

Abstract
A novel form of cell-to-cell communication involving the formation and shedding of large vesicular structures, called microvesicles (MVs), from the surfaces of highly aggressive forms of human cancer cells has been attracting increasing amounts of attention. This is in large part due to the fact that MVs contain a variety of cargo that is not typically thought to be released from cells including cell-surface receptor tyrosine kinases, cytosolic and nuclear signaling proteins and RNA transcripts. MVs, by sharing their contents with other cells, can greatly impact cancer progression by increasing primary tumor growth,1–3 as well as by promoting the development of the pre-metastatic niche.4 We have recently shown that the small GTPase RhoA is critical for MV biogenesis in human cancer cells. Moreover, we have now obtained evidence that implicates the highly related small GTPases, Rac and Cdc42, in regulating the loading of specific cargo into MVs, as well as in the shedding of MVs from cancer cells. Thus, linking the Rho family of small GTPases to MV biogenesis has begun to shed some light on a new and unexpected way that these signaling proteins contribute to human cancer progression.
Keywords: microvesicles, oncosomes, cancer, cell communication, tissue transglutaminase, glutaminase, Rho, GTPase, Warburg effect, transformation
Microvesicles: A Novel Form of Cell Communication
Cell-to-cell communication is a fundamental cellular process that has important consequences in development, tissue homeostasis, and, when de-regulated, in promoting human disease states, such as cancer.5,6 One of the most common and best studied mechanisms of cell communication is paracrine signaling, where diffusible factors (i.e., growth factors and pro-inflammatory cytokines) secreted by one cell bind to their corresponding receptors expressed on the surface of a nearby cell. Depending on the diffusible factor, this results in the induction of a specific set of signaling events within the recipient cell that determines its fate. A case in point involves epidermal growth factor (EGF). Many normal cell types express the EGF-receptor, and when cultures of these cells are stimulated with EGF, the ligand-bound EGF-receptor becomes activated and triggers signaling pathways that promote their growth and survival.7 However, increases in EGF and EGF-receptor expression are also hallmarks of human brain, breast and lung cancers.7,8 These findings, coupled with the fact that overexpressing the EGF-receptor in normal fibroblasts is sufficient to induce ligand-dependent transformation,9 underscores the importance of paracrine signaling in cancer progression.
However, with the recent discovery of microvesicles (MVs), a new and unexpected twist in the paracrine signaling paradigm has emerged. MVs are large (0.1–2.0 µM in diameter) vesicular structures that are formed and shed directly from the surfaces of cells, especially by aggressive forms of human cancer cells (when shed by cancer cells MVs are sometimes referred to as oncosomes).1,10,11Figure 1A shows an example of a high grade MDAMB231 breast cancer cell that is heavily decorated with MVs. One of the main reasons that MVs are attracting increasing amounts of attention has to do with their contents. Rather than containing just growth factors and pro-inflammatory cytokines, MVs contain a plethora of cargo that are not typically thought to be released from cells including cell surface receptor tyrosine kinases, cytosolic signaling proteins, RNA transcripts, as well as microRNAs (Fig. 1B).1–3,10,11 Interestingly, MVs can be transferred between cancer cells, an outcome that potentiates the growth and transformed properties of the recipient cells.1,3 Moreover, we have recently shown that MVs shed by MDAMB231 breast cancer cells or U87 brain tumor cells, when isolated and then added to cultures of normal fibroblasts or mammary epithelial cells, cause the recipient cells to acquire a transformed phenotype, as read-out by their ability to exhibit anchorage-independent growth and an overall survival advantage (Fig. 1C).2 The transforming capabilities of the MVs derived from the MDAMB231 and the U87 cells are transient in nature and require a continuous bathing of the recipient cells with freshly prepared MVs in order for the cells to maintain their transformed characteristics. When considering this unique form of paracrine signaling in the context of a cancer patient, MVs might contribute to cancer progression by causing the normal cells that surround a tumor (i.e., the tumor microenvironment) to become transformed. Thus, the resulting tumor mass would not be solely due to the growth of the cancer cells, as generally believed, but would also include the expansion of the cells in the tumor microenvironment that become “transiently” transformed through the continuous supply of MVs provided by the primary cancer cells. Consistent with this idea, when we co-injected mitotically arrested human MDAMB231 breast carcinoma cells, which are unable to proliferate but retain the ability to generate MVs, with normal mouse fibroblasts into nude mice, tumors comprised primarily of cells of mouse fibroblastic origin formed.2,12

Figure 1. Highly aggressive forms of human cancer cells generate and shed MVs. (A) A scanning electron microscopy (SEM) image of a human MDAMB231 breast cancer cell covered with MVs. (B) A list highlighting some of the contents that have been identified in MVs. (C) Schematic showing that MVs generated and shed from a cancer cell (Donor Cell) can be transferred to a recipient normal cell, conferring upon the recipient cell the characteristics of a transformed cell (i.e., providing the cell with growth and survival advantages). PM stands for plasma membrane.
