between individual samples and to increase the overall number of samples tested. We selected patients with advanced disease under the assumption that these patients may have higher levels of ovarian cancer proteins in their sera than patients with early stage disease, thus increasing the likelihood of detecting ovarian cancer specific biomarkers. There is some evidence that the serum protein profile may change with disease progression [45, 46], which may limit the utility of late stage samples for the discovery of early stage biomarkers. However, a recent analysis of 21 tumor biomarkers in a large cohort of patients demonstrated consistent biomarker expression across stages within a particular ovarian cancer subtype, suggesting that differential expression of biomarkers is related to ovarian cancer subtype and not stage [47]. Regardless, it will be important to validate the candidate biomarkers identified using a large cohort of individual serum samples, which are age-matched and include samples from women with early stage cancers and benign gynecological conditions, as well as healthy controls. Moreover, the candidate biomarkers detected herein may prove useful for the detection of disease recurrence.
Supplementary Material
Acknowledgments
We would like to thank the Gynecologic Oncology Group (GOG) Tissue Bank for the serum samples. We also thank Thomas McGowan of the University of Minnesota Center for Mass Spectrometry and Proteomics, and Dr. Yanji Xu of the University of Minnesota Supercomputing Institute for computational support, and Dr. Timothy Griffin (University of Minnesota) for critical reading of the manuscript. This work was supported by grants from the Minnesota Ovarian Cancer Alliance, the National Institutes of Health/National Cancer Institute (R01CA106878), Cancurables, and the Minnesota Medical Foundation. The MS and DIGE analyses were performed at the Center for Mass Spectrometry and Proteomics at the University of Minnesota, which is supported in part by grants from the National Science Foundation (9871237, 0215759 and CHE0078192) and the National Institutes of Health (RRR15808).
Abbreviations
- DIGE
Differential-In-Gel-Electrophoresis
- MARS
multiple affinity removal system
- AACT
alpha-1 antichymotrypsin
- LRG1
leucine rich alpha-2 glycoprotein 1
Footnotes
Conflict of Interest Statement: The authors have declared no conflict of interest.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Jacobs IJ, Menon U. Mol Cell Proteomics. 2004;3:355–366. doi: 10.1074/mcp.R400006-MCP200. [DOI] [PubMed] [Google Scholar]
- 3.Bast RC, Jr, Klug TL, St John E, Jenison E, Niloff JM, Lazarus H, Berkowitz RS, et al. N Engl J Med. 1983;309:883–887. doi: 10.1056/NEJM198310133091503. [DOI] [PubMed] [Google Scholar]
- 4.Bast RC, Jr, Urban N, Shridhar V, Smith D, Zhang Z, Skates S, Lu K, et al. Cancer Treat Res. 2002;107:61–97. doi: 10.1007/978-1-4757-3587-1_3. [DOI] [PubMed] [Google Scholar]
- 5.Zurawski VR, Jr, Orjaseter H, Andersen A, Jellum E. Int J Cancer. 1988;42:677–680. doi: 10.1002/ijc.2910420507. [DOI] [PubMed] [Google Scholar]
- 6.Williams TI, Toups KL, Saggese DA, Kalli KR, Cliby WA, Muddiman DC. J Proteome Res. 2007;6:2936–2962. doi: 10.1021/pr070041v. [DOI] [PubMed] [Google Scholar]
