

Abstract
Background
Established ARDS is often refractory to treatment. Clinical trials have demonstrated modest treatment effects, and mortality remains high. Ventilator strategies must be developed to prevent ARDS.
Hypothesis
Early ventilatory intervention will block progression to ARDS if the ventilator mode: 1) maintains alveolar stability and 2) reduces pulmonary edema formation.
Methods
Yorkshire Pigs (38–45kg) were anaesthetized and subjected to "2-hit" Ischemia-Reperfusion and Peritoneal Sepsis. Following injury, animals were randomized into two groups: Early Preventative Ventilation (Airway Pressure Release Ventilation- APRV) vs. Non-Preventative Ventilation (NPV) and followed for 48hr. All animals received anesthesia, antibiotics, and fluid/vasopressor therapy per Surviving Sepsis Campaign. Ventilation parameters: 1) NPV Group - Tidal volume (Vt): 10cc/kg + PEEP- 5 cm/H2O volume-cycled mode, 2) APRV Group - Vt: 10–15 cc/kg; Phigh, Plow, Thigh, Tlow were titrated for optimal alveolar stability. Physiologic data and plasma were collected throughout the 48hr study period, followed by BAL and necropsy.
Results
APRV prevented development of ARDS (p<0.001 vs NPV) by PaO2/FiO2 ratio. Quantitative histological scoring showed APRV prevented lung tissue injury (p<0.001 vs. NPV). BALF showed APRV lowered total protein and IL-6, while preserving surfactant proteins A & B (p<0.05 vs. NPV). APRV significantly lowered lung water (p<0.001 vs. NPV). Plasma IL-6 concentrations were similar between groups.
Conclusions
Early preventative mechanical ventilation with APRV blocked ARDS development, preserved surfactant proteins, and reduced pulmonary inflammation and edema, despite systemic inflammation similar to NPV. These data suggest early preventative ventilation strategies stabilizing alveoli and reducing pulmonary edema can attenuate ARDS after ischemia-reperfusion-sepsis.
Keywords: Sepsis, Shock, ARDS, ALI, Ventilator Induced Lung Injury, Airway Pressure Release Ventilation
INTRODUCTION
The Acute Respiratory Distress Syndrome (ARDS) is a lung condition that can be caused by systemic inflammation or lung injury, affecting nearly 200,000 critically ill patients annually (1). Despite thirty years of research, ARDS claims the lives of 40% of affected patients and is associated with costs of in excess of $150,000 per case (1–3). Low tidal volume ventilation (LTV), the present ‘gold standard’ of management (4), represents a supportive intervention that limits progression rather than altering the underlying pathophysiology of ARDS. LTV improves survival by a modest 9% (5). A new approach is therefore needed in order to improve patient outcome and reduce the grave human toll taken by this disease.
The current perception of ARDS pathophysiology is limited by the characterization of ARDS in a binary construct: it is viewed by clinicians as either being present or absent (1, 6). Consequently, therapy to treat ARDS is not implemented until all features of the disease are already present (7). However, recent population data suggest ARDS is a hospital-acquired phenomenon in which 67% of ARDS patients develop the disease in hospital usually within 30 hours of admission (8). Patients appear to be developing ARDS during those 30 hours prior to showing signs of respiratory compromise. These studies suggest the development of ARDS represents a progression of pathology in the lung between the time of insult and the time at which ARDS criteria are met. The concept of a gradual progression from moderate lung injury to ARDS vs. the "all or none" phenomenon is important clinically for several reasons. First, fulminant disease is resistant to treatment and thus prevention of ARDS is key (2, 4). Second, ARDS may have preclinical stages, similar to some cancers, offering a window of opportunity to prevent ARDS before the full disease expression (8, 9). Third, patients at risk are hospitalized during this process (8). Clinicians therefore have an opportunity to intervene earlier, potentially reversing disease progression prior to clinical manifestations when ARDS criteria are met.
During the onset of ARDS there are two processes that contribute significantly to the development of disease and represent potential excellent targets for early preventative therapy: high permeability pulmonary edema and alveolar instability (the repetitive expansion and collapse of alveoli with tidal ventilation that causes atelectrauma) (10–12). We hypothesized a preventative ventilatory intervention would be successful in blocking the progression from injury to ARDS if the ventilator mode was capable of: 1) maintaining alveolar stability, thereby preventing the injurious process of atelectrauma; and 2) reducing pulmonary edema formation, which is the driving force of pathology in ARDS (10, 11, 13). We refer to this strategy as early, preventative Stabilizing Alveolar Ventilation and Edema reduction (SAVE). Any ventilatory mode that both stabilizes alveoli by minimizing intratidal collapse and reduces edema in the alveolar space would fall under the SAVE category. The key to this novel strategy is not the ventilatory mode itself, but rather the early intervention aimed at preventing the progression of disease and targeting the central components of alveolar instability and pulmonary edema.
In this study, we applied SAVE in the form of Airway Pressure Release Ventilation (APRV) immediately following injury, in a translational porcine model of sepsis and ischemia-induced ARDS. In this setting, SAVE dramatically reduced lung pathology and prevented the development of fulminant ARDS.
MATERIALS & METHODS
All techniques and procedures described have been fully approved by the Committee for the Human Use of Animals at Upstate Medical University. Animals: Healthy female Yorkshire pigs (30–50kg) were anesthetized using continuous infusion of ketamine/xylazine/propofol to maintain a surgical plane of anesthesia. Animals were continuously monitored and cared for by the investigators for the full 48-hour duration of the experiment. Surgical Preparation: Under sterile conditions, animals underwent tracheostomy, arterial and venous catheterization, as well as bladder catheterization. Baseline (BL) measurements were taken after surgical preparation and prior to injury. Injury: Lung injury was induced using a “2-hit” model as previously described (14, 15). Briefly: 1) Ischemia/Reperfusion (IR): The superior mesenteric artery (SMA) was clamped for 30 min to induce intestinal ischemia. 2) Peritoneal Sepsis (PS): stool was harvested from a cecotomy and mixed with blood to create a fecal clot which was implanted in the abdomen prior to abdominal closure. Time Zero (T0) measurements were taken immediately after the induction of injury (i.e. removal of clamp and placement of fecal clot) upon closure of the abdomen. Treatment Groups: One hour following injury (T1), animals were randomized into two groups: Airway Pressure Release Ventilation (APRV, n = 3) or Non- Preventative Ventilation (NPV n = 5). Non- Preventative Ventilation Group animals were connected to a Galileo™ ventilator (Hamilton Medical, Reno, NV) with initial settings: tidal volume (Vt) 10 cc/kg, respiratory rate (RR) 15 breaths/minute, FiO2 of 21% and positive end-expiratory pressure (PEEP) of 5 cmH2O. FiO2 was adjusted to maintain adequate oxygenation throughout the study. Respiratory rate was titrated to maintain PaCO2 at 35–45 mmHg. APRV Group The technique of administering APRV below has been described previously (16) and is critical for APRV functioning as a SAVE mode. APRV administered without the specific techniques described will not stabilize alveoli or reduce edema, and may actually produce significant lung injury. Animals were connected to a Dräger™ Evita XL ventilator (Dräger, Lubeck, Germany) with initial settings during surgical preparation identical to the NPV group. At one hour post-injury (T1), the ventilator mode was switched to Airway Pressure Release Ventilation (APRV) with the following initial settings: High Pressure (Phigh) was initially set at the Plateau Pressure from the volume cycled setting (15–25 cmH2O). Low Pressure (Plow) was set at 0 cmH2O to minimize expiratory resistance and maximize Peak Expiratory Flow Rate (PEFR). Duration of Phigh (Thigh) was set at 3.9–6 seconds to equal 90–95 % of the respiratory cycle. Duration of Plow (Tlow) was set at 0.4–0.55 seconds to achieve a Termination of Peak Expiratory Flow Rate (T-PEFR) that was equal to 75% of PEFR. This was calculated based on the angle of deceleration noted on the expiratory flow waveform (16). Release volumes were titrated to a range of 10cc/kg by adjusting Phigh and Tlow in order to ensure that the APRV animals were not inadvertently receiving 'protective' low-tidal volume ventilation. The Phigh, Thigh, Tlow and FiO2 were titrated throughout the study to minimize lung de-recruitment, limit airway pressures, optimize the efficiency of CO2 clearance, and minimize dead space ventilation. These adjustments were based on interpretation of expiratory flow waveform, static compliance, calculated resistance, and arterial blood gases.
