

Abstract
Ideal reporter genes for temporal transcription programs have short half-lives that restrict their detection to the window in which their transcripts are present and translated. In an effort to meet this criterion for reporters of transcription in individual living cells, we adapted the ubiquitin fusion strategy for programmable N-end rule degradation to generate an N-degron version of green fluorescent protein (GFP) with a half-life of ~7 min. The GFP variant we used here (designated GFP*) has excellent fluorescence brightness and maturation properties, which make the destabilized reporter well suited for tracking the induction and attenuation kinetics of gene expression in living cells. These attributes are illustrated by its ability to track galactose and pheromone induced transcription in S. cerevisiae. We further show that the fluorescence measurements using the short-lived N-degron GFP* reporter gene accurately predict the transient mRNA profile of the prototypical pheromone induced FUS1 gene.
Keywords: Reporter genes, green fluorescent protein, N-end rule pathway, pheromone-induced transcription, galactose-regulated transcription
Introduction
Genetically encoded fluorescent reporter proteins have made it possible to investigate mechanisms controlling temporal and spatial responses to various environmental stimuli in individual living cells. To make these reporters more suitable for monitoring transient events, destabilized versions of fluorescent proteins have been generated using strategies based on fusions to destabilization domains or peptides that direct protein destruction by C-tail-specific proteases (Andersen, et al., 1998; Hackett, et al., 2006; Li, et al., 1998; Mateus and Avery, 2000). We previously exploited the ubiquitin fusion strategy for programmable N-end rule degradation developed by Varshavsky and colleagues (Bachmair, et al., 1986; Bachmair and Varshavsky, 1989; Gonda, et al., 1989; Park, et al., 1992; Suzuki and Varshavsky, 1999) to generate a family of cyan fluorescent protein (CFP) reporters with half-lives ranging from 75 to < 10 min (Hackett, et al., 2006). These “N-degron” CFP variants were engineered into convenient plasmid constructs with features to enable their expression from upstream activating sequences (UAS) of choice and to facilitate their targeted integration to the URA3-TIM9 intergenic region of Chromosome V (Hackett, et al., 2006). Using the pheromone inducible FUS1 UAS to drive expression, we provided proof that the short-lived N-degron CFP reporter can track a transient pheromone induced transcriptional response (Hackett, et al., 2006). Subsequently, we developed a mathematical model that takes into account the half-life and maturation time of the reporters so that actual transcript profiles can be inferred the from the fluorescence data (Wang, et al., 2008). Although the short-lived N-degron CFP reporter performed reasonably well, it is nevertheless limited in its utility because of its poor fluorescence intensity and the undesirable characteristic of a long delay between the emergence of the reporter protein and its detectable fluorescence.
With the intention of improving the performance of short-lived reporters, we applied the same N-degron strategy using a green fluorescent protein (GFP) variant, GFP* (GFP-F64L,S65T,V163A) (Harkins, et al., 2001). The S65T substitution alone results in a GFP derivative with six-fold greater brightness and four-fold faster maturation kinetics than the wild-type GFP from Aequorea victoria (Heim, et al., 1995). The additional mutations in GFP* were incorporated to further improve brightness and confer thermostability (Harkins, et al., 2001; Straight, et al., 1998; Cormack et al., 1996). N-degron GFP* expression driven from the carbon source-regulated GAL1 promoter was used to assess intrinsic characteristics of the reporter including half-live and time to detect protein and fluorescence. The N-degron GFP* proteins were also expressed under control of the pheromone-induced FUS1 promoter to assess their suitability as reporters of transient transcription.
Materials and methods
Recombinant DNA procedures and plasmid constructions
Bacterial transformations, bacterial DNA preparation, and DNA restriction enzyme digestions were performed by standard methods (Sambrook, et al., 1989). Plasmids used in these studies are listed in Table 1. Those not previously reported are described below.
Table 1.
