

Abstract
Phenotypes are determined by a complex series of physical (e.g. protein-protein) and functional (e.g. gene-gene or genetic) interactions (GI)1. While physical interactions can indicate which bacterial proteins are associated as complexes, they do not necessarily reveal pathway-level functional relationships1. GI screens, in which the growth of double mutants bearing two deleted or inactivated genes is measured and compared to the corresponding single mutants, can illuminate epistatic dependencies between loci and hence provide a means to query and discover novel functional relationships2. Large-scale GI maps have been reported for eukaryotic organisms like yeast3-7, but GI information remains sparse for prokaryotes8, which hinders the functional annotation of bacterial genomes. To this end, we and others have developed high-throughput quantitative bacterial GI screening methods9, 10.
Here, we present the key steps required to perform quantitative E. coli Synthetic Genetic Array (eSGA) screening procedure on a genome-scale9, using natural bacterial conjugation and homologous recombination to systemically generate and measure the fitness of large numbers of double mutants in a colony array format. Briefly, a robot is used to transfer, through conjugation, chloramphenicol (Cm) - marked mutant alleles from engineered Hfr (High frequency of recombination) 'donor strains' into an ordered array of kanamycin (Kan) - marked F- recipient strains. Typically, we use loss-of-function single mutants bearing non-essential gene deletions (e.g. the 'Keio' collection11) and essential gene hypomorphic mutations (i.e. alleles conferring reduced protein expression, stability, or activity9, 12, 13) to query the functional associations of non-essential and essential genes, respectively. After conjugation and ensuing genetic exchange mediated by homologous recombination, the resulting double mutants are selected on solid medium containing both antibiotics. After outgrowth, the plates are digitally imaged and colony sizes are quantitatively scored using an in-house automated image processing system14. GIs are revealed when the growth rate of a double mutant is either significantly better or worse than expected9. Aggravating (or negative) GIs often result between loss-of-function mutations in pairs of genes from compensatory pathways that impinge on the same essential process2. Here, the loss of a single gene is buffered, such that either single mutant is viable. However, the loss of both pathways is deleterious and results in synthetic lethality or sickness (i.e. slow growth). Conversely, alleviating (or positive) interactions can occur between genes in the same pathway or protein complex2 as the deletion of either gene alone is often sufficient to perturb the normal function of the pathway or complex such that additional perturbations do not reduce activity, and hence growth, further. Overall, systematically identifying and analyzing GI networks can provide unbiased, global maps of the functional relationships between large numbers of genes, from which pathway-level information missed by other approaches can be inferred9.
Keywords: Genetics, Issue 69, Molecular Biology, Medicine, Biochemistry, Microbiology, Aggravating, alleviating, conjugation, double mutant, Escherichia coli, genetic interaction, Gram-negative bacteria, homologous recombination, network, synthetic lethality or sickness, suppression
Protocol
1. Constructing Hfr Cavalli Donor Mutant Strains by Recombineering15, 16
The steps for constructing the eSGA donor stains are described below. Briefly, we use targeted λ- Red mediated homologous recombination16 of amplified selectable DNA marker cassette fragments generated by PCR to create non-essential gene deletion mutants (section 1.1) or essential gene hypomorphic mutant donor strains (section 1.2) that are then used as 'queries' to define GI networks.
Note: During the technology development process, we assessed the effectiveness of using Hfr-mediated conjugative transfer for combining mutations using a well-defined Hfr donor strain (Hfr Cavalli, Hfr Hayes, and Hfr 3000). We examined (i) the ability to efficiently make donor mutants using the recombineering method pioneered by Yu et al. (2000)16, (ii) the relative efficiencies of conjugative DNA transfer of different chromosomal markers, and (iii) the effects of query gene position and chromosomal orientation relative to the Hfr transfer locus, oriT. We found that λ-Red-mediated homologous recombination efficiency was much higher in Hfr Cavalli than in Hfr Hayes or Hfr3000. If required, P1 transduction or a Psuedo Hfr strain17, can be used to create mutant donors. We have successfully adapted all these methods for making and screening large numbers of donors. Since in our trial conjugation experiments, the overall efficiency of transfer and the number of ex-conjugants observed was significantly greater with Hfr Cavalli, this particular strain background was chosen for donor construction and large-scale eSGA. All procedures for eSGA screens using Hfr Cavalli donors are described.
- Amplifying the DNA fragment for subsequent recombineering to delete a non-essential gene Employ two-step nested PCR amplification (Figure 1) to create a gene deletion cassette for replacing the target open reading frame with a Cm resistance marker from the pKD3 plasmid (GI: 15554330; protein id: AAL02033.1)15. The marker is flanked by the FRT-sites11 allowing the removal of the resistance gene, if necessary, in follow up experiments. Note: We do nested two-step PCR to remove the plasmid from the amplified product that is used subsequently to transform the bacterial cells. Removing the plasmid (in the first PCR) and then creating the gene knockout cassette in the second PCR is necessary because transformation with a plasmid is much more efficient than recombineering, and the goal is to obtain - as much as possible - only strains that have successfully replaced the targeted genomic DNA with the selectable marker. An alternative to nested PCR is to perform a restriction digest of the plasmid outside of the region amplified in the second PCR step. Then the product of the restriction digest is purified and used as a template in the nested PCR step (1.1.2). This option is also simple, but requires more work up time.
- Use about 45 ng of pKD3 as a template in PCR with forward F1 and reverse R1 primers to produce a 1,070 bp product. Purify the PCR fragment using the Qiagen PCR purification protocol, and dilute the product in sterile distilled water to 5 ng/μl concentration.
- Using the purified PCR product as a template, set up a second PCR with gene-specific (e.g. knockout) nested primers, F2 and R2, which have (i) 45 nt of gene-specific homology regions at the 5'-ends to allow for homologous recombination immediately upstream and downstream of the target gene to replace it with the Cm resistance marker and (ii) 20 nt at the 3'-ends to prime the synthesis of the Cm marker. The size of the produced fragment is 1,123 bp. The gene-specific homology regions are designed to remove the target open reading frame while leaving any adjacent or partially overlapping coding segments intact. After this amplification, proceed to step 1.3.