- 7.Kachman MT, Wang H, Schwartz DR, Cho KR, Lubman DM. Anal Chem. 2002;74:1779–1791. doi: 10.1021/ac011159c. [DOI] [PubMed] [Google Scholar]
- 8.Wang H, Kachman MT, Schwartz DR, Cho KR, Lubman DM. Proteomics. 2004;4:2476–2495. doi: 10.1002/pmic.200300763. [DOI] [PubMed] [Google Scholar]
- 9.Wang H, Kachman MT, Schwartz DR, Cho KR, Lubman DM. Electrophoresis. 2002;23:3168–3181. doi: 10.1002/1522-2683(200209)23:18<3168::AID-ELPS3168>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 10.Jones MB, Krutzsch H, Shu H, Zhao Y, Liotta LA, Kohn EC, Petricoin EF., 3rd Proteomics. 2002;2:76–84. [PubMed] [Google Scholar]
- 11.Bengtsson S, Krogh M, Szigyarto CA, Uhlen M, Schedvins K, Silfversward C, Linder S, et al. J Proteome Res. 2007 doi: 10.1021/pr060593y. [DOI] [PubMed] [Google Scholar]
- 12.Zhang H, Liu AY, Loriaux P, Wollscheid B, Zhou Y, Watts JD, Aebersold R. Mol Cell Proteomics. 2007;6:64–71. doi: 10.1074/mcp.M600160-MCP200. [DOI] [PubMed] [Google Scholar]
- 13.Gong Y, Li X, Yang B, Ying W, Li D, Zhang Y, Dai S, et al. J Proteome Res. 2006;5:1379–1387. doi: 10.1021/pr0600024. [DOI] [PubMed] [Google Scholar]
- 14.Brand J, Haslberger T, Zolg W, Pestlin G, Palme S. Proteomics. 2006;6:3236–3242. doi: 10.1002/pmic.200500864. [DOI] [PubMed] [Google Scholar]
- 15.Liu T, Qian WJ, Mottaz HM, Gritsenko MA, Norbeck AD, Moore RJ, Purvine SO, et al. Mol Cell Proteomics. 2006;5:2167–2174. doi: 10.1074/mcp.T600039-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okano T, Kondo T, Kakisaka T, Fujii K, Yamada M, Kato H, Nishimura T, et al. Proteomics. 2006;6:3938–3948. doi: 10.1002/pmic.200500883. [DOI] [PubMed] [Google Scholar]
- 17.Dowling P, O'Driscoll L, Meleady P, Henry M, Roy S, Ballot J, Moriarty M, et al. Electrophoresis. 2007;28:4302–4310. doi: 10.1002/elps.200700246. [DOI] [PubMed] [Google Scholar]
- 18.Yu KH, Rustgi AK, Blair IA. J Proteome Res. 2005;4:1742–1751. doi: 10.1021/pr050174l. [DOI] [PubMed] [Google Scholar]
- 19.Kakisaka T, Kondo T, Okano T, Fujii K, Honda K, Endo M, Tsuchida A, et al. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;852:257–267. doi: 10.1016/j.jchromb.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Byrne JC, Downes MR, O'Donoghue N, O'Keane C, O'Neill A, Fan Y, Fitzpatrick JM, et al. J Proteome Res. 2009;8:942–957. doi: 10.1021/pr800570s. [DOI] [PubMed] [Google Scholar]
- 21.Blum H, Beier H, Gross HJ. Electrophoresis. 1987;8:93–99. [Google Scholar]
- 22.Shevchenko A, Wilm M, Vorm O, Mann M. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 23.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 24.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. Anal Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 25.Ohashi T, Erickson HP. Arch Biochem Biophys. 1998;360:223–232. doi: 10.1006/abbi.1998.0957. [DOI] [PubMed] [Google Scholar]
- 26.Ahmed N, Barker G, Oliva KT, Hoffmann P, Riley C, Reeve S, Smith AI, et al. Br J Cancer. 2004;91:129–140. doi: 10.1038/sj.bjc.6601882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahmed N, Oliva KT, Barker G, Hoffmann P, Reeve S, Smith IA, Quinn MA, et al. Proteomics. 2005;5:4625–4636. doi: 10.1002/pmic.200401321. [DOI] [PubMed] [Google Scholar]
- 28.Mueller WK, Handschumacher R, Wade ME. Obstet Gynecol. 1971;38:427–435. [PubMed] [Google Scholar]