Plasmids
| Plasmid | Description | Source |
|---|---|---|
| YEpGFP*-BUD8 | GFP*-BUD8 in YEplac181 | (Harkins, et al., 2001) |
| pFA6a GFP(S65T) His3MX6 |
GFP(S65T)-His3MX6 in pFA6a | (Longtine, et al., 1998) |
| pNC824 | FUS1-UBIY in pUC118 | (Hackett, et al., 2006) |
| pNC820 | FUS1-UBIM in pUC118 | (Hackett, et al., 2006) |
| apNC951 | URA3-GAL1-UBIYdkCFP-SpHis5-TIM9 in pUC118 | (Hackett, et al., 2006) |
| apNC952 | URA3-GAL1-UBIMdkCFP-SpHis5-TIM9 in pUC118 | (Hackett, et al., 2006) |
| bpNC1011 | 3 tandem copies of GFP* in pFA6a His3MX6 | See text |
| pNC1124 | URA3-GAL1-UBIYdkGFP*-SpHis5-TIM9 in pUC118 | See text |
| pNC1125 | URA3-GAL1-UBIMdkGFP*-SpHis5-TIM9 in pUC118 | See text |
| pNC1136 | FUS1-UBIYdkGFP*-SpHis5-TIM9 in pUC118 | See text |
| pNC1137 | FUS1-UBIMdkGFP*-SpHis5-TIM9 in pUC118 | See text |
The selectable marker in the CFP cassette plasmids described in Hackett et al. was incorrectly specified as His3MX6. The sequence is that of S. pombe His5 (SpHis5).
GFP* sequence encodes GFP with amino acid substitutions F64L, S65T, and V163A . GFP*also has a silent substitution (CAT to CAC) at the H77 codon that destroys the an NdeI restriction site.
The plasmid pNC1011 has 3 tandem copies of GFP* (F64L, S65T, V163A) that replace the single copy of GFP(S65T) in pFA6a GFP(S65T)-His3MX6 (Harkins, et al., 2001; Longtine, et al., 1998). In addition to the specified amino acid substitutions, the GFP* variant has a silent substitution of the His77 codon (CAT to CAC) that destroys an NdeI restriction enzyme recognition site. pNC1011 was constructed by the following series of manipulations. The GFP* coding region flanked by different restriction enzyme sites was amplified using the plasmid YEpGFP*-BUD8 (Harkins, et al., 2001) as template and oligonucleotide primer pairs 737(PacI)/738(BglII), 739(BamHI)/738(BglII), and 739(BamHI)/740(TAG-AscI) (Table 2). The three PCR products were cloned into pCRTopoII (Life Technologies, Grand Island, NY) to generate pNC1000, pNC1003 and pNC1006, respectively. The BamHI-BglII GFP* fragment from pNC1006 was ligated to BglII digested pNC1000 to generate pNC1009, which carries two tandem in frame copies of GFP* joined by a BglII/BamHI fusion junction. A 778 bp BamHI fragment of pNC1003 encompasses a copy of GFP* that includes a TAG stop codon. This fragment was ligated to BglII digested pNC1009 to add the terminal in frame copy of GFP* in the plasmid pNC1010. A 2162 bp PacI-AscI fragment that has the three tandem copies of GFP* from pNC1010 was ligated to the 4046 bp PacI-AscI pFA6a backbone of pFA6a GFP(S65T) His3MX6 to generate pNC1011 (Longtine, et al., 1998).
Table 2.
Oligonucleotides
| Oligo | Sequence (5′ to 3′) | Purpose |
|---|---|---|
| 737 |
TTA ATT AAC ATG AGT AAA GGA GAA GAA C PacI |
Forward primer for PCR amplification of GFP* |
| 738 | GCC GCA AGA TCT TTT GTA TAG TTC BglII |
Reverse primer for PCR amplification of GFP* |
| 739 |
GGA TCC ATG AGT AAA GGA GAA GAA C BamHI |
Forward primer for PCR amplification of GFP* |
| 740 |
GGC GCG CCC TAT TTG TAT AGT TCA TCC ATG CC AscI |
Reverse primer for PCR amplification of GFP* |
| 899 | GCC GTT GGA CGG TTC TTA TG | Forward qPCR primer for GAL1 mRNA |
| 900 | GAC CAG TCC GAC ACA GAA GGA T | Reverse qPCR primer for GAL1 mRNA |
| 952 | TCA GTG GAG AGG GTG AAG GT | Forward qPCR primer for GFP mRNA |
| 953 | GTT GGC CAT GGA ACA GGT AG | Reverse qPCR primer for GFP mRNA |
| 1002 | GCG TCC AAT TAG GGA AGA CA | Forward qPCR primer for FUS1 mRNA |
| 1003 | AAC TTT TTC ACC CAG CGA GA | Reverse qPCR primer for FUS1 mRNA |
| 1004 | GAG GTT GCT GCT TTG GTT ATT GA | Forward qPCR primer for ACT1 mRNA |
| 1005 | ACC GGC TTT ACA CAT ACC AGA AC | Reverse qPCR primer for ACT1 mRNA |
The plasmids pNC1124 and pNC1125 have the UBIYΔkGFP* and UBIMΔkGFP* alleles, respectively, under control of the GAL1 promoter. The two plasmids were constructed by ligating the 1176 bp NcoI fragment from pNC1011 to the 6425 bp NcoI fragment of pNC951 and pNC952, respectively. This manipulation serves to replace the segment of the fluorescent protein with CFP specific amino acids with those for GFP*. The resulting plasmids have the His5 gene from Schizosaccharomyces pombe (SpHis5) as a selectable marker for Saccharomyces cerevisiae his3 mutant strains and flanking sequences that allow targeted integration of the GAL1 driven reporters to the URA3-TIM9 intergenic region of Chromosome V.