- Amplifying a tagging cassette for creating an essential gene hypomorphic mutation
- To construct hypomorphic donor mutant strains, add a C-terminal sequential peptide affinity (SPA) fusion tag, which often slightly impacts essential protein function by destabilizing transcript abundance (i.e. protein copy number) or protein folding/activity. To this end, create a selectable SPA-tag (SPA-Cm) DNA template by converting the Kan resistance marker in pJL14818 to Cm using recombineering between identical nucleotide sequences flanking the Kan marker in pJL148 and Cm marker in pKD315. The resulting template consists of in-frame SPA-tag followed by a Cm resistance marker, and is suitable for PCR amplification, recombineering and subsequent eSGA12. Note: Essential genes are potentially the most interesting for gene function studies. However, because these genes cannot be deleted, innovative methods had to be developed for investigating the functional relationships of essential genes. Several suitable highly successful approaches have been developed initially in yeast. For example, Davierwala et al. (2005)19 performed GI screens using a panel of 575 temperature-sensitive essential gene mutants, and found that essential genes exhibited about five times the number of GIs as nonessential genes. Likewise, Breslow et al. (2008)20 developed a 'decreased abundance by mRNA perturbation' (DAmP) approach to generate a library of hypomorphic alleles covering ~82% of essential yeast genes to study pathways and complexes. The DAmP approach modifies the 3'end of the expressed mRNA, reducing the level of the target gene6, likely due to mRNA destabilization. Similar to the DAmP approach6, our strategy has been to exploit the subtle effects of perturbing the 3' end of the expressed mRNA of essential proteins. As we describe below, the tag by itself does not likely affect protein folding or function, but the subtle perturbation of the mRNA by the tag is sufficient, when combined with environmental stressors or other mutations, to reveal functionally informative gene-environment 13 and gene-gene 9, 12 interactions. Firstly, to date, we used SPA-tags to purify ~3,000 essential and non-essential E. coli proteins21-23, with over 80% of the 307 E. coli essential proteins being represented among our SPA-tagged strains8. Because the tag was successfully used to isolate known protein complexes, which were reciprocally validated (i.e. the same complex could be isolated by tagging and purifying different proteins from within the same complex); and the isolated complexes were biologically relevant, we reason that the tags do not usually affect protein folding or function12. Similarly, our unpublished observations revealed that the tag by itself does not usually affect strain growth. However, we reasoned that altering the 3'-UTR by integrating the tag and the marker cassette, as was reported for DAmP strains6, could destabilize certain transcripts on the RNA level or, on rare occasions, impede the proper folding or function of the fusion proteins. Therefore, we predicted that combining such perturbed alleles of essential genes with other functionally relevant mutations elsewhere in the genome would result in informative GIs, which indeed was observed9, 12. Furthermore, SPA-tagged alleles of essential genes exhibited gene-environment interactions (e.g. drug hypersensitivity) according to the phenotypic profiling work by Nichols et al. (2011)13, who subjected a subset of SPA-tagged essential strains to a variety of growth conditions. The same study confirmed that most (54 out of 58 tested) SPA-tagged essential strains showed impaired fitness in one or more culture conditions13, which supports the notion that the SPA-tag often creates mild hypomorphic alleles in essential genes that are useful for genome-wide GI screen applications including eSGA.
- Amplify the tagging cassette from the SPA-Cm template using gene-specific primers S1 and S2, each containing (i) a 45 nt gene-specific region for homologous recombineering at the 5' end, followed by (ii) a 27 bp SPA-Cm specific sequence at the 3' end.
- Confirming and purifying the PCR product
- Confirm that the tagging cassette was correctly amplified by subjecting 2-5 μl of the PCR product to agarose gel electrophoresis and purify the remaining DNA following the manufacturer's standard PCR purification protocol (Qiagen). Elute the PCR product in 30 μl of sterile distilled water and dilute to ~50 ng/μl concentration. The purified product can be used immediately in transformation (step 1.5) or stored at -20 °C for up to 6 months until use.
- Preparing Hfr Cavalli competent cells for donor construction
- Inoculate one ml of the saturated overnight culture of an Hfr Cavalli strain into 70 ml of fresh Luria-Bertani (LB) medium with 35 μl of 50 μg/ml ampicillin in a 250 ml flask. Incubate the culture at 32 °C with shaking at 220 rpm until an optical density (OD) 600 nm of ~0.5-0.6 is obtained (~2 hr).
- Transfer the culture to a water bath for heat induction of the λred recombination system16 at 42 °C for 15 min with shaking at 160 rpm. Stop the induction by transferring the culture to a chilled ice-slurry water bath for 10-20 min at 160 rpm. Make sure to keep the cells cold from this point until after transformation; use tubes and cuvettes that have been chilled on ice for at least 15 min prior to use.
- Divide the culture equally into 2 pre-chilled 50 ml polypropylene tubes (~35 ml culture per tube) and centrifuge at 4,400 x g for 6 min at 4 °C.
- Discard the supernatant, and re-suspend the cell pellet by gentle inversion in ~50 ml of ice-cold sterile distilled water. Centrifuge the cells again at 4,400 x g for 6 min at 4 °C. Discard the supernatant and re-suspend each cell pellet in 20 ml of ice-cold sterile 10% glycerol and centrifuge again as described above.
- Decant the supernatant and re-suspend the cell pellet in 500 μl of ice-cold 10% glycerol. Aliquot the prepared competent cells in 50 μl volumes into individual, pre-chilled 1.5 ml micro-centrifuge tubes for immediate transformation. The cells may be frozen on dry ice or in liquid nitrogen and stored at -80 °C for up to 3 months. However, transformation efficiency is highest with freshly prepared competent cells.