Antibiotics: Broad-spectrum antibiotics (Ampicillin 2 grams IV [Bristol Myers Squibb, Princeton, NJ] and Metronidazole 500mg IV [Baxter, Deerfield, IL]) given following abdominal closure and every 12 hours until the end of the study. Resuscitative Protocol: Animals were treated with intravenous fluid resuscitation and vasopressors in a protocol adapted from the Early Goal Directed Therapy (EGDT) strategy (17). This strategy is considered standard-of-care for management of hemodynamic collapse in sepsis. Maintenance intravenous fluid requirements were calculated by body weight and given via continuous infusion. Ringer’s Lactate was used for maintenance and resuscitation. Hemodynamic parameters were assessed to determine the need for resuscitative fluid bolus according to parameters described by Rivers et al(17). According to EGDT guidelines, continuous infusion of Norepinephrine was started when the animal was no longer responsive to fluid bolus. Epinephrine was added when Norepinephrine was no longer effective. Physiologic Measurements: Hemodynamic parameters were measured (Agilent, CMS-2001™ System M1176A, with Monitor M1094B Böbingen, Germany) using Edwards transducers (Pressure Monitoring Kit [PXMK1183], Edwards Lifesciences, Irvine, CA). Pulmonary parameters were measured or calculated by the Dräger™ ventilator (Lubeck, Germany). Blood was drawn every 6 hours for ELISA quantification of IL-6 levels in systemic circulation. Measurement of blood gases and chemistries were made with Roche Blood gas analyzer (Cobas b221, Basel, Switzerland) every 3 hours. Clinical pathology and blood cultures were performed by the Upstate Medical University pathology laboratory facility. Necropsy: At necropsy the lungs, liver, spleen kidney and small intestine were removed and preserved in formalin. Lungs were inflated to 25 cm H2O using stepwise increases in PEEP prior to submersion in formalin. Bronchoalveolar Lavage Fluid (BALF): At necropsy, the right middle lobe of the lung was lavaged with 60mL of normal saline. Concentration of total protein in BALF was determined by BCA method. The concentration of IL-6 was determined in the plasma and BALF using ELISA according to the manufacturer’s recommendations. Western blot analysis of surfactant proteins A & B (SP-A & SP-B) expression in the BALF as well as Sodium/Potassium Adenosine Triphosphatase (Na, K-ATPase) in lung tissue were performed as previously described (18, 19).
Quantitative Histology: The quantitative histological assessment of the lung was based on image analysis of 120 photomicrographs (10 per animal), made at high-dry magnification following a validated, unbiased, systematic sampling protocol (14, 20). Each photomicrograph was scored using a 4-point scale for each of 5 parameters: atelectasis, fibrinous deposits, and blood in air space, vessel congestion, alveolar wall thickness, and leukocytes. The histologist performing the analysis was blinded to the treatment groups and samples. Statistics: Repeated Measures ANOVA with pig number and treatment as random effects was performed to compare differences within and between treatment groups for continuous parameters, probability values <0.001 were considered significant. Post hoc Tukey’s tests were performed on continuous data at specific time points only if significance was found in the group*time effect using RM ANOVA. Categorical data were compared using an unpaired Student’s t-test. Quantitative histology data was analyzed using Mann-Whitney U test after testing for normality. Probability values <0.05 were considered significant. All analyses were performed using JMP version 5.1.1 (Cary, NC).
RESULTS
Hemodynamics & Laboratory Data
All animals in both groups developed polymicrobial bacteremia in blood cultures, as previously described (20). No differences between groups were found in hemoglobin/hematocrit, white blood cell count, platelets, liver enzymes, creatinine, electrolytes, or coagulation parameters. There was a significant time trend towards anemia, leukopenia, thrombocytopenia and coagulopathy in both groups, consistent with severe sepsis. Both groups demonstrated a similar degree of hyperlactatemia and base deficit consistent with shock. Plasma IL-6 levels increased over the 48-h course of the experiment in both groups from 0 pg/mL at baseline for both groups up to 2045±650 pg/mL for NPV at 48 hours and 1779±458 for APRV at 48 hours, this difference was not significant.
All animals demonstrated hemodynamics consistent with severe septic shock: progressive hypotension, tachycardia, and high resuscitative fluid and vasopressor requirements. Mean arterial pressure, central venous pressure, pulmonary artery pressure, and thermodilutional cardiac output were similar between groups.
Survival
This study was designed and powered to detect a difference in ARDS incidence between groups as the primary outcome and not differences in mortality. Therefore statistical significance in mortality is not achieved. 100% of APRV animals survived the full 48 hour duration of the experiment. NPV animals suffered a 40% mortality, with 60% of animals surviving the full 48 hour experiment. These differences were not statistically significant.
Sequential Organ Failure Assessment (SOFA)
To give a sense of shock severity SOFA scores were calculated (21). Scores were similarly elevated in both groups with no differences between APRV & NPV. SOFA score of NPV at 24h was 2.2 ± 0.45 vs. 4.0 ± 2.65 for APRV; SOFA score of NPV at 48h was 7.4 ± 0.89 vs. 6.0 ± 3.21 for APRV. Both groups had increase in SOFA score reflecting worsening clinical status over time.
Fluid Balance
All animals in both groups required aggressive fluid resuscitation and had significantly positive fluid balances (36.54L ± 1.02L). There were no differences between groups in fluids infused, urine output or fluid balance.
Wet: Dry (W:D) Weight ratio
In order to assess the degree of high permeability edema we assessed W:D weight ratio. In the lung the NPV group had significantly higher W:D weight ratio (8.82±0.76) than the APRV group (6.73±0.45) (p<0.0001). There were no differences between groups in W:D weight ratios for liver, kidney, intestine or spleen.