The plasmids pNC1136 and pNC1137 are the same as pNC1124 and pNC1125 except they have the FUS1 promoter driving the UBIYΔkGFP* and UBIMΔkGFP* reporter genes, respectively. The GAL1 -UBIY or GAL1-UBIM cassette region of pNC1136 and pNC1137 was replaced with the 676 bp XhoI-BamHI FUS1-UBIY or FUS1-UBIM cassette from pNC824 or pNC820, respectively, by ligating the isolated fragments to the 6297 bp XhoI-BamHI fragment of pNC1124.
Yeast genetic procedures, strains and culture conditions
Unless otherwise specified, yeast growth media and genetic manipulations were as described in Amberg, Burke, and Strathern (Amberg, et al., 2005). Yeast transformations and targeted integrations were done using standard procedures (Gietz, et al., 1995; Rothstein, 1983). All integrations were confirmed by PCR analysis of genomic DNA.
Strains used in these analyses are C699-181 (GAL1-UBIMΔkGFP*), C699-183 (PGAL1-UBI-YΔkGFP*), C699-198 (FUS1-UBIMΔkGFP*), and C699-199 (PFUS1-UBI-YΔkGFP*). These four strains were derived by integrating the SacI-SalI reporter cassette from pNC1125, pNC1124, pNC1137 and pNC1136, respectively, at the URA3-TIM9 intergenic region of Chromosome V of strain C699-94 (MATa ade2-1 bar1Δ::hisG can1-100 his3-11,15 LEU2 trp1-1 ura3-1) (Hackett, et al., 2006). For all experiments, these strains were grown in liquid cultures at 30°C using synthetic complete medium supplemented with 40 mg/ L adenine sulfate. The specific strains and manipulation of carbon sources for analysis of reporter degradation rates or induction profiles are described below.
To characterize the intrinsic properties of MΔkGFP* and YΔkGFP*, we exploited strains C699-181 and C699-183 in which the reporter alleles are regulated by the galactose inducible and glucose repressible GAL1 UAS. To characterize galactose induction profiles, the strains were inoculated to a density of 1x106 cells/ml in synthetic complete medium containing 3% (v/v) glycerol, 1% (v/v) ethanol, and 0.2% (w/v) glucose. After an overnight incubation, cultures deplete the glucose in the medium, adapt to utilization of glycerol/ethanol and reach early log phase density (~2 × 107 cell/ml and at least 60% budded). A sample was removed from the culture for determination of uninduced reporter gene expression. After addition of galactose (2% w/v, final concentration) to the remaining culture, samples were removed at indicated times for analysis of the mRNA, protein, and fluorescence induction profiles. For half-life determinations, the strains were grown in synthetic complete medium with 2% galactose (w/v) as the sole carbon source to a density of 1-2 × 107 cells/ml. Samples were removed for determination of the steady-state amounts of mRNA, protein, or fluorescence (t=0). After inhibition of further transcription from the GAL1 UAS by the addition of glucose (2% w/v, final concentration) to the remaining cultures, degradation kinetics of mRNA and protein were determined from analysis of samples removed at specified time intervals.
To monitor pheromone induction profiles we used strains C699-198 and C699-199 that express MΔkGFP* and YΔkGFP*, respectively, from the pheromone inducible FUS1 UAS. The strains were grown in synthetic complete medium with 2% glucose as the carbon source to a density of 5 – 7 × 106 cells/ ml. Samples were removed for determination of basal reporter gene mRNA and fluorescence. After addition of mating pheromone α-factor (1 μM final concentration) to the remaining culture, samples were removed at indicated times for the analysis of mRNA and fluorescence profiles.
RNA preparation and quantification
10 ml samples were removed from cultures immediately before and at indicated times after inhibition (addition of glucose) or induction (addition of galactose) of GAL1 UAS transcription by changing carbon source or induction of FUS1 UAS transcription by addition of mating pheromone. Cells were collected from these samples by vacuum filtration on to filters (0.45 μm HAWG, Millipore). The filters were placed in 1.5 ml eppendorf tubes, cooled in a dry ice/ethanol bath and stored at −80°C until all samples were collected for RNA extraction.