- Transformation
- Add 100 ng of purified PCR product from step 1.3.1 to the competent cells. Flick the tube and allow suspension to sit on ice for 5 min.
- Transfer the suspension of ice-cold competent cells and DNA to a pre-chilled electroporation cuvette. Electroporate cell mixture with the 2.5 kV, 25 μF, 200 ohms setting in a 2 mm gap cuvette (i.e. if using a different cuvette, please see the manufacturer's instructions for electroporation settings), and immediately add 1 ml of room temperature SOC medium. Transfer the electroporated cells in SOC medium into a 15 ml culture tube and incubate at 32 °C for 1 hr with orbital shaking at 220 rpm.
- After incubation, centrifuge the cells at 4,400 x g for 5 min at room temperature. Remove approximately 850 μl of the supernatant, and re-suspend the cell pellet in the remaining liquid.
- Spread the cells on pre-warmed LB plates containing 17 μg/ml Cm, and incubate at 32 °C overnight. Pick two individual transformant colonies and streak out on LB-Cm plates for mutant confirmation. We recommend avoiding largest transformant colonies in order to reduce the possibility of picking strains with rare suppressor mutations. Also streak the same transformants on LB-Kan to confirm that the strains are not Kan resistant. Note: Since the Cm resistance marker is used to make the donor strains, we don't expect the donor mutants to be Kan resistant. By streaking the transformants onto both LB-Kan and LB-Cm plates, we ensure that the donor mutation itself did not somehow cause Kan resistance. If this step were not performed and the target mutations were to affect the ability of the strain to survive on Kan, double mutants would not be made effectively using eSGA since donor strains would survive all selection steps. This is merely a precautionary logical step to catch unexpected phenotypes or any other issues that may interfere with the completion of the eSGA screens. If this step were skipped, other controls would still identify failed screens. However, an earlier detection of a donor that is resistant to Kan would eliminate a series of unproductive and unnecessary steps. Other possible reasons for a strain to grow on Kan could include for example expired antibiotic as well as the use of a wrong PCR product or a strain for the transformation. All these issues can be identified and dealt with early on in the procedure with the aid of this control, which is not required, but recommended for eSGA.
- Confirming the non-essential gene deletion (step 1.6.1) or essential gene hypomorphic mutation (section 1.6.2) Note: When confirming the gene deletion by PCR, genomic DNA from a wild type strain should be used as a control.
- Confirming non-essential gene deletion
- To confirm gene deletion by PCR, isolate the genomic DNA from the transformed, putative knockout strains grown in liquid LB-Cm medium, following the manufacturer's instructions (Promega).
- To confirm successful recombineering, amplify the DNA from each transformant with three different sets of knockout confirmation primers. Perform the reactions with ~150 ng of genomic DNA template, and confirm the correct fragment sizes by running the PCR-synthesized products on an agarose gel.
- The first primer set consists of a 20-nt flanking primer, located 200 bp upstream of the targeted region (KOCO-F), and Cm-R primer, which is complementary to the Cm cassette sequence. This amplification is expected to produce a 445 nt amplicon in the mutant, but not in the wild type strain (Figure 1).
- The second set includes a forward primer (Cm-F), annealing to the Cm cassette sequence, and a 20-nt reverse flanking confirmation primer (KOCO-R), which is designed to anneal 200 bp downstream of the 3' end of the deleted gene. This amplification reaction is expected to produce a 309 nt amplicon in the mutant, but not in the wild type strain (Figure 1).
- The third PCR contains KOCO-F and KOCO-R primers. This reaction is required to confirm that the selected strain is not a merodiploid, with one gene locus having been replaced by the cassette and another duplicated but otherwise wild-type gene copy still present. This amplification from a correct deletion mutant is expected to produce a 1.4 kb product (Figure 1). Note: When the target gene has about the same length in base pairs as the gene deletion cassette, one will not notice a difference between a locus which has been replaced by the cassette, and a locus which has not. For a definitive confirmation in such cases, we recommend using additional primers to specifically amplify a DNA segment within the deleted gene. In this case, if the gene has been deleted, an amplification product will be observed only in the wild type control strain and not in the mutant. These primers can be manually or computationally designed to specifically amplify a segment of DNA within the deleted region. We typically amplify 140 bp regions.
- Confirming essential gene hypomorphic mutation
- To confirm the hypomorphic mutation by PCR, use gene-specific 20-nt forward (KOCO-F) and reverse primers (KOCO-R) located 200 bp upstream and downstream, respectively, of the SPA-tag insertion site (Figure 2). Note: This amplification step is used as a control to ensure the absence of an untagged copy of the target essential gene. When only a tagged version is present, a single band 310 bp larger than the SPA-Cm amplicon is observed. A 400 bp product would signal the presence of the untagged version of the gene.
- In parallel to the PCR confirmation, use Western blotting to verify the in-frame insertion of the SPA-tag using an anti-FLAG M2 antibody specific to the FLAG-epitopes of the SPA-tag21.
- Storing the confirmed donor strain
- Transfer the overnight culture of the confirmed recombinant donor mutant strain to the labeled cryovials, supplement with glycerol to 15% final concentration, and mix well. For long-term storage, keep the cryovials at -80 °C.
2. Arraying E. coli F- Recipient Mutants for eSGA Screens
Replica-pin the E. coli Keio single non-essential gene deletion mutant collection generated by Mori et al. (3,968 strains11; F- BW25113; can be obtained from Open Biosystems) robotically to twenty-four 384-well microplates containing 80 μl of liquid LB media per well, supplemented with 50 μg/ml Kan. Note: For systematically assessing GIs with essential genes, the recipient Keio collection11 can be supplemented with the F- Kan-marked essential gene hypomorphic mutant strains with C-terminal SPA-tags22, 23.