It is worth mentioning that the impact of MVs in cancer progression is not limited to paracrine-mediated effects on cell growth and survival, but that MVs also can have important consequences in metastasis. For example, MVs have been shown to stimulate the vascularization of tumors through the recruitment of endothelial cells to promote the formation of new blood vessels.4 Moreover, MVs are stable in the circulation of cancer patients1,3 and travel to secondary sites where they prepare the recipient tissue for accepting a circulating (metastatic) cancer cell.4
Tissue Transglutaminase and Fibronectin are Critical Mediators of MVs-Induced Cellular Transformation
Because the MVs from MDAMB231 breast cancer cells or U87 brain tumor cells were capable of conferring upon normal cells the properties of transformed cells, we then wanted to determine the contents of the MVs that were responsible for mediating this effect. Proteomics analyses performed on the MVs isolated from these two different cancer cell lines revealed 39 common proteins, which included several key metabolic enzymes, such as the M2 isoform of pyruvate kinase (see below), members of the heat shock protein family and RNA and DNA binding proteins.2 While any of these proteins could potentially contribute to the biological actions of the MVs, it was the presence of two additional proteins in the MVs, namely tissue transglutaminase (tTG) and fibronectin, which caught our attention.
tTG is a dual functioning enzyme that can bind and hydrolyze GTP, similar to other classical GTP-binding proteins, as well as catalyze an enzymatic transamidation (cross-linking) reaction which results in the formation of covalent linkages between two proteins or between a protein and a polyamine.13 tTG expression and its enzymatic crosslinking activity are frequently upregulated in a variety of human cancers, with the highest levels of tTG expression being consistently detected in the more advanced and aggressive tumors.14–16 Moreover, work done by our laboratory and by others has shown that tTG participates in cancer progression by promoting cell growth and survival, as well as through enhancing the migration and invasive activities exhibited by cancer cells.14–17
The connections between tTG and malignant transformation prompted us to consider whether the ability of MVs to transform normal cells was dependent on tTG. We found that depleting the MDAMB231 or U87 cancer cell-derived MVs of tTG by siRNA inhibited the transforming capabilities of the MVs.2 Likewise, treating isolated MVs with the tTG crosslinking inhibitor, T101, prior to adding them to normal fibroblasts, also blocked the ability of the MVs to transform recipient cells, suggesting that the enzymatic cross-linking activity of tTG was crucial for this outcome. Indeed, this appears to be the case, as we then went on to show that tTG, by crosslinking the fibronectin present in the MVs, leads to the formation of a fibronectin species (a dimer) with unique signaling capabilities. In particular, the crosslinked fibronectin in the MVs (acting as a ligand) was able to engage integrins expressed on the surfaces of the recipient cells (functioning as a receptor) and excessively activated them. This, in turn, potentiated integrin-dependent mitogenic and survival signaling in the recipient cells that led to their transformation.