Total RNA was extracted from each sample using a modified version of the hot acidic phenol-chloroform method developed by Collart and Oliviero (Collart and Oliviero, 2001). After adding 400 μL of TES buffer [10 mM Tris-Cl (pH 7.5), 10 mM EDTA, 0.4% SDS, 4% RNAsecure (Ambion, Inc)] to each tube, the samples were briefly vortexed and centrifuged to separate the suspended cells from the filters, which were then discarded. 400 μL of phenol pH 4.5 (MP) was added to each cell suspension. Samples were vortexed vigorously, incubated at 65°C for 1 hour with intermittent vortexing, and then placed on ice for 5 min. Each mixture was applied to a phase lock gel tube (5 Prime) to separate the aqueous and organic phases by centrifugation. The aqueous phase was removed to a clean phase lock tube and extracted with an equal volume of chloroform. The aqueous phase was transferred to a clean eppendorf tube and ethanol precipitated. The precipitated RNA samples were suspended in 100 μl RNase-free water. The quality and concentration of the RNA for subsequent real-time quantitative PCR (qPCR) analysis was assessed based on 260nm/280nm and 260nm /230nm absorbance ratios.
First-strand cDNA synthesis for qPCR analysis was generated from total RNA using the SuperScript® III First-Strand Synthesis System (Invitrogen). qPCR reactions using 1/20 of total cDNA as template were completed using primers specific to FUS1 mRNA, GAL1 mRNA or GFP* mRNA and the ACT1 reference mRNA (Table 2). Changes in gene expression relative to ACT1 were assessed using the 2-ΔΔCT method of analysis (Livak and Schmittgen, 2001). To validate application of this method, we determined that ΔCT for the different primer pairs was essentially constant using serial dilutions of the cDNA template (from 3 - 0.1 the amount in standard reactions). qPCR reactions were carried out in triplicate for each cDNA sample using SYBR GreenER qPCR supermix (Invitrogen) and the Applied Biosystems 7900HT Fast Real-Time PCR system. Reported values for amounts of the different mRNA species are the average of determinations made on samples from three or more independent cultures.
Protein extract preparation and immune blotting conditions
10 ml samples were removed from cultures as described for mRNA preparation. Whole-cell protein extracts were prepared using the method of Mattison et al. (Mattison, et al., 1999). Protein concentrations were determined by the BCA assay using the BCA assay kit from Pierce. Equal amounts of protein (40 μg for the MΔkGFP* reporter strains and 60 μg for the YΔkGFP* reporter strains) were loaded for each sample and fractionated on 12% SDS-PAGE gels. The fractionated proteins were transferred to PVDF membranes. Each membrane was divided into two portions at the 43 kDa molecular size marker. GFP* was detected on the lower molecular size portion of the membrane by using anti-GFP monoclonal antibody (1:1000, Living Colors) with goat anti-mouse-IgG-HRP conjugated secondary antibody (1:10,000; Jackson Immuno Research). As an internal loading reference, yeast α-tubulin was detected on the higher molecular size portion of the membrane by using rat anti-tubulin primary antibody (1:200; Accurate Chemicals) with rabbit anti-rat IgG-HRP-conjugated secondary antibody (1:10,000; Jackson Immuno Research). Immunoreactive species were detected by ECL plus and were exposed to X-ray film. Signal intensities for GFP* and tubulin were quantified from scanned blots using NIH Image J software.
Fluorescence microscopy and imaging analysis
Agarose slabs were made by heating up 4% agarose in synthetic complete medium and allowing it to cool on glass slides. ~10 μL of the culture were placed onto the cooled agarose and allowed to settle for several minutes without completely drying out. To measure fluorescence, 100 msec exposures where taken of four different z-axis positions centered on the cell.For capturing fluorescent images we used an epifluorescent microscope (Nikon TE 2000 inverted microscope) using a Hammatsu OrcaII Monochrome camera. The resulting image stack was projected onto a single plane by adding up the contribution from the individual planes. We then subtracted the background from the projection using the standard algorithm background subtraction in ImageJ (http://rsbweb.nih.gov/ij/). We measured the total fluorescence and area of the cell by manually bounding the cell and adding up all pixels inside those bounds. The mean fluorescence value for each sample was then calculated by averaging the amount of fluorescence in at least 25 individual cells for each individual repeat.