To make room for the border control strain, which will act as a positive control, aid in within and between plate normalizations as well as in automated colony quantizations, remove the inoculated media from the outermost wells of each plate from the above step and transfer to new plates, again leaving the outermost wells empty. Note: The border control strain JW5028 from the Keio collection11 is deleted for a small pseudo gene that should not, and has not in our whole-genome screens, exhibit GIs with any donors. Therefore, this is a good positive control strain to identify rare instances when conjugation, pinning, or selections have failed, resulting in absent or sporadic border strain growth. For instance, if the border control colonies do not grow after the double mutant selection, the query strain may not be capable of conjugation, conjugation may have been attempted in a condition that prevented it from taking place (e.g. on an antibiotic-containing plate), or a wrong antibiotic may have been added to the selection plate. Similarly, if very few, sporadic colonies are observed on the whole plate, including the border, conjugation or pinning may have failed and the screen needs to be repeated. If this sporadic border growth is reproducible, it may indicate consistently poor donor conjugation and thus instances where double mutants are not observed are possibly due to conjugation failure, not true GIs. Such screens would need to be discarded from further analysis. Furthermore, by being present on all plates, the border control strain allows the colony quantization software to automatically determine colony positions on the double mutant plates and thus colony positions do not have to be demarcated manually. Lastly, the border control strain aids in the normalization of colony sizes within (e.g. different rows within a plate) and between (e.g. different recipient plates pinned with the same donor at different times) plates.
Likewise, create negative control spots by removing any two strains from a different location within each plate (not border) and transfer the strains to a new plate. Fill these empty wells with LB media containing 17 μg/ml of Cm. These negative control spots are expected to be empty in the recipient and therefore double mutant plates and help ensure that there was no processing or plate handling errors when numbering, imaging, or pinning the plates. Note: These empty spots are required negative controls. Negative controls help identify instances when selection fails - when negative control strains grow during selection. We suggest to not only select unique negative control spots for each of the recipient plates, but to also choose negative control spots within the same quadrant of the plate (e.g. lower right). This way, the pattern of negative control spots would identify possible plate mishandling errors (e.g. plates being mislabeled or being pinned upside down, where the upper left colony is pinned in lower right position).
Inoculate the Keio strain JW502811, with a pseudogene deletion, into a 500 ml flask with 200 ml LB, containing 50 μg/ml of Kan. Based on our whole-genome eSGA screens, the growth of this particular mutant is the closest to wild type BW25113 and it is thus used as a border control.
Grow the arrayed strains as well as the JW5028 culture overnight at 32 °C with 190 rpm orbital shaking to an OD of ~0.4-0.6 at 600 nm. After the overnight growth, fill in the border wells with approximately 80 μl of JW5028 overnight culture.
The assembled plates may be used in step 3.2. Alternatively, for long-term storage, supplement each well in the recipient plates with 15% glycerol, mix the media and glycerol, and keep the plates at -80 °C.
3. High-density Strain Mating Procedure for Generating E. coli Double Mutants
Grow the Hfr donor strain, bearing the query mutation, marked with Cm, overnight at 32 °C in rich LB-Cm liquid medium (17 μg/ml of Cm) with shaking at 220 rpm.
Pin the ordered recipient mutant collection in 384-colony density onto solid LB plates supplemented with 50 μg/ml of Kan. In parallel, pin the donor query mutant strain in 384-colony density onto the same number of LB plates, supplemented with 17 μg/ml of Cm. Incubate the plates overnight at 32 °C. Note: The recipient plates used for pinning may require up to 36 hr for obtaining sufficiently large colonies for subsequent steps. When average border colony size is about 2 mm in diameter (or greater), the plates are ready to be pinned.
To conjugate the strains, pin the donor from a 384-colony overnight donor plate onto solid LB plate. Subsequently pin a single 384-colony recipient plate over the freshly-pinned donor on the conjugation plate. Continue pinning until all donor-recipient plates have been conjugated by pinning onto solid LB conjugation plates. Incubate the pinned conjugation plates at 32 °C for 16 to 24 hr. Note: Individual single mutant donor strains may show gene-to-gene variability in mating efficiency because of the potential effects of the mutations on conjugation, DNA transfer efficiency, or fitness. Growth can be stopped at any time between 16 and 24 hr, when sufficiently large colonies have been obtained for subsequent pinning steps. Conjugations lasting 16 to 24 hr produce sufficient numbers of ex-conjugants for reproducible eSGA results even for the mutants of late transferring genes or mutants with slightly lower fitness.
Pin each 384-density conjugation plate onto a single solid LB plate, containing Cm (17 μg/ml) and Kan (50 μg/ml) until all conjugation plates have been pinned. Incubate the freshly-pinned first selection plates for 16-36 hr at 32 °C. Note: In regards to the selection steps, different amounts of time may be necessary for different donor screens. Since all double mutants in a given screen have the same donor mutation, on average, a slower growing donor mutant gives rise to slower growing double mutants. Thus, depending on the donor fitness, selection steps may require between 16 and 36 hr. The double mutant growth can be stopped when the double mutants are sufficiently large for the subsequent step either pinning after the first selection or imaging after the second. This usually translates to border control colonies on average with at least 2 mm diameters.
Re-pin each first selection plate onto a second, double drug (Cm and Kan), selection plate in 1,536-colony format, such that each first selection colony is represented by four colonies on the second selection plate. Incubate the plates for 16-36 hr at 32 °C. Photograph the final double mutant selection plates to quantitatively measure the growth fitness of the mutant and to analyze the interactions between gene pairs (Figure 3). Note: Although the frequency of merodiploidy (partial chromosomal duplications) is low at about 1/1,000 cells24, merodiploidy can mask GI. Hence, we recommend including biological replicates in all eSGA screens. Fortunately, commercial disposable plates and plastic pinning pads can be reused up to three times by sterilizing the materials as follows: Agar from the plates is discarded and the plates as well as pads are sterilized by soaking them in 10% bleach overnight, followed by a rinse with distilled water, washing in 70% ethanol, and air-drying the plasticware in a flow hood under ultraviolet light. The sterile pads and plates are stored in sealed sterile plastic bags until use.