RhoA Signaling Regulates MV Biogenesis in Human Cancer Cells
The mechanisms underlying the formation and shedding of MVs by cells is currently poorly understood. What is known is that MVs are generated via a mechanism that does not require the classical secretory pathway in cells that involves the processing, sorting and transport of proteins to be secreted through the endoplasmic-reticulum and Golgi apparatus.1,2,10,11 Thus, other mechanisms likely exist that promote MV biogenesis. Because MVs cause such dramatic morphological changes to cells (Fig. 1A), we felt that proteins that regulated cell shape via rearrangements of the actin cytoskeleton might be likely participants in their formation. The Rho family of small GTPases, including RhoA, Rac and Cdc42, were originally identified as major regulators of actin dynamics,18 and so we asked whether ectopically expressing activated forms of these small GTPases in the human cervical carcinoma HeLa cell line could induce MV formation. HeLa cells were particularly useful in this regard, because MV formation is tightly regulated and induced by EGF stimulation.2,12 We found that a constitutively active RhoA mutant, but not activated mutants of Cdc42 or Rac, potently induced MV formation in HeLa cells, whereas knocking-down RhoA inhibited their formation. Surprisingly, we also found that an activated form of RhoC, which is 85% identical to RhoA and is upregulated in invasive and metastatic cancers,19 was ineffective at inducing MV formation in cells, suggesting that the signaling mechanism leading to MV biogenesis is highly specific.12 How this specificity is achieved is currently being investigated in the laboratory and may have to do with temporal and/or spatial processes that uniquely regulate the signaling activities of RhoA.
What are the effectors of RhoA that are important for inducing MV biogenesis? While several targets for RhoA have been identified, one of them, Rho-associated, coiled-coil containing protein kinase (ROCK), has been shown to influence cytoskeletal changes.20 This widely expressed serine/threonine kinase has also been linked to enhanced cancer cell migration and tumor growth, and so it is not surprising that inhibitors against ROCK have been generated and examined for use as possible cancer therapies.20,21 Using the ROCK inhibitor, Y-27632, we found that the ability of HeLa cells expressing an activated form of RhoA to induce MV formation in HeLa cells was dependent on ROCK activation.12 Moreover, the ability of MDAMB231 breast cancer cells or U87 brain tumor cells to generate MVs was sensitive to Y-27632 treatment, suggesting that ROCK may function as a general regulator of MV biogenesis (Fig. 2).
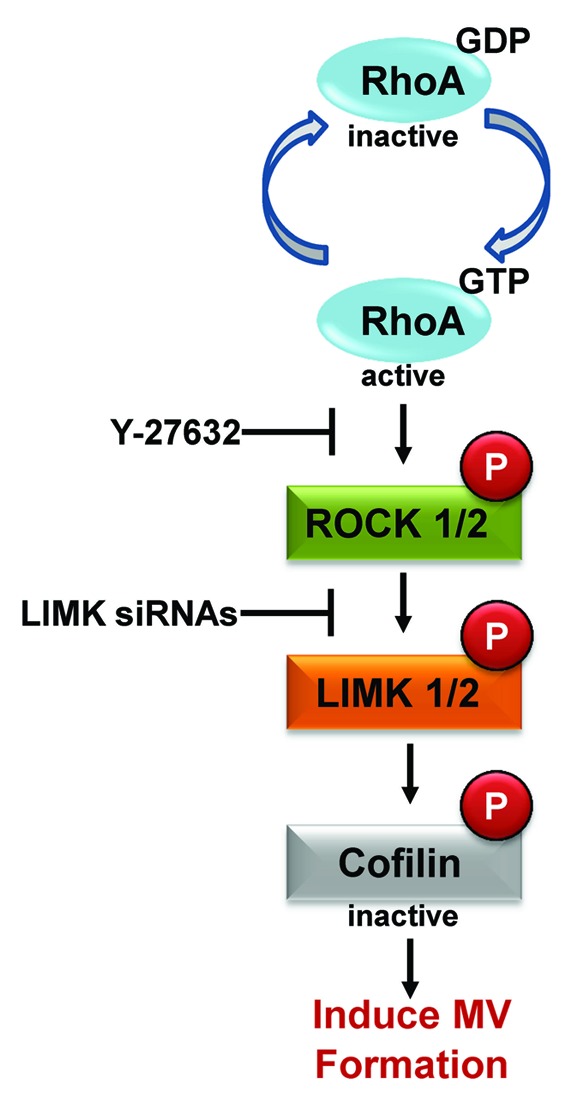
Figure 2. RhoA-dependent signaling induces MV biogenesis. Schematic showing the RhoA signaling pathway that is responsible for promoting MV formation in human cancer cells. The pathway culminates with the phosphorylation of cofilin, which inhibits its actin-severing activity. This results in increased actin polymerization and the generation of MVs. The ROCK inhibitor, Y-27632, and LIMK siRNAs (for knocking-down LIMK expression) are particularly useful reagents for blocking RhoA-induced MV formation in cancer cells.