Lung Injury
The development of lung injury in the present study was determined by PaO2/FiO2 (P/F) ratio, and static compliance (Cstat) (Figures 1a & 1b). The APRV group had normal P/F ratios and normal Cstat throughout the 48-hour experiment (Figure 1a & 1b). In contrast animals in the NPV group developed progressive, severe lung injury with declining P/F ratio and Cstat over the course of the study (Figure 1a & 1b). The P/F ratio in the NPV animals dropped to meet Acute Lung Injury (ALI) criteria (<300) by 30 hours and ARDS criteria (<250) by 39 hours (Figure 1a) (1). This progression to lung injury in the NPV group was corroborated by steadily declining Cstat to less than 50% of baseline by the end of the study, reflecting pulmonary mechanics characteristic of ARDS.
Figure 1.


a. PaO2/FiO2 (P/F) Ratio Non Preventative Ventilation group n=5 (dashed line) Airway Pressure Release Ventilation group n=3 (bold line). Data mean ± SEM (*= p< 0.05). APRV P/F ratio remained within normal limits (>300) throughout the 48h study. NPV P/F ratio decreased below Acute Lung Injury (ALI) criteria (<300) by 36 h and subsequently well below ARDS criteria (<200) by 39h.
b. Static Compliance (Cstat) Non Preventative Ventilation group n=5 (dashed line) Airway Pressure Release Ventilation group n=3 (bold line). Data mean ± SEM (*= p< 0.05). Both groups begin with normal Cstat at baseline. While APRV maintains normal levels throughout the 48h study NPV demonstrates a progressive decline in Cstat reflecting 'stiffening' lungs and worsening lung injury.
Pulmonary Data
No significant differences were found in Peak Pressure (Ppeak), PEEP, PaCO2 and Minute Volume (MV) between groups. Ppeak rose from 15–20cm H2O for both groups at baseline to >~30 cm H2O for both groups by the end of the experiment. MV rose from 4 L/min for both groups at baseline to 6 L /min for both groups by the end of the experiment. PaCO2 was 35–40 mm Hg at baseline for both groups, in the NPV group it increased to 54.7± 7.9 mm Hg by 48 h whereas in the APRV group it remained 38.7 ± 1.7 mm Hg at 48 hours; however, this difference was not statistically significant. Mean Airway Pressure (Pmean) was significantly higher in APRV than in the NPV group: Pmean began at 7 cm H2O for both groups at baseline and was 20.7± 2.6 cm H2O at 48 hours for NPV vs. 30.0 ± 4.0 cm H2O at 48 h for APRV (p>0.0001). Tidal Volume (VT) began at ~10cc/kg for both groups at baseline and remained 10cc/kg throughout the 48-hour course of the study for both groups.
BALF Data
Total protein concentration in the BALF was lower in the APRV group (p<0.05 vs. NPV) (Figure 2). BALF IL-6 levels at the end of the study were increased in the NPV group (p<0.05 vs. APRV) (Figure 2).
Figure 2. Bronchoalveolar Lavage Data.

Total Protein & IL-6. Bronchoalveolar lavage fluid (BALF) Total Protein (left sided vertical axis) and IL-6 (right sided vertical axis) concentrations. □= Airway Pressure Release Ventilation group (n=3) ■ = Non Preventative Ventilation group (n=5). Total protein and IL-6 concentrations are significantly higher in BALF of NPV (*=p<0.05 vs. APRV) reflecting greater pulmonary vascular permeability and lung inflammation.
Alveolar stability, one of two key components of the SAVE strategy, is dependent on surfactant composition and function. Surfactant composition was assessed by measuring surfactant proteins-A & -B (SP-A & SP-B) in BALF by western blot. SP-A was significantly lower in NPV (p<0.05 vs. APRV; Figure 3a). Three out of five NPV animals showed a fine monomer of SP-A protein; the remaining two did not show SP-A monomer band. All of the APRV animals exhibited SP-A monomer bands with higher density (p<0.05) (Figure 3a). SP-B protein expression in the BALF from NPV was markedly lower compared with APRV but not statistically significant (p=0.08) (Figure 3b). These data suggest degradation of both SP-A and SP-B occurred in the NPV animals.
Figure 3.


a & b. Western Blot of Surfactant Protein A and B in BALF Each animal is labeled by treatment group (APRV vs NPV). Figure 3a shows that APRV preserved Surfactant Protein A abundance relative to NPV (*=p<0.05).
b shows SP-B protein expression in the BAL fluid from NPV was markedly lower compared with APRV however this was not statistically significant (p=0.08).
The difference in W:D weight ratio of lungs between groups shows APRV significantly alters extravascular fluid handling in the lung. This occurs at least in part via greater expression of the Na, K-ATPase pump at the alveolar surface, which could facilitate fluid transport and effect a difference in pulmonary edema between groups. Na, K-ATPase was quantified in the cryopreserved lung tissue. Beta pump/actin expression in the NPV group was 1.0 ± 0.24, in the APRV group was 1.16 ± 0.16. Alpha pump/actin expression in the NPV group was 1.0 ± 0.19 in the APRV group it was 1.29 ± 0.21. Data are relative to the beta-actin loading control. There were no significant differences in the relative abundance of Na, K-ATPase between groups.
Gross Pathology
The lungs of NPV treated animals demonstrated severe inflammation, heterogeneous atelectasis, airway edema pouring from the bronchial openings, and hemorrhagic areas (Fig 4A). By comparison the APRV treated animals exhibited normal, pink, homogenously well-inflated lung tissue with no evidence of inflammation (Figure 4B). The cut surface of the representative APRV lung specimen shows no bronchial edema (Figure 4B).
Figure 4.