4. Processing Data and Deriving GI Scores
Measure the colonies on each plate in batch mode or individually using a specialized colony imaging software 3, 9. Note: A suitable Java-based high-throughput colony imaging and scoring application is now freely available for public access (http://srcollins77.users.sourceforge.net/). The software works on different platforms (i.e. Mac or PC) and can be launched either as an executable or from a terminal window.
Normalize the raw colony size measurements by correcting for systematic biases within and between plates such as plate edge effects, inter-plate variation effects, uneven image lighting, artifacts due to physical curvature of the agar surface, competition effects for neighboring colonies and possible pinning defects, as well as differences in growth time9. These systematic artifacts are independent of the double mutant fitness and should be corrected as they give rise to spurious growth fitness estimates.
The colonies in edge rows and columns tend to be larger due to lower competition for nutrients than found in the center of the plate. Thus, to correct for this effect, scale the sizes of edge colonies to be such that the median size in that row or column is equal to the median size of the colonies in the center of the plate.
Analyze the results statistically to take into account the reproducibility and the standard variance from the median sizes of the replicate colony measurements. At least three independent biological replicate experiments are necessary to evaluate the experimental reproducibility.
Finally, use the normalized median colony growth fitness sizes to generate a GI score (S) for each gene pair using the following formula:
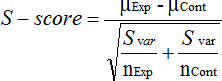 Where, Svar = (varExp x (nExp-1) + varCont x (nCont -1)) / (nExp + nCont -2); varExp = the maximum variance of the normalized colony sizes for the double mutant; varCont = median of the variances in the normalized double mutant colony sizes from the reference set; nExp = number of measurements of double mutant colony sizes; nCont = the median number of experimental replicates over all the experiments; μExp = median normalized colony sizes of the double mutants; and μCont = median of normalized colony sizes for all double mutants arising from the single donor mutant strain. The S-score reflects both the statistical confidence of a putative digenic interaction as well as the biological strength of the interaction. Strong positive S-scores indicate alleviating effects, suggesting that the interacting genes participate in the same pathway9, while significant negative S-scores reflect synthetic sickness or lethality, which is often suggestive of membership in parallel redundant pathways9. For determining the pathway-level functional relationships, a single S-score for each pair of tested genes is produced from the final dataset. Note: In our previous study9, we found that recombination tends to occur less frequently between genes within 30 kbp of each other. Therefore, for downstream analysis, following normalization and score generation, we remove interactions between genes within 30 kbp of each other. In addition to GIs themselves, functional relationships between pairs of genes can be investigated by looking at how similar the GI profiles of the two genes are. The GI profile of a gene is the set of all interactions of that gene with all other genes in the genome outside of the gene's linkage area. Functionally similar genes tend to have higher correlations of genetic interaction profiles12, 25. Thus, by calculating correlation coefficients for all genes in the experimental dataset one can use eSGA for investigating functional relationships even between genes lying close to each other on the chromosome.
Where, Svar = (varExp x (nExp-1) + varCont x (nCont -1)) / (nExp + nCont -2); varExp = the maximum variance of the normalized colony sizes for the double mutant; varCont = median of the variances in the normalized double mutant colony sizes from the reference set; nExp = number of measurements of double mutant colony sizes; nCont = the median number of experimental replicates over all the experiments; μExp = median normalized colony sizes of the double mutants; and μCont = median of normalized colony sizes for all double mutants arising from the single donor mutant strain. The S-score reflects both the statistical confidence of a putative digenic interaction as well as the biological strength of the interaction. Strong positive S-scores indicate alleviating effects, suggesting that the interacting genes participate in the same pathway9, while significant negative S-scores reflect synthetic sickness or lethality, which is often suggestive of membership in parallel redundant pathways9. For determining the pathway-level functional relationships, a single S-score for each pair of tested genes is produced from the final dataset. Note: In our previous study9, we found that recombination tends to occur less frequently between genes within 30 kbp of each other. Therefore, for downstream analysis, following normalization and score generation, we remove interactions between genes within 30 kbp of each other. In addition to GIs themselves, functional relationships between pairs of genes can be investigated by looking at how similar the GI profiles of the two genes are. The GI profile of a gene is the set of all interactions of that gene with all other genes in the genome outside of the gene's linkage area. Functionally similar genes tend to have higher correlations of genetic interaction profiles12, 25. Thus, by calculating correlation coefficients for all genes in the experimental dataset one can use eSGA for investigating functional relationships even between genes lying close to each other on the chromosome.
Representative Results
GIs reveal functional relationships between genes. Similarly, since genes in the same pathway display similar GI patterns and the GI profile similarity represents the congruency of phenotypes, we can group functionally related genes into pathways by clustering their GI profiles. Integrating GI and GI correlation networks with physical interaction information or other association data, such as genomic context (GC) relationships can also reveal the organization of higher-order functional modules that define core biological systems (and system crosstalk) in bacteria. For example, as with yeast4, 6, 7, 26, E. coli gene pairs exhibiting alleviating interactions that also have highly correlated GI profiles tend to encode proteins that are either physically associated (e.g. form a complex; Figure 4a) or that act coherently in a common biochemical pathway (e.g. components in a linear cascade). A representative example is shown in Figure 4b, where the components of the functionally redundant Isc and Suf pathways, which jointly participate in the essential Fe-S biosynthesis process, form distinct clusters that are linked together by extensive aggravating interactions (i.e. synthetic lethality). A statistical measure (e.g. hypergeometric distribution function27) can be used on GI or GI correlation data to find significant enrichment for interactions between and within pathways (Figure 4c). Cluster analysis can also be applied to functional networks derived using eSGA to predict the functions of genes lacking annotations5, 6, 9, 28-30 (Figure 4d). Since clustering algorithms vary, however, putative functional assignments determined through clustering require independent experimental verification.