The additional steps in the RhoA-ROCK signaling pathway needed to influence actin dynamics have been well established and include the sequential phosphorylation of LIM kinase (LIMK) and cofilin.20,22 We went on to show that these same players are also needed for MV biogenesis (Fig. 2), suggesting that these two cellular processes are intimately linked. In line with this idea, we have demonstrated by immunofluorescence that the membranes of MVs from cancer cells contain filamentous actin.2,12 Moreover, increases in the expression and activation of ROCK and LIMK have been linked to human cancer progression,20–22 raising the intriguing possibility that perhaps some of the transforming potential of these kinases is due to their abilities to promote MV formation. This further suggests that targeting ROCK or LIMK to inhibit MV production may provide a novel therapeutic strategy for the treatment of human cancers (Fig. 2).
Role of Metabolism in MV Biogenesis
Recently, we have found that the biogenesis of MVs in cancer cells is linked to the metabolic changes that have been suggested to be essential for malignant transformation and cancer progression. One of the most striking features of cancer cells is the well-known “Warburg effect,” whereby the limited amounts of pyruvate that are generated from the penultimate step of the glycolytic pathway, namely the conversion of phosphoenolpyruvate to pyruvate by the M2 isoform of pyruvate kinase, are primarily converted to lactic acid, rather than being used to generate citrate in the mitochondria.23,24 Because of the Warburg effect, cancer cells have developed alternative inputs into the citric acid cycle, with one mechanism being through elevations in glutamine metabolism. Specifically, cancer cells exhibit an accelerated conversion of glutamine to glutamate by activated forms of glutaminase, followed by the generation of α-ketoglutarate from glutamate by glutamate dehydrogenase, with α-ketoglutarate then entering the citric acid cycle. Recently, we demonstrated that the activation of glutaminase in cancer cells occurs downstream from hyper-activated Rho GTPase-dependent signals to NFκB.25 The activation of glutaminase can be blocked through a specific allosteric inhibitor of the enzyme, a bromo-dibenzophenanthridine called 968. A distinct allosteric inhibitor of this important metabolic enzyme, designated BPTES (for bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide], has also been identified and characterized.26 Interestingly, we have recently shown that treatment of cancer cells with either of these inhibitors can block MV biogenesis (Fig. 3A and B). This would suggest that the formation and loading of MVs within cancer cells is linked to their metabolic capability. Moreover, because Rho GTPases are both responsible for driving the maturation of MVs by signaling to the actin cytoskeleton,12 and for activating glutamine metabolism in cancer cells,25 it will be interesting to see whether these two sets of signaling outcomes are highly coordinated in transformed cells.
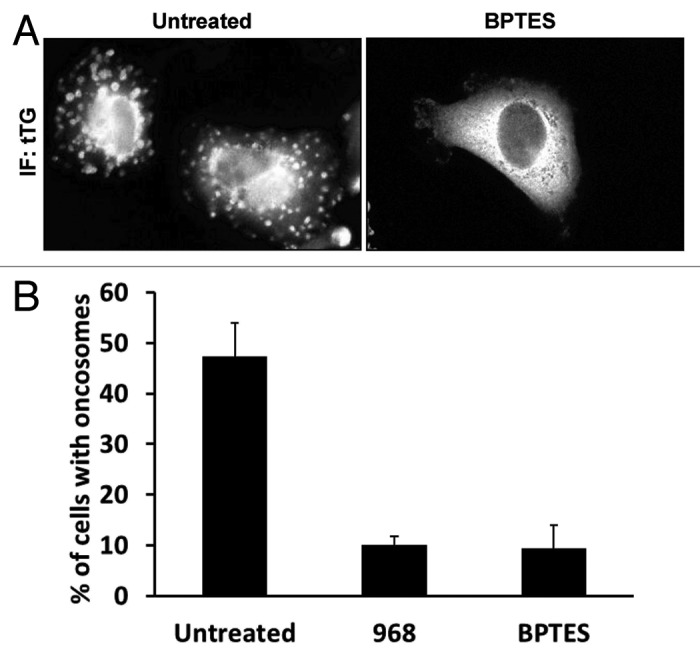
Figure 3. The altered metabolism exhibited by human cancer cells is important for MV biogenesis. MDAMB231 breast cancer cells were left untreated or were treated with the glutaminase C inhibitors, 968 and BPTES, for 2 d prior to being fixed. The cells were subjected to immunofluorescence using a tTG antibody (IF: tTG) to label the MVs on the surfaces of the cells. (A) Representative images of MDAMB231 cells treated without (Untreated) or with BPTES. Note the lack of detectable MVs on the BPTES-treated cell. (B) Quantification of MV production by MDAMB231 cells treated without (Untreated) or with 968 or BPTES. The experiments were done three times and the results from each experiment were averaged together and graphed. The error bars indicate standard deviation.