- 29.Ye B, Cramer DW, Skates SJ, Gygi SP, Pratomo V, Fu L, Horick NK, et al. Clin Cancer Res. 2003;9:2904–2911. [PubMed] [Google Scholar]
- 30.Zhao C, Annamalai L, Guo C, Kothandaraman N, Koh SC, Zhang H, Biswas A, et al. Neoplasia. 2007;9:1–7. doi: 10.1593/neo.06619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Z, Bast RC, Jr, Yu Y, Li J, Sokoll LJ, Rai AJ, Rosenzweig JM, et al. Cancer Res. 2004;64:5882–5890. doi: 10.1158/0008-5472.CAN-04-0746. [DOI] [PubMed] [Google Scholar]
- 32.Bini L, Magi B, Marzocchi B, Cellesi C, Berti B, Raggiaschi R, Rossolini A, et al. Electrophoresis. 1996;17:612–616. doi: 10.1002/elps.1150170333. [DOI] [PubMed] [Google Scholar]
- 33.Weivoda S, Andersen JD, Skogen A, Schlievert PM, Fontana D, Schacker T, Tuite P, et al. J Immunol Methods. 2008;336:22–29. doi: 10.1016/j.jim.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gillott DJ, Al-Rumaih HM, Leung KY, Eldib A, Grudzinskas JG. Fertil Steril. 2008;90:761–768. doi: 10.1016/j.fertnstert.2007.01.094. [DOI] [PubMed] [Google Scholar]
- 35.Ferrero S, Gillott DJ, Remorgida V, Anserini P, Ragni N, Grudzinskas JG. Arch Gynecol Obstet. 2009;279:365–371. doi: 10.1007/s00404-008-0741-1. [DOI] [PubMed] [Google Scholar]
- 36.Su GL, Simmons RL, Wang SC. Crit Rev Immunol. 1995;15:201–214. doi: 10.1615/critrevimmunol.v15.i3-4.10. [DOI] [PubMed] [Google Scholar]
- 37.Hummelshoj T, Fog LM, Madsen HO, Sim RB, Garred P. Mol Immunol. 2008;45:1623–1632. doi: 10.1016/j.molimm.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 38.Gortzak-Uzan L, Ignatchenko A, Evangelou AI, Agochiya M, Brown KA, St Onge P, Kireeva I, et al. J Proteome Res. 2008;7:339–351. doi: 10.1021/pr0703223. [DOI] [PubMed] [Google Scholar]
- 39.Faca VM, Ventura AP, Fitzgibbon MP, Pereira-Faca SR, Pitteri SJ, Green AE, Ireton RC, et al. PLoS ONE. 2008;3:e2425. doi: 10.1371/journal.pone.0002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borgono CA, Diamandis EP. Nat Rev Cancer. 2004;4:876–890. doi: 10.1038/nrc1474. [DOI] [PubMed] [Google Scholar]
- 41.Bertenshaw GP, Yip P, Seshaiah P, Zhao J, Chen TH, Wiggins WS, Mapes JP, et al. Cancer Epidemiol Biomarkers Prev. 2008;17:2872–2881. doi: 10.1158/1055-9965.EPI-08-0464. [DOI] [PubMed] [Google Scholar]
- 42.Goswami B, Rajappa M, Sharma M, Sharma A. Int J Gynecol Cancer. 2008;18:591–599. doi: 10.1111/j.1525-1438.2007.01089.x. [DOI] [PubMed] [Google Scholar]
- 43.Ogata Y, Heppelmann CJ, Charlesworth MC, Madden BJ, Miller MN, Kalli KR, Cilby WA, et al. J Proteome Res. 2006;5:3318–3325. doi: 10.1021/pr060344+. [DOI] [PubMed] [Google Scholar]
- 44.Jackson D, Craven RA, Hutson RC, Graze I, Lueth P, Tonge RP, Hartley JL, et al. Clin Cancer Res. 2007;13:7370–7379. doi: 10.1158/1078-0432.CCR-07-0747. [DOI] [PubMed] [Google Scholar]
- 45.Lowenthal MS, Mehta AI, Frogale K, Bandle RW, Araujo RP, Hood BL, Veenstra TD, et al. Clin Chem. 2005;51:1933–1945. doi: 10.1373/clinchem.2005.052944. [DOI] [PubMed] [Google Scholar]
- 46.Lee CJ, Ariztia EV, Fishman DA. Crit Rev Clin Lab Sci. 2007;44:87–114. doi: 10.1080/10408360600778885. [DOI] [PubMed] [Google Scholar]
- 47.Kobel M, Kalloger SE, Boyd N, McKinney S, Mehl E, Palmer C, Leung S, et al. PLoS Med. 2008;5:e232. doi: 10.1371/journal.pmed.0050232. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.