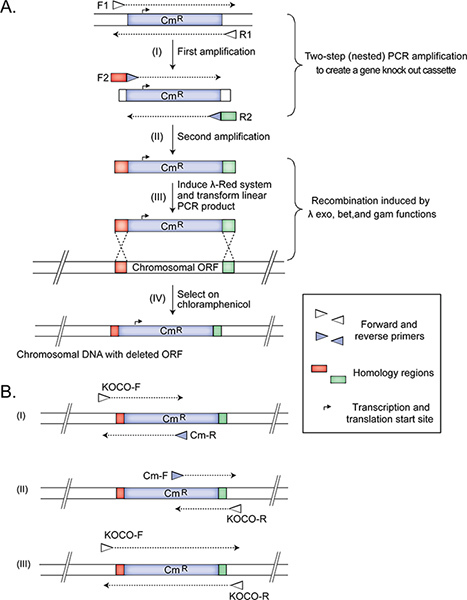 Figure 1. The construction and confirmation of Hfr Cavalli non-essential single gene deletion donor strains. The panels (adapted from14) illustrate the deletion of E. coli chromosomal ORF (A) and the three primer sets used for confirming (B) the correctly generated mutant Hfr Cavalli donor strain. Amplifications from a correctly constructed deletion donor strain should produce (i) 445 bp, (ii) 309 bp, and (iii) 1.4 kb products. See protocol text for details. Click here to view larger figure.
Figure 1. The construction and confirmation of Hfr Cavalli non-essential single gene deletion donor strains. The panels (adapted from14) illustrate the deletion of E. coli chromosomal ORF (A) and the three primer sets used for confirming (B) the correctly generated mutant Hfr Cavalli donor strain. Amplifications from a correctly constructed deletion donor strain should produce (i) 445 bp, (ii) 309 bp, and (iii) 1.4 kb products. See protocol text for details. Click here to view larger figure.
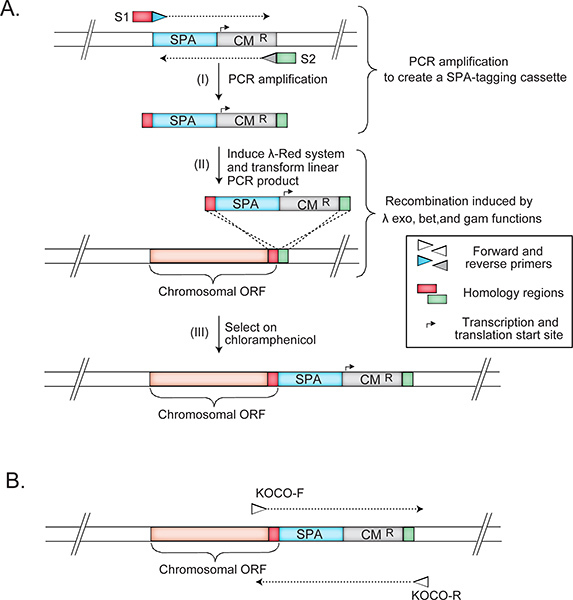 Figure 2. The construction and confirmation strategies of essential hypomorphic donor mutant strains in an Hfr Cavalli genetic background. The panels illustrate the creation of hypomorphic mutations of essential genes through recombineering technology (A) and the primer sets (B) used for the PCR confirmation of essential gene hypomorphic mutations. See protocol text for details.
Figure 2. The construction and confirmation strategies of essential hypomorphic donor mutant strains in an Hfr Cavalli genetic background. The panels illustrate the creation of hypomorphic mutations of essential genes through recombineering technology (A) and the primer sets (B) used for the PCR confirmation of essential gene hypomorphic mutations. See protocol text for details.
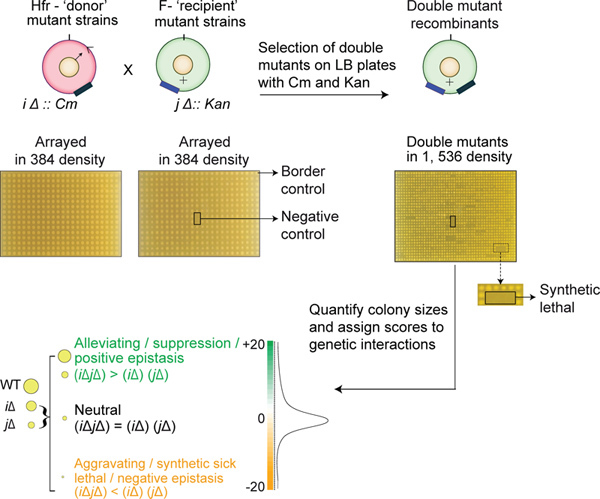 Figure 3. Schematic summary of key eSGA steps. The Hfr donor (marked with chloramphenicol resistance, CmR) and recipient F- (marked with kanamycin resistance, KanR) mutant strains are grown in 384-colony density on LB-Cm and LB-Kan plates, respectively. The donor and the recipient strains are conjugated by pinning them over each other onto an LB plate, which is then incubated overnight at 32 °C. Consequently, the conjugants are pinned onto plates containing both Kan and Cm to select double mutants. The double mutant plates are digitally imaged and the growth fitness of the colony sizes is quantitatively scored to identify aggravating and alleviating interactions.
Figure 3. Schematic summary of key eSGA steps. The Hfr donor (marked with chloramphenicol resistance, CmR) and recipient F- (marked with kanamycin resistance, KanR) mutant strains are grown in 384-colony density on LB-Cm and LB-Kan plates, respectively. The donor and the recipient strains are conjugated by pinning them over each other onto an LB plate, which is then incubated overnight at 32 °C. Consequently, the conjugants are pinned onto plates containing both Kan and Cm to select double mutants. The double mutant plates are digitally imaged and the growth fitness of the colony sizes is quantitatively scored to identify aggravating and alleviating interactions.