Future Directions: How is Cargo Loaded into MVs?
Proteomics analyses performed on MVs isolated from human breast cancer (MDAMB231) cells and human glioblastoma (U87) cells show that these two sets of vesicles contain a number of interesting signaling proteins and metabolic enzymes, many of which are shared between them.2 Thus, an important question concerns how the cargo for cancer cell MVs is selected and loaded. We know that the loading of MVs with their cargo does not occur through conventional secretory pathways, and consequently the shedding of cargo-laden MVs from cancer cells is not susceptible to standard secretory inhibitors.1,2,10,11 Preliminary results from our laboratory suggest that the loading of proteins into these vesicles may be under the regulatory control of the Rac GTPase. If this indeed turns out to be the case, it would present an interesting picture whereby various key steps in the biogenesis of MVs from cancer cells are under the control of Rho family GTPases, with the structural formation of the vesicles being driven by RhoA signaling through the actin cytoskeleton while, the loading of cargo is being directed by Rac, and the ultimate shedding of the vesicles from the cell surface potentially being triggered by Cdc42 (see below). Figure 4 (left side) highlights how we think different members of the Rho family of small GTPases contribute to MV formation, loading and shedding.

Figure 4. Schematic depicting the lifecycle of a MV. Activation of RhoA in a cancer (Donor) cell initiates MV formation by inducing a “budding” event at a particular site along the plasma membrane. Our preliminary studies suggest that the loading of specific cargo into the MV involves Rac, whereas the release or shedding of the MV from the cancer cell is directed by Cdc42. The shed MV contains transforming cargo, including tTG and the extracellular matrix protein, fibronectin. tTG crosslinks fibronectin to itself within the shed MV, generating a fibronectin dimer that exhibits enhanced signaling capabilities when it engages integrins that are expressed on the surface of a normal recipient cell. The signaling induced by the crosslinked fibronectin-integrin complex aberrantly regulates signaling pathways that promote cell growth and survival, causing the recipient cell to acquire a transformed phenotype.
Distinctions Between MVs and Membrane Blebs: How are MVs Shed from the Surfaces of Cancer Cells?
Some interesting questions also exist regarding the distinctions between MVs and membrane-blebs that are formed as an outcome of disruptions to the membrane-actomyosin interactions, leading to their rapid protrusion from the plasma membrane.27 The fact that an apparently common RhoA-signaling pathway is involved in the formation of membrane blebs28 and MVs12 would seem to imply that the differences between these two membrane processes lies in the underlying mechanisms that enable MVs to be shed from the cell surface, rather than undergoing retraction back into the cell. While at the present time, we know relatively little about the mechanisms of MV shedding, an intriguing clue came from our finding that there are remarkably few MVs present along the surfaces of diffuse B-lymphoma (Dbl)-transformed fibroblasts, as viewed by the immunofluorescence staining of actin that forms at the base of these vesicles, or the MV marker protein, tTG. This was surprising given that Dbl is an activator (guanine nucleotide exchange factor or GEF) for the RhoA GTPase.29 However, Dbl also strongly activates Cdc42,30 which therefore raised the possibility that Cdc42 either antagonizes the formation of MVs or stimulates the shedding of these vesicles, thus leaving fewer vesicles behind on the cell surface. Recently, we have found that the latter appears to be the case, as an activated mutant of Cdc42 capable of constitutive GDP-GTP exchange strongly increased the amount of MVs shed into the medium from HeLa cells. These findings suggest that both the maturation and shedding of MVs are under the control of distinct Rho GTPase-dependent signaling pathways. An important goal of future studies will be to determine the downstream signaling partner/effector(s) that mediates the regulatory effects of activated Cdc42 on MV shedding, as well as to see whether this signaling pathway is hyper-activated in the more aggressive cancer cells.