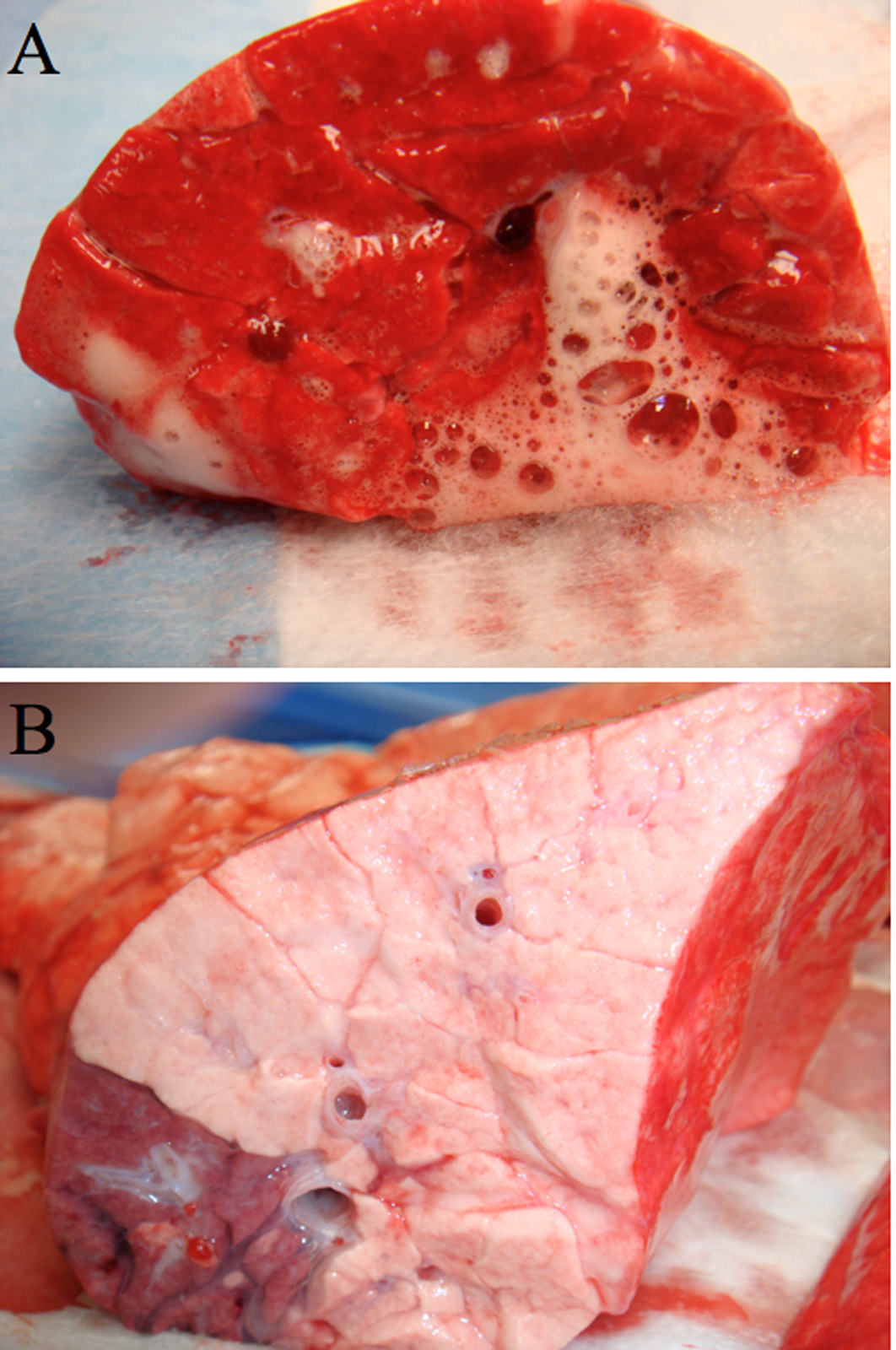
A & B: Gross Pathology- Cut Surface of right lower lobe of lung of representative animals from NPV group (4A) and APRV group (4B). NPV (A) shows severe inflammation, atelectasis, edema pouring from the bronchial openings, and hemorrhagic areas. APRV (B) shows normal pink, homogenously well-inflated lung tissue with no inflammation and no bronchial edema.
Quantitative & Descriptive Histology
The quantitative histological technique described in the methods section allows an unbiased comparison of degree of tissue injury to the lungs between the two treatment groups. The histological lesions analyzed are considered pathognomonic for ARDS when the clinical context is appropriate (1). These lesions are: atelectasis, fibrin deposits, capillary congestion, leukocyte infiltration, intra-alveolar hemorrhage, and alveolar wall thickness. Significant differences were observed between groups in all histological lesions except atelectasis. APRV resulted in 78% improvement in fibrinous exudates, 83% improvement in intra-alveolar hemorrhage, 61% improvement in capillary congestion, 83% improvement in alveolar wall thickness, and 33% improvement in leukocytic infiltration (p<0.0001 vs. NPV; SDC Table 1).
Table 1.
Quantitative Histology
| Lesion | NPV (N=40) | APRV (N=30) | t-Test |
|---|---|---|---|
| Atelectasis | 0.03 ± 0.02 | 0.00 ± 0.02 | |
| Fibrinous Deposits | 1.75 ± 0.16 | 0.4 ± 0.19 | § |
| Hemorrhage in air space | 1.88 ± 0.16 | 0.33 ± 0.19 | § |
| Capillary congestion | 2.15 ± 0.15 | 0.83 ± 0.17 | § |
| Thickened alveolar walls | 1.95 ± 0.14 | 0.33 ± 0.17 | § |
| Cellular infiltration | 2.93 ± 0.09 | 1.97 ± 0.11 | § |
=p<0.0001 following unpaired t-test.
Histological section of the lung of a representative NPV animal (Fig 5A) shows classic stigmata of ARDS atelectasis, fibrinous exudates, intra-alveolar hemorrhage, congested capillaries, thickened alveolar walls and leukocytic infiltrates. A histological section of the lung of a representative APRV animal (Figure 5B) shows preservation of normal pulmonary architecture and none of the lesions characteristic of ARDS. Notably, APRV animals exhibit prominent perivascular edema cuffs and dilated lymphatics as compared to NPV animals. Furthermore, APRV animals exhibit normal alveolar patency whereas NPV animals exhibit intra-alveolar fibrin as well as alveolar collapse.
Figure 5. Histology.