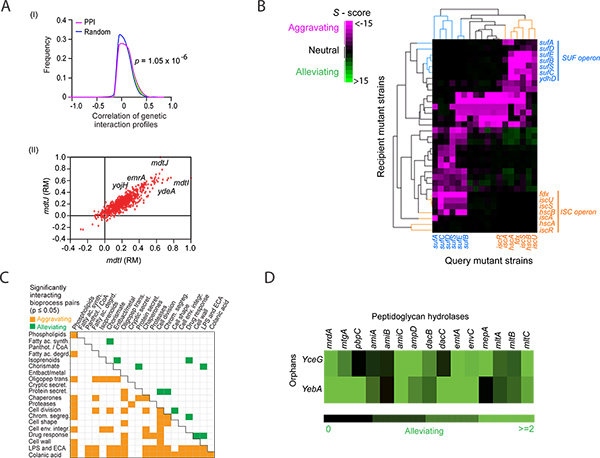 Figure 4. Representative computational analyses of genetic interaction (GI) scores for determining pathway-level functional relationships. (A) Panel I, Distribution of correlation coefficients between the GI profiles of gene pairs encoding proteins linked by protein-protein interactions (PPI) versus randomly drawn gene pairs. The p-value was computed using the two-sample Kolmogorov-Smirnov (KS) test; Panel II, Scatter plot of correlated genetic profiles from rich media (RM) for two transporters (mdtI, mdtJ) that form a heterodimeric complex required for spermidine excretion. (B) The hierarchical clustering of the GI sub-network adapted from Butland et al.9 highlights the functional connectivity between components of the previously known Isc and Suf pathways with similar GI patterns. Pink represents aggravating (negative S-score) interactions, green represents alleviating (positive S-score) interactions and black represents absence of GI. A predicted novel component of the SUF pathway, ydhD, displays GIs with the members of the Isc pathway. (C) Pathway cross-talk recorded among cell envelope processes that are significantly enriched for aggravating or alleviating GI according to the hypergeometric enrichment analysis27. (D) A GI sub-network predicts the role of two functionally unannotated genes, yceG and yebA, in peptidoglycan splitting based on their pattern of strong alleviating interactions with well-known cell division peptidoglycan hydrolases. The figures shown in panels A, C and D are adapted from Babu et al. (2011)12. Click here to view larger figure.
Figure 4. Representative computational analyses of genetic interaction (GI) scores for determining pathway-level functional relationships. (A) Panel I, Distribution of correlation coefficients between the GI profiles of gene pairs encoding proteins linked by protein-protein interactions (PPI) versus randomly drawn gene pairs. The p-value was computed using the two-sample Kolmogorov-Smirnov (KS) test; Panel II, Scatter plot of correlated genetic profiles from rich media (RM) for two transporters (mdtI, mdtJ) that form a heterodimeric complex required for spermidine excretion. (B) The hierarchical clustering of the GI sub-network adapted from Butland et al.9 highlights the functional connectivity between components of the previously known Isc and Suf pathways with similar GI patterns. Pink represents aggravating (negative S-score) interactions, green represents alleviating (positive S-score) interactions and black represents absence of GI. A predicted novel component of the SUF pathway, ydhD, displays GIs with the members of the Isc pathway. (C) Pathway cross-talk recorded among cell envelope processes that are significantly enriched for aggravating or alleviating GI according to the hypergeometric enrichment analysis27. (D) A GI sub-network predicts the role of two functionally unannotated genes, yceG and yebA, in peptidoglycan splitting based on their pattern of strong alleviating interactions with well-known cell division peptidoglycan hydrolases. The figures shown in panels A, C and D are adapted from Babu et al. (2011)12. Click here to view larger figure.
Discussion
We have outlined a step-wise protocol for using robotic eSGA screening to investigate bacterial gene functions at a pathway level by interrogating GI. This approach can be used to study individual genes as well as entire biological systems in E. coli. Carefully executing the experimental steps described above, including all appropriate controls, and rigorously analyzing and independently validating the GI data are key aspects for the success of eSGA in making new functional discoveries. In addition to eSGA, a conceptually similar method for studying GI in E. coli, termed GIANT-coli17, can be used to illuminate novel functional relationships that are often missed by other approaches, such as proteomic23 or phenotypic13 screens alone. Nevertheless, as with any genome-wide approach, limitations do exist on the applicability of eSGA or GIANT-coli, for example to other bacterial species. This is partly because of the unavailability of genome-wide single gene deletion mutant strain collections for most bacterial species, particularly for species where targeted mutagenesis is hindered by a low natural frequency of homologous recombination. In such cases, gene disruptions using methods such as random trackable mutagenesis-based assays like TnSeq31 can potentially be used for investigating bacterial gene functions. For an overview of emerging alternate bacterial genetic screening procedures, see review paper by Gagarinova and Emili (2012)32.
Although GI methods often reveal many interesting connections suggestive of novel mechanistic links, the integration of these data with other information such as phenotypic profiling13, physical interaction networks22, 23, and GC-inferred associations can be particularly informative. For example, as with GI networks, GC associations, which are based on the conservation of gene order (operons), bacterial gene fusions, operon recombination frequencies derived from intergenic distances of predicted operons across genomes, as well as phylogenetic profiling33 can provide additional insights into how a bacterial cell organizes protein complexes into functional pathways to mediate and coordinate major cellular processes12, 23.
E. coli is a workhorse model for understanding the molecular biology of other Gram-negative bacteria. To this end, the eSGA GI data can be used in conjunction with comparative genomics information (e.g. phylogenetic profiling, gene expression profiling) to investigate the evolutionary conservation of functional relationships discovered by eSGA across other proteobacterial species and Prokaryotic taxa. Furthermore, the interaction data generated for E. coli may be used to gain insight into the pathway architecture of microbes discovered by metagenomics, for which functional annotations are lacking. Since many genes are widely conserved across all microbes23, and because antibiotic sensitivity can be enhanced by certain genetic perturbations34, the functional relationships illuminated by eSGA may even potentially be exploited for designing innovative combination drug therapies.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was supported by funds from Genome Canada, the Ontario Genomics Institute, and the Canadian Institutes of Health Research grants to J.G. and A.E. AG is a recipient of Vanier Canada Graduate Scholarship.