How Do MVs Engage Recipient Cells and Transfer Their Contents?
One of the most important aspects of MV function, and what will certainly be an extremely interesting area of future research, concerns how MVs, upon being shed from cancer cells, are able to engage “recipient” cells and transfer their contents in a manner that results in fundamental changes in cellular phenotypes. It is this remarkable capability that makes cancer cell-derived MVs so interesting and explains why they are gaining a great deal of attention in the cancer biology community. One can imagine that at a primary tumor site, cancer cells are shedding MVs that are then able to engage non-transformed (normal) fibroblasts or epithelial cells in the immediate environment, thereby changing these cells, upon transferal of the cargo from MVs, in a manner that has a significant influence on the tumor microenvironment (Fig. 4). Moreover, because MVs are stable in the circulation,1,3 it is also attractive to envisage scenarios where these vesicles, upon reaching distant secondary sites, help contribute to the pre-metastatic niche and thereby have an important impact on cancer metastasis.4
One potential mechanism by which MVs engage their target recipient cells is through the interactions between MV-associated fibronectin and integrins.2 Therefore, an important question concerns whether a similar mechanism is at play in other cancer cells; for example, in the ability of MVs shed from glioblastomas in cell culture to engage and enhance the transformed characteristics of other (recipient) glioblastomas.1,3 It also will be extremely interesting to see how the engagement of recipient cells by MVs is coupled to the transfer of their contents into recipient cells. Is this the outcome of some type of fusion event or are other mechanisms at work, such as endocytosis? Moreover, we will want to know what the consequences are for such transfer events, i.e., what does it mean to transfer signaling receptors to recipient cells, vs. metabolic enzymes, or RNA transcripts? The answers to these questions await a host of future studies and promise to keep this field extremely active and at the forefront of research by the cancer biology community during the next few years.
Acknowledgments
This work was supported by grants from the National Institutes of Health. We also would like to acknowledge Ms. Cindy Westmiller for her expert secretarial assistance.
Glossary
Abbreviations:
- BPTES
bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide
- Dbl
diffuse B-lymphoma
- EGF
epidermal growth factor
- LIMK
LIM kinase
- MV
microvesicle
- ROCK
Rho-associated, coiled-coil containing protein kinase
- tTG
tissue transglutaminase
Footnotes
Previously published online: www.landesbioscience.com/journals/smallgtpases/article/20755
References
- 1.Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, et al. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. 2008;10:619–24. doi: 10.1038/ncb1725. [DOI] [PubMed] [Google Scholar]
- 2.Antonyak MA, Li B, Boroughs LK, Johnson JL, Druso JE, Bryant KL, et al. Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Proc Natl Acad Sci U S A. 2011;108:4852–7. doi: 10.1073/pnas.1017667108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Skog J, Würdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10:1470–6. doi: 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peinado H, Lavotshkin S, Lyden D. The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts. Semin Cancer Biol. 2011;21:139–46. doi: 10.1016/j.semcancer.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 5.Schauer IG, Sood AK, Mok S, Liu J. Cancer-associated fibroblasts and their putative role in potentiating the initiation and development of epithelial ovarian cancer. Neoplasia. 2011;13:393–405. doi: 10.1593/neo.101720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schneider MR, Wolf E. The epidermal growth factor receptor ligands at a glance. J Cell Physiol. 2009;218:460–6. doi: 10.1002/jcp.21635. [DOI] [PubMed] [Google Scholar]
- 7.Eccles SA. The epidermal growth factor receptor/Erb-B/HER family in normal and malignant breast biology. Int J Dev Biol. 2011;55:685–96. doi: 10.1387/ijdb.113396se. [DOI] [PubMed] [Google Scholar]