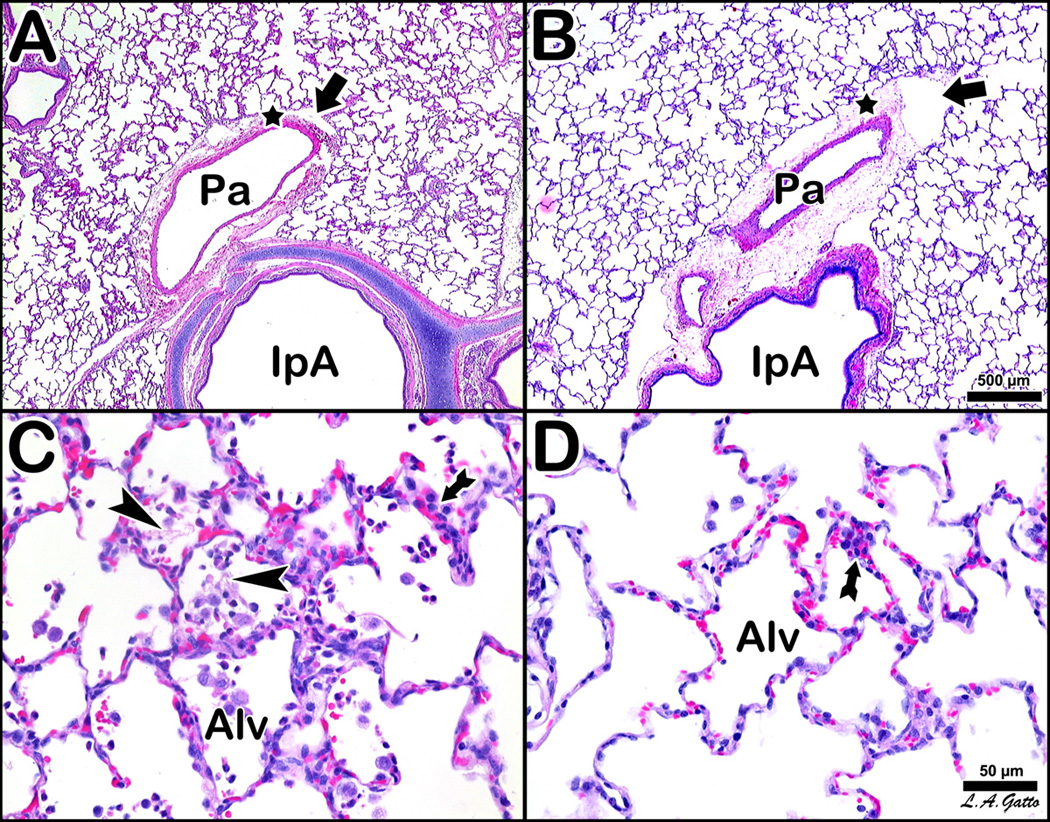
Histological comparison of 4 pigs, two NPV (A and C) vs. two APRV (B and D), at low magnification (A and B, bar=500 µm) and high magnification (C and D, bar=50 µm). The NPV animals (A and C) show classic stigmata of ARDS atelectasis, fibrinous exudates, intra-alveolar hemorrhage, congested capillaries, thickened alveolar walls and leukocytic infiltrates. The APRV animal (B and D) show preservation of nearly normal pulmonary architecture. The NPV pig at low magnification (A) shows an intrapulmonary airway (IpA) accompanied by a branch of the pulmonary artery (Pa), which exhibits an edematous cuff (star) with conspicuous lymphatic vessels (arrow). The APRV counterpart (B) is marked by a more substantial cuff with more prominent lymphatic vessels. Alveoli are notably more patent in APRV. The NPV pig at high magnification (C) shows lumina of alveoli (Alv) occupied by fibrin deposits (arrowheads) and wandering cells, while alveolar walls show pronounced cellular infiltration (arrow). APRV at high magnification (D) shows comparable cellular infiltration (arrow), although alveoli (Alv) are patent and clear.
DISCUSSION
The current study demonstrates SAVE ventilation in the form of APRV applied in an early preventative strategy successfully stops the development of ARDS in our clinically applicable lung injury model. To our knowledge this is the first study in which all ARDS-related lung injury was completely prevented by early preventative ventilatory intervention. SAVE intervention applied one hour after injury preserved oxygenation, lung compliance, and SP-A & SP-B abundance. SAVE also significantly reduced pulmonary edema, pulmonary vascular permeability (BALF Total Protein), lung inflammation (BALF IL-6), and histopathologic lung injury score. Lung function and architecture were so preserved by APRV they were similar to the values of our historic naive controls (22). The timeframe for manifestation of ARDS among NPV animals in this study was 39–42 hours, which is consistent with recent population data (8) (7)further demonstrating the fidelity of our experimental model to the human disease.
I. Prevention of ARDS
The most novel aspect of this approach is the concept that ARDS can be prevented using the mechanical ventilator. Prevention of any disease depends on a clear understanding of the natural history of the disease, identification of reversible early events in the pathogenesis, and modulation of those events prior to clinical manifestations (23). A classic example of this approach is the management of colorectal cancer. The understanding that colon cancer progresses from preclinical adenoma to carcinoma allowed evolution of routine screening colonoscopies in high-risk patients, thereby revolutionizing the management of colon cancer from treatment to prevention (24). Within the intensive care setting both thromboprophylaxis for venous thromboembolism (VTE), and stress ulcer prophylaxis are preventative maneuvers that significantly improved outcomes for these complications of critical illness (25). The present study provides compelling evidence ARDS, like VTE and gastric stress ulceration, may be preventable. Our finding ARDS is potentially preventable using an immediate post-injury SAVE strategy suggests progression of this disease can be attenuated in its preclinical pathogenesis. To take full therapeutic advantage of these findings we must depart from the binary concept of ARDS as either present or absent and begin to target the populations at risk for ARDS with early preventative ventilation strategies.
ARDS is notoriously difficult to treat once the diagnosis is made. The consistently poor clinical performance of various treatments for many decades suggests that the timing of intervention is too late in the pathogenesis of ARDS to succeed. Three recent reviews of the literature show that nearly all clinical trials in ARDS have had unimpressive outcomes over the past 30 years (2, 4, 26). Only low tidal volume ventilation received a relatively high recommendation; yet this standard-of-care intervention only reduced mortality by 9% (5). Since treatment of established ARDS is so difficult and has not proven efficacious over the years, we must investigate approaches that prevent this disease from developing. Recent data suggest in most cases ARDS develops while the patient is in the hospital. This finding offers physicians the opportunity to intervene earlier and prevent the disease (8). The present study builds on this compelling clinical data by suggesting that ARDS is in fact preventable by the early application of ventilation techniques that stabilize the alveolus and reduce pulmonary edema.
II. Alveolar Stability
Alveolar instability is an important driver of lung injury in the surfactant-depleted lung (10, 11, 13). Surfactant depletion happens early in ARDS onset as edema fluid leaks into the alveolus, disrupting the surfactant layer and deactivating surfactant (27). Surfactant depletion-induced alveolar instability causes atelectrauma with tidal ventilation, which is a significant contributor to alveolar shear stress and eventual ARDS (13). Therefore we designed SAVE to target alveolar instability directly, since blocking alveolar instability and atelectrauma could help prevent progression to ARDS.
The ventilator mode APRV was selected for the early SAVE strategy in our study. The specific technique of APRV used is critical as not all APRV settings fit the SAVE category. We postulate that the mechanisms of APRV’s success in this study are derived from airway pressure (P) sustained over a period of time (T), or the Pressure-Time Profile (PTP). Maintaining a prolonged PTP keeps the alveoli stable for a greater portion of the ventilatory cycle. In fact, the method of APRV used in this study maintains the Phigh for more than 90% of the respiratory cycle (16). These pulmonary mechanics support that APRV minimizes atelectrauma and stabilizes alveoli. The relative preservation of SP-A & SP-B in APRV vs. NPV (Figures 3a & 3b) further supports that APRV maintains alveolar stability.
III. Pulmonary Edema Reduction
Pulmonary edema from increased vascular permeability is the key pathophysiologic event associated with the development of ARDS (12, 27). APRV nearly eliminated accumulation of pulmonary edema as assessed by lung W:D weight ratio and also reduced lung vascular permeability as indicated by BALF Total Protein (Fig 2). These data suggest that lung distending pressure sustained over time (prolonged PTP) changes the dynamics of pulmonary fluid flux and reduces edema formation in the alveolar space. In contrast pulmonary edema accumulated in the NPV group (short PTP) as assessed by lung W:D weight ratio and vascular permeability was significantly elevated (Fig 2). The mechanisms for APRV-induced reduction in edema may include increasing interstitial hydrostatic pressure, reducing capillary transmural pressure, and reducing permeability surface area (28–30). A prolonged PTP may reverse the pressure gradient for edema flowing out of the capillaries and into the alveolus. Investigations in high-permeability lung injury have shown pressurization of the alveolus with PEEP can reduce permeability and thereby reduce extravascular lung water (29, 31), which corroborates our findings using PTP. Others have shown that a prolonged PEEP, effecting a long PTP, not only reduces extravascular lung water but also causes redistribution of edema from alveoli into perivascular cuffs (29–31). Qualitative histology in the present study revealed that animals treated with APRV showed normally inflated alveoli with edematous perivascular cuffs and interlobular septae corroborating the findings of Malo et al that prolonged PTP moves edema from the airways into extravascular cuffs (30).