References
- Bandyopadhyay S, Kelley R, Krogan NJ, Ideker T. Functional maps of protein complexes from quantitative genetic interaction data. PLoS Comput. Biol. 2008;4:e1000065. doi: 10.1371/journal.pcbi.1000065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Myers CL, Andrews B, Boone C. Charting the genetic interaction map of a cell. Curr. Opin. Biotechnol. 2011;22:66–74. doi: 10.1016/j.copbio.2010.11.001. [DOI] [PubMed] [Google Scholar]
- Costanzo M, et al. The genetic landscape of a cell. Science. 2010;327:425–4231. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedler D, et al. Functional organization of the S. cerevisiae phosphorylation network. Cell. 2009;136:952–963. doi: 10.1016/j.cell.2008.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roguev A, et al. Conservation and rewiring of functional modules revealed by an epistasis map in fission yeast. Science. 2008;322:405–4010. doi: 10.1126/science.1162609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuldiner M, et al. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell. 2005;123:507–519. doi: 10.1016/j.cell.2005.08.031. [DOI] [PubMed] [Google Scholar]
- Wilmes GM, et al. A genetic interaction map of RNA-processing factors reveals links between Sem1/Dss1-containing complexes and mRNA export and splicing. Mol. Cell. 2008;32:735–746. doi: 10.1016/j.molcel.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu M, et al. Systems-level approaches for identifying and analyzing genetic interaction networks in Escherichia coli and extensions to other prokaryotes. Mol. Biosyst. 2009;12:1439–1455. doi: 10.1039/B907407d. [DOI] [PubMed] [Google Scholar]
- Butland G, et al. coli synthetic genetic array analysis. Nat. Methods. 2008;5:789–7895. doi: 10.1038/nmeth.1239. [DOI] [PubMed] [Google Scholar]
- Typas A, et al. Regulation of peptidoglycan synthesis by outer-membrane proteins. Cell. 2010;143:1097–10109. doi: 10.1016/j.cell.2010.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006;2:2006–200008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu M, et al. Genetic interaction maps in Escherichia coli reveal functional crosstalk among cell envelope biogenesis pathways. PLoS Genet. 2011;7:e1002377. doi: 10.1371/journal.pgen.1002377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols RJ, et al. Phenotypic landscape of a bacterial cell. Cell. 2011;144:143–156. doi: 10.1016/j.cell.2010.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu M, Gagarinova A, Greenblatt J, Emili A. Array-based synthetic genetic screens to map bacterial pathways and functional networks in Escherichia coli. Methods Mol Biol. 2011;765:125–153. doi: 10.1007/978-1-61779-197-0_9. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 2000;97:6640–665. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, et al. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2000;97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Typas A, et al. quantitative analyses of genetic interactions in. E. coli. Nat. Methods. 2008;5:781–787. doi: 10.1038/nmeth.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeghouf M, et al. Sequential Peptide Affinity (SPA) system for the identification of mammalian and bacterial protein complexes. J. Proteome Res. 2004;3:463–468. doi: 10.1021/pr034084x. [DOI] [PubMed] [Google Scholar]
- Davierwala AP, et al. 2005. pp. 1147–1152.
- Breslow DK, et al. A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat. Methods. 2008;5:711–718. doi: 10.1038/nmeth.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu M, et al. Sequential peptide affinity purification system for the systematic isolation and identification of protein complexes from Escherichia coli. Methods Mol. Biol. 2009;564:373–400. doi: 10.1007/978-1-60761-157-8_22. [DOI] [PubMed] [Google Scholar]
- Butland G, et al. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature. 2005;433:531–537. doi: 10.1038/nature03239. [DOI] [PubMed] [Google Scholar]
- Hu P, Janga SC, Babu M, Diaz-Mejia JJ, Butland G, et al. Global functional atlas of Escherichia coli encompassing previously uncharacterized proteins. PLoS Biol. 2009;7 doi: 10.1371/journal.pbio.1000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RP, Roth JR. Tandem genetic duplications in phage and bacteria. Annu. Rev. Microbiol. 1977;31:473–505. doi: 10.1146/annurev.mi.31.100177.002353. [DOI] [PubMed] [Google Scholar]
- Boone C, Bussey H, Andrews BJ. Exploring genetic interactions and networks with yeast. Nat. Rev. Genet. 2007;8:437–449. doi: 10.1038/nrg2085. [DOI] [PubMed] [Google Scholar]
- Collins SR, et al. Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature. 2007;446:806–8010. doi: 10.1038/nature05649. [DOI] [PubMed] [Google Scholar]
- Le Meur N, Gentleman R. Modeling synthetic lethality. Genome Biol. 2008;9:R135. doi: 10.1186/gb-2008-9-9-r135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AH, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- Tong AH, et al. Global mapping of the yeast genetic interaction network. Science. 2004;303:808–813. doi: 10.1126/science.1091317. [DOI] [PubMed] [Google Scholar]
- Wong SL, et al. Combining biological networks to predict genetic interactions. Proc. Natl. Acad. Sci. U.S.A. 2004;101:15682–15687. doi: 10.1073/pnas.0406614101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Opijnen T, Bodi KL, Camilli A. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat. Methods. 2009;6:767–772. doi: 10.1038/nmeth.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagarinova A, Emili A. Genome-scale genetic manipulation methods for exploring bacterial molecular biology. Mol. Biosyst. 2012;8:1626–1638. doi: 10.1039/c2mb25040c. [DOI] [PubMed] [Google Scholar]
- Dewey CN, et al. Positional orthology: putting genomic evolutionary relationships into context. Brief Bioinform. 2011;12:401–412. doi: 10.1093/bib/bbr040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Onge RP, et al. Systematic pathway analysis using high-resolution fitness profiling of combinatorial gene deletions. Nat. Genet. 2007;39:199–206. doi: 10.1038/ng1948. [DOI] [PMC free article] [PubMed] [Google Scholar]