- 8.Mendelsohn J, Baselga J. Epidermal growth factor receptor targeting in cancer. Semin Oncol. 2006;33:369–85. doi: 10.1053/j.seminoncol.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 9.Moscatello DK, Montgomery RB, Sundareshan P, McDanel H, Wong MY, Wong AJ. Transformational and altered signal transduction by a naturally occurring mutant EGF receptor. Oncogene. 1996;13:85–96. [PubMed] [Google Scholar]
- 10.Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol. 2009;19:43–51. doi: 10.1016/j.tcb.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 11.Muralidharan-Chari V, Clancy JW, Sedgwick A, D’Souza-Schorey C. Microvesicles: Mediators of extracellular communication during cancer progression. J Cell Sci. 2010;123:1603–11. doi: 10.1242/jcs.064386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li B, Antonyak MA, Zhang J, Cerione RA. RhoA triggers a specific signaling pathway that generates transforming microvesicles in cancer cells. Oncogene. 2012 doi: 10.1038/onc.2011.636. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Folk JE. Transglutaminases. Annu Rev Biochem. 1980;49:517–31. doi: 10.1146/annurev.bi.49.070180.002505. [DOI] [PubMed] [Google Scholar]
- 14.Hwang JY, Mangala LS, Fok JY, Lin YG, Merritt WM, Spannuth WA, et al. Clinical and biological significance of tissue transglutaminase in ovarian carcinoma. Cancer Res. 2008;68:5849–58. doi: 10.1158/0008-5472.CAN-07-6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li B, Antonyak MA, Druso JE, Cheng L, Nikitin AY, Cerione RA. EGF potentiated oncogenesis requires a tissue transglutaminase-dependent signaling pathway leading to Src activation. Proc Natl Acad Sci U S A. 2010;107:1408–13. doi: 10.1073/pnas.0907907107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan L, Siegel M, Choi K, Khosla C, Miller CR, Jackson EN, et al. Transglutaminase 2 inhibitor, KCC009, disrupts fibronectin assembly in the extracellular matrix and sensitizes orthotopic glioblastomas to chemotherapy. Oncogene. 2007;26:2563–73. doi: 10.1038/sj.onc.1210048. [DOI] [PubMed] [Google Scholar]
- 17.Boroughs LK, Antonyak MA, Johnson JL, Cerione RA. A unique role for heat shock protein 70 and its binding partner tissue transglutaminase in cancer cell migration. J Biol Chem. 2011;286:37094–107. doi: 10.1074/jbc.M111.242438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 19.Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406:532–5. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]
- 20.Sahai E, Ishizaki T, Narumiya S, Treisman R. Transformation mediated by RhoA requires activity of ROCK kinases. Curr Biol. 1999;9:136–45. doi: 10.1016/S0960-9822(99)80067-0. [DOI] [PubMed] [Google Scholar]
- 21.Hahmann C, Schroeter T. Rho-kinase inhibitors as therapeutics: from pan inhibition to isoform selectivity. Cell Mol Life Sci. 2010;67:171–7. doi: 10.1007/s00018-009-0189-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scott RW, Olson MF. LIM kinases: function, regulation and association with human disease. J Mol Med (Berl) 2007;85:555–68. doi: 10.1007/s00109-007-0165-6. [DOI] [PubMed] [Google Scholar]
- 23.Erickson JW, Cerione RA. Glutaminase: a hot spot for regulation of cancer cell metabolism? Oncotarget. 2010;1:734–40. doi: 10.18632/oncotarget.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–37. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 25.Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010;18:207–19. doi: 10.1016/j.ccr.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson MM, McBryant SJ, Tsukamoto T, Rojas C, Ferraris DV, Hamilton SK, et al. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES) Biochem J. 2007;406:407–14. doi: 10.1042/BJ20070039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ridley AJ. Life at the leading edge. Cell. 2011;145:1012–22. doi: 10.1016/j.cell.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 28.Gadea G, de Toledo M, Anguille C, Roux P. Loss of p53 promotes RhoA-ROCK-dependent cell migration and invasion in 3D matrices. J Cell Biol. 2007;178:23–30. doi: 10.1083/jcb.200701120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hart MJ, Eva A, Zangrilli D, Aaronson SA, Evans T, Cerione RA, et al. Cellular transformation and guanine nucleotide exchange activity are catalyzed by a common domain on the dbl oncogene product. J Biol Chem. 1994;269:62–5. [PubMed] [Google Scholar]
- 30.Hart MJ, Eva A, Evans T, Aaronson SA, Cerione RA. Catalysis of guanine nucleotide exchange on the CDC42Hs protein by the dbl oncogene product. Nature. 1991;354:311–4. doi: 10.1038/354311a0. [DOI] [PubMed] [Google Scholar]