The present study demonstrated that APRV-induced reduction of airspace edema was associated with a reduction in other downstream events associated with ARDS. Without airspace edema there is no disruption of surfactant, which prevents alveolar instability-induced atelectrauma, a process critical for the development of fulminant ARDS (10, 13, 27, 32–34). SAVE ventilation preserved SP-A and SP-B to near normal levels in the BALF of the APRV versus the near-total loss of SP-A & B in the NPV group suggesting improved surfactant function in the APRV animals. Thus, preventing alveolar flooding with SAVE eliminated the pathologic sequelae of airspace edema and speaks to the powerful potential of this novel strategy to reverse the progression to ARDS. Derailing the ARDS cascade early reinforces the importance of timing in interventions such as mechanical ventilation. Once alveolar edema and instability develop, the mechanical ventilator may propagate rather than prevent lung dysfunction (35).
IV. New Understandings of ARDS Onset
In the current paradigm ARDS pathogenesis begins after diagnostic criteria for ARDS have been met and has been categorized into three phases: 1) Exudative, 2) Fibroproliferative, and 3) Fibrotic repair and recovery (1). The success of early intervention in the present study has led us to conceptualize the onset of ARDS as a staged process (Figure 6). We hypothesize that there are three distinct Pre-ARDS stages based on the position of edema with respect to the airspace. Stage-1 (Occult-ARDS) is characterized by interstitial edema in alveolar walls and in vascular cuffs without alveolar flooding or clinical symptoms; this stage is reversible with SAVE. Stage-2 (Pseudo-ARDS), characterized by alveolar flooding with moderate surfactant deactivation that causes alveolar instability. All of the clinical symptoms of ARDS are present, except that hypoxemia is not refractory if SAVE is applied. Stage-3 (Fulminant-ARDS) is characterized by complete alveolar flooding with edema, total surfactant deactivation, and all diagnostic criteria of ARDS are met (5, 7, 26); in this stage, hypoxemia cannot be reversed with SAVE. We are currently investigating the pathophysiology of each stage and the mechanisms of SAVE-induced prevention of ARDS.
Figure 6. Hypothetical Staging of ARDS Onset.
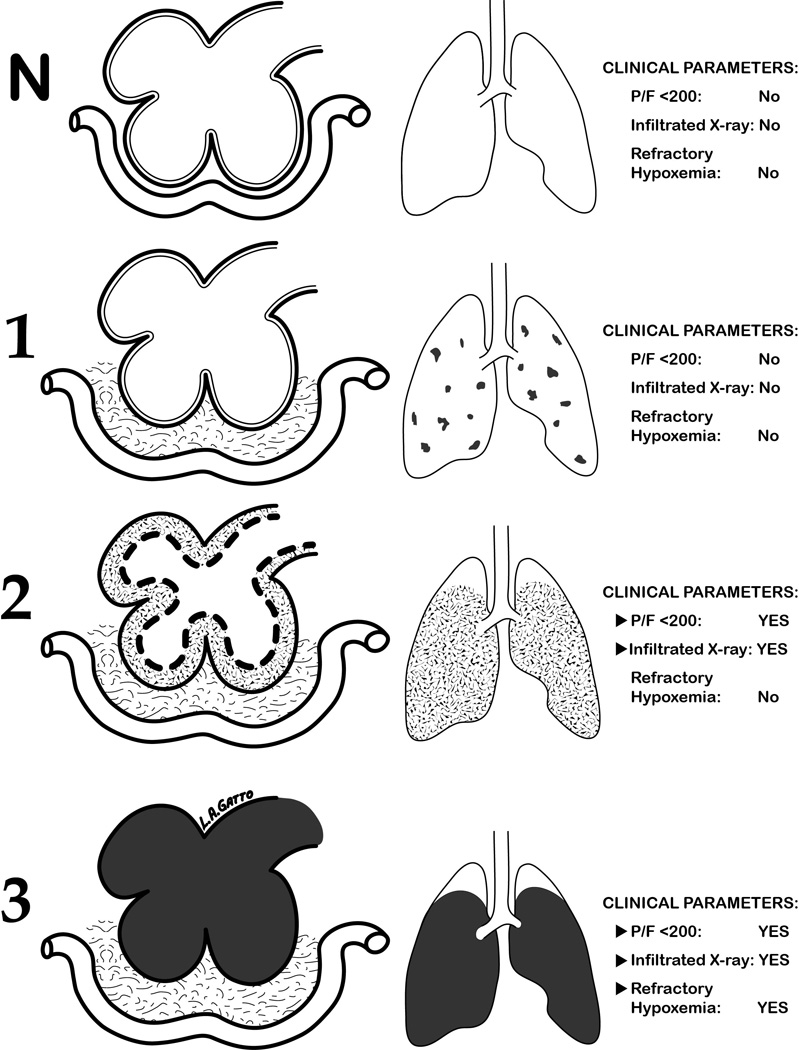
Diagram of ARDS development from Normal (N) to Fulminant ARDS (3) with clinical parameters at each Stage. Column 1 = diagram of alveoli, interstitial space and capillary; Column 2 = the percent of the entire lung that these lesions occupy; and Column 3 = the clinical presentation at each stage. N) Normal Alveoli no interstitial or alveolar edema; 1) Stage-1 (Occult-ARDS) interstitial edema in vascular cuffs (grey) without alveolar flooding or serious clinical symptoms; 2) Stage-2 (Pseudo-ARDS) interstitial edema (light grey) and partial flooding of alveoli (dark grey) with moderate surfactant deactivation (dotted lines) causing alveolar instability and hypoxemia. Pseudo-ARDS has all of the clinical features of Fulminant ARDS except hypoxemia is not refractory if SAVE ventilation is applied; and 3) Stage-3 (Fulminant-ARDS) interstitial edema (light grey) and complete alveolar flooding with edema (black) with total surfactant deactivation and all clinical features as defined by the ARDS consensus conference including refractory hypoxemia even if SAVE is applied. Stage-3 is the currently the only description of ARDS.
Summary
Since we were investigating a preventative intervention this study was designed and powered to detect a difference in ARDS incidence between groups as its primary outcome. Mortality was not an outcome detected by this study so while the APRV animals had a 100% survival compared to the NPV 40% survival rate this difference was not significant. Only 3 animals were included in the APRV group. Although this is a small number of animals all key parameters attained statistical significance as compared with the NPV group (0% ARDS incidence for APRV vs 100% for NPV) therefore the study was stopped to minimize the number of animals used, a key requirement of IACUC. The present study clearly demonstrates timing-based early ventilatory intervention with stabilizing alveolar ventilation and edema reduction (SAVE) strategy, in the form of APRV, completely prevents ARDS in a clinically relevant animal model. There are distinctly attractive aspects to this treatment strategy since APRV is readily available on most commercial ventilators and can be implemented with appropriate technique (16) as soon as patients are selected based on well-known risk factors for developing ARDS (8). While mechanistic details of this type of treatment regimen and of our proposed ARDS stages still need to be elucidated, we believe that the future of ARDS treatment can be best summed up by a recent quote from Villar and Slutsky, “ARDS is no longer a syndrome that must be treated, but is a syndrome that should be prevented (35).
Acknowledgements
The investigators would like to thank our patients and Oisin McGinty for teaching us what is possible when new thinking is applied to careful observation and Robert Cooney for his patience and guidance.
List of Abbreviations
- ARDS
Acute Respiratory Distress Syndrome
- LTV
Low Tidal Volume Ventilation
- SAVE
Stabilizing Alveolar Ventilation & Edema Reduction
- APRV
Airway Pressure Release Ventilation
- PS
Peritoneal Sepsis
- I/R
Ischemia Reperfusion
- NPV
Non preventative ventilation
- PEEP
Positive End Expiratory Pressure
- IL-6
Interleukin 6
- EGDT
Early Goal Directed Therapy
- ELISA
Enzyme-linked immunosorbent assay
- BALF
Bronchoalveolar Lavage Fluid
- SP-A
Surfactant Protein A
- SP-B
Surfactant Protein B
- ANOVA
Analysis of Variance
- SOFA
Sequential Organ Failure Assessment
- Ppeak
Peak Pressure
- Cstat
static compliance
- Pmean
Mean Airway Pressure
- Na, K-ATPase
Sodium, Potassium Adenosine Triphosphatase
- VTE
Venous Thromboembolism
- PTP
Pressure Time Profile
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This work will be presented at the annual EAST conference in 2012 as a finalist in the Raymond H. Alexander, MD Resident Paper Competition
Author Contacts & Contributions-
Shreyas Roy MD CM1- Corresponding Author, experimental design, experimentation, data collection & analysis, tissue analysis, literature review, manuscript composition and editing
Benjamin Sadowitz MD1(sadowitbd@upstate.edu)- experimental design, experimentation, data collection, tissue analysis, manuscript composition and editing
Penny Andrews2(Plandrews@intensivecareonline.com)- experimental design, experimentation, data collection, tissue analysis, manuscript composition and editing
Louis Gatto PhD3(louis.gatto@cortland.edu)- data analysis, tissue analysis, histological analysis & quantification, manuscript editing
William Marx DO4(marxw@upstate.edu)- experimental design, data analysis, literature review, manuscript editing
Lin Ge PhD1(GeL@upstate.edu)- data analysis, tissue analysis, BALF analysis, western blots of surfactant protein, IL-6 Data, total protein analysis, construction of figures, editing
Guirong Wang PhD1(wangg@upstate.edu)- experimental design, data analysis, tissue analysis, BALF analysis, western blots of surfactant protein, IL-6 Data, total protein analysis, construction of figures, editing
David Dean PhD5(David_Dean@urmc.rochester.edu)- experimental design, tissue analysis, sodium potassium adenosine triphosphatase data, editing
Xin Lin PhD5 - tissue analysis, sodium potassium adenosine triphosphatase data, editing
Auyon Ghosh BSc1(auyon.ghosh@gmail.com)- experimentation, data collection & analysis, statistical analysis
Michael Kuhn BA1(michael.kuhn315@gmail.com)- experimentation, data collection, data analysis
Joshua Satalin BA1(satalinj@upstate.edu)- administration of experiments, experimentation, data collection, tissue analysis, data analysis, statistics, construction of figures, editing
Kathy Snyder BA1(snyderk@upstate.edu)- experimental design, administration of experiments experimentation, data collection, tissue analysis, data analysis, editing
Yoram Vodovotz PhD6(vodovotzy@upmc.edu)- experimental design, literature review, editing
Gary Nieman BA1(niemang@upstate.edu)- experimental design, experimentation, data collection & analysis, tissue analysis, literature review, manuscript composition and editing
Nader Habashi MD2(nmhabashi@gmail.com)- experimental design, experimentation, data collection & analysis, tissue analysis, literature review, manuscript composition and editing
References
- 1.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342(18):1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 2.Cepkova M, Matthay MA. Pharmacotherapy of acute lung injury and the acute respiratory distress syndrome. J Intensive Care Med. 2006;21(3):119–143. doi: 10.1177/0885066606287045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herridge MS, Tansey CM, Matte A, Tomlinson G, Diaz-Granados N, Cooper A, et al. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med. 2011;364(14):1293–1304. doi: 10.1056/NEJMoa1011802. [DOI] [PubMed] [Google Scholar]
- 4.McIntyre RC, Jr, Pulido EJ, Bensard DD, Shames BD, Abraham E. Thirty years of clinical trials in acute respiratory distress syndrome. Crit Care Med. 2000;28(9):3314–3331. doi: 10.1097/00003246-200009000-00034. [DOI] [PubMed] [Google Scholar]
- 5.Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med. 2000;342(18):1301–1308. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- 6.Artigas A, Bernard GR, Carlet J, Dreyfuss D, Gattinoni L, Hudson L, et al. The American-European Consensus Conference on ARDS, part 2. Ventilatory, pharmacologic, supportive therapy, study design strategies and issues related to recovery and remodeling. Intensive Care Med. 1998;24(4):378–398. doi: 10.1007/s001340050585. [DOI] [PubMed] [Google Scholar]
- 7.Acute lung injury and the acute respiratory distress syndrome in Ireland: a prospective audit of epidemiology and management. Crit Care. 2008;12(1):R30. doi: 10.1186/cc6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shari G, Kojicic M, Li G, Cartin-Ceba R, Alvarez CT, Kashyap R, et al. Timing of the onset of acute respiratory distress syndrome: a population-based study. Respir Care. 2011;56(5):576–582. doi: 10.4187/respcare.00901. [DOI] [PubMed] [Google Scholar]
- 9.Navarrete-Navarro P, Rivera-Fernandez R, Rincon-Ferrari MD, Garcia-Delgado M, Munoz A, Jimenez JM, et al. Early markers of acute respiratory distress syndrome development in severe trauma patients. J Crit Care. 2006;21(3):253–258. doi: 10.1016/j.jcrc.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 10.Schiller HJ, McCann UG, 2nd, Carney DE, Gatto LA, Steinberg JM, Nieman GF. Altered alveolar mechanics in the acutely injured lung. Crit Care Med. 2001;29(5):1049–1055. doi: 10.1097/00003246-200105000-00036. [DOI] [PubMed] [Google Scholar]
- 11.Carney D, DiRocco J, Nieman G. Dynamic alveolar mechanics and ventilator-induced lung injury. Crit Care Med. 2005;33(3 Suppl):S122–S128. doi: 10.1097/01.ccm.0000155928.95341.bc. [DOI] [PubMed] [Google Scholar]
- 12.Ware LB. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Semin Respir Crit Care Med. 2006;27(4):337–349. doi: 10.1055/s-2006-948288. [DOI] [PubMed] [Google Scholar]
- 13.Otto CM, Markstaller K, Kajikawa O, Karmrodt J, Syring RS, Pfeiffer B, et al. Spatial and temporal heterogeneity of ventilator-associated lung injury after surfactant depletion. J Appl Physiol. 2008;104(5):1485–1494. doi: 10.1152/japplphysiol.01089.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kubiak B, Albert SP, Gatto LA, Vieau CJ, Roy SK, Snyder KP, Maier KG, Nieman GF. A Clinically Applicable Porcine Model of Septic and Ischemia/Reperfusion-Induced Shock and Multiple Organ Injury. Journal of Surgical Research. 2010;166(1):59–69. doi: 10.1016/j.jss.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kubiak B, Roy S, Albert S, Gatto L, Synder K, Vieau C, et al. COL-3 prevents histologic intestine damage in a clinically applicable porcine model of multiple organ dysfunction syndrome (MODS) Critical Care Medicine. 2009;37(12) [Google Scholar]
- 16.Habashi NM. Other approaches to open-lung ventilation: airway pressure release ventilation. Crit Care Med. 2005;33(3 Suppl):S228–S240. doi: 10.1097/01.ccm.0000155920.11893.37. [DOI] [PubMed] [Google Scholar]
- 17.Rivers E, Nguyen B, Havstad S, Ressler J, Muzzin A, Knoblich B, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345(19):1368–1377. doi: 10.1056/NEJMoa010307. [DOI] [PubMed] [Google Scholar]
- 18.Machado-Aranda D, Adir Y, Young JL, Briva A, Budinger GR, Yeldandi AV, et al. Gene transfer of the Na+,K+-ATPase beta1 subunit using electroporation increases lung liquid clearance. Am J Respir Crit Care Med. 2005;171(3):204–211. doi: 10.1164/rccm.200403-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang G, Taneva S, Keough KM, Floros J. Differential effects of human SP-A1 and SP-A2 variants on phospholipid monolayers containing surfactant protein B. Biochim Biophys Acta. 2007;1768(9):2060–2069. doi: 10.1016/j.bbamem.2007.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kubiak BD, Albert SP, Gatto LA, Snyder KP, Maier KG, Vieau CJ, et al. Peritoneal Negative Pressure Therapy Prevents Multiple Organ Injury in a Chronic Porcine Sepsis and Ischemia/Reperfusion Model. Shock. 2010;34(5):525–534. doi: 10.1097/SHK.0b013e3181e14cd2. [DOI] [PubMed] [Google Scholar]
- 21.Minne L, Abu-Hanna A, de Jonge E. Evaluation of SOFA-based models for predicting mortality in the ICU: A systematic review. Crit Care. 2008;12(6):R161. doi: 10.1186/cc7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steinberg J, Halter J, Schiller H, Gatto L, Carney D, Lee HM, et al. Chemically modified tetracycline prevents the development of septic shock and acute respiratory distress syndrome in a clinically applicable porcine model. Shock. 2005;24(4):348–356. doi: 10.1097/01.shk.0000180619.06317.2c. [DOI] [PubMed] [Google Scholar]
- 23.Gordon RS., Jr An operational classification of disease prevention. Public Health Rep. 1983;98(2):107–109. [PMC free article] [PubMed] [Google Scholar]
- 24.Muller AD, Sonnenberg A. Prevention of colorectal cancer by flexible endoscopy and polypectomy. A case-control study of 32,702 veterans. Ann Intern Med. 1995;123(12):904–910. doi: 10.7326/0003-4819-123-12-199512150-00002. [DOI] [PubMed] [Google Scholar]
- 25.Parrillo J. Critical Care Medicine: Principles of Diagnosis and Management in the Adult. Philadephia, PA: Mosby; 2008. [Google Scholar]
- 26.Brower RG, Fessler HE. Another "negative" trial of surfactant. Time to bury this idea? Am J Respir Crit Care Med. 2011;183(8):966–968. doi: 10.1164/rccm.201101-0018ED. [DOI] [PubMed] [Google Scholar]
- 27.Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;163(6):1376–1383. doi: 10.1164/ajrccm.163.6.2004035. [DOI] [PubMed] [Google Scholar]
- 28.Pare PD, Warriner B, Baile EM, Hogg JC. Redistribution of pulmonary extravascular water with positive end-expiratory pressure in canine pulmonary edema. Am Rev Respir Dis. 1983;127(5):590–593. doi: 10.1164/arrd.1983.127.5.590. [DOI] [PubMed] [Google Scholar]
- 29.Russell JA, Hoeffel J, Murray JF. Effect of different levels of positive end-expiratory pressure on lung water content. J Appl Physiol. 1982;53(1):9–15. doi: 10.1152/jappl.1982.53.1.9. [DOI] [PubMed] [Google Scholar]
- 30.Malo J, Ali J, Wood LD. How does positive end-expiratory pressure reduce intrapulmonary shunt in canine pulmonary edema? J Appl Physiol. 1984;57(4):1002–1010. doi: 10.1152/jappl.1984.57.4.1002. [DOI] [PubMed] [Google Scholar]
- 31.Ruiz-Bailen M, Fernandez-Mondejar E, Hurtado-Ruiz B, Colmenero-Ruiz M, Rivera-Fernandez R, Guerrero-Lopez F, et al. Immediate application of positive-end expiratory pressure is more effective than delayed positive-end expiratory pressure to reduce extravascular lung water. Crit Care Med. 1999;27(2):380–384. doi: 10.1097/00003246-199902000-00046. [DOI] [PubMed] [Google Scholar]
- 32.Imai Y, Parodo J, Kajikawa O, de Perrot M, Fischer S, Edwards V, et al. Injurious mechanical ventilation and end-organ epithelial cell apoptosis and organ dysfunction in an experimental model of acute respiratory distress syndrome. JAMA. 2003;289(16):2104–2112. doi: 10.1001/jama.289.16.2104. [DOI] [PubMed] [Google Scholar]
- 33.Slutsky AS. Lung injury caused by mechanical ventilation. Chest. 1999;116(1 Suppl):9S–15S. doi: 10.1378/chest.116.suppl_1.9s-a. [DOI] [PubMed] [Google Scholar]
- 34.Bredenberg CE, Nieman GF, Paskanik AM, Hart AK. Microvascular membrane permeability in high surface tension pulmonary edema. J Appl Physiol. 1986;60(1):253–259. doi: 10.1152/jappl.1986.60.1.253. [DOI] [PubMed] [Google Scholar]
- 35.Villar J, Slutsky AS. Is acute respiratory distress syndrome an iatrogenic disease? Crit Care. 2010;14(1):120. doi: 10.1186/cc8842. [DOI] [PMC free article] [PubMed] [Google Scholar]