We assessed the extent of photobleaching by imaging fluorescence of GAL1-UBIMΔkGFP* (C699-181) and GAL1-UBIYΔkGFP* (C699-183) cells that were grown in synthetic complete medium with 2% galactose (w/v) as the sole carbon source to allow maximal steady state reporter gene expression. Images of cells on agarose slabs where taken one after another as quickly as possible for a total of fifty images and fluorescence intensities determined as before. These measurements gave us an upper bound of photobleaching for both MΔkGFP* and YΔkGFP* of < 0.2% per image for our microscope settings. This rate puts the upper bound for total photobleaching throughout our experiment to be <2.6%. However the total photobleaching in our experiments with YΔkGFP* is likely less than this as the shorted-lived reporter has a high turnover rate (~1/7 min) and we only take pictures every 15 min. Autofluorescence was accounted for by quantifying the fluorescence of cells not expressing GFP* (parental strain C699-94) and subtracting that average value from the GFP* expressing cells (strain C699-181, C699-183, C699-198, or C699-199).
Model fitting and mRNA back-prediction
Here we describe our method of back-predicting the FUS1 mRNA profile from the fluorescence data of the GFP* reporter. Our approach is similar to that of Wang et al. (Wang, et al., 2008) except we assume that oxidation of GFP* is rate limiting as has been empirically determined by measurements on several GFP variants (Miyawaki, et al., 2003). This assumption allows us to use a quasi-equilibrium approach to reduce the number of equations and parameters needed for fitting. We justify the simplification based on comparison of galactose induction profiles for fluorescence and protein (see results).
The temporal profile of the GFP reporter is related to mRNA profile by:
| (1) |
where k is the combined rate of translation and maturation and Kdeg is the GFP* protein degradation rate. In all fitting, we held Kdeg fixed at the measured value corresponding to a half-life of 7 min. Any arbitrary system of ordinary differential equations with linear reaction rates can be solved by a weighted sum of exponentials, whose arguments are the eigenvalues. Of course it is possible that the system contains highly nonlinear reaction rates, however, this method is a good first approximation. Thus the equation for the mRNA is of the form
| (2) |
Where the λn and bn parameters are similar to the eigenvalues and the eigenvalue weights of the mRNA induction, respectively. The parameter b0 is set by the initial condition. With this functional form for the mRNA profile, equation 1 has a straightforward, analytical, solution. We then estimated the free parameters by performing a nonlinear least squares fit to this analytical solution of the GFP* profile to our data on the YΔkGFP* (short-lived reporter) pheromone induction profile driven by the FUS1 promoter. We found that the free parameters were constrained best when we used, at most, 2 exponentials to describe the mRNA profile. Our estimate of the parameters were b0 = − 808, b1 = 818, b2 = −8.8, λ1 = 2.9E-5 min−1, λ2 = 0.1 min−1 (note the parameters bn are unit-less). We also estimated the uncertainty in back-predicting the mRNA profile that is due to the variability of the observed GFP* reporter profile. To define the range we added and subtracted the standard error of the GFP* reporter profile to the mean of the same profile. We then fit our model to the result, and estimated the profile and parameters of the +/-1 standard error (max/min) mRNA induction time course. For the max profile the estimated parameters where b0 = -579, b1 = 589, b2 = −6, λ1 = 3.9E-5 min−1, λ2 = 0.1 min−1. For the min profile the estimated parameters where b0 = −930, b1 = 938, b2 = −10, λ1 =2.4E-5 min−1, λ2 =0.1 min−1. For all fitting we used the Matlab (MathWorks) built in function “lsqnonlin”.
Results
Reporter gene construction strategy
We exploited the previously described ubiuqitin-N-degron tagging strategy to express green fluorescent protein (GFP) reporters that have different half-lives (Hackett, et al., 2006). The GFP* protein (GFP-F64L,S65T,V163A) we chose for these reporters is a rapidly maturing and bright amino acid substitution variant of GFP from Aequorea victoria (Harkins, et al., 2001). The S65T substitution confers six-fold greater brightness and four-fold faster maturation kinetics compared with the wild-type GFP (Heim, et al., 1995). The two additional substitutions found in GFP* improve brightness compared with the wild-type or S65T variant at temperatures above 28° C (Harkins, et al., 2001; Straight, et al., 1998; Cormack et al., 1996). The oxidative step in fluorescence maturation is known to be rate limiting (Miyawaki, et al., 2003). We infer de novo fluorescence development for GFP* is rapid (14-16 min) based on the times reported for GFP-S65T (16 min), EGFP-F64L,S65T (14 min) and GFPuv3-F64L, S65T, F99S, M153T, V163A (15 min) (Iizuka, et al., 2011).
The N-degron is a bipartite sequence that becomes exposed at the N-terminus because the ubiquitin moiety is cleaved off the nacent fusion protein by the deubiquitinating enzymes during translation. This exposed tag targets proteins to the proteasome for destruction with efficiencies that are determined by the identity of the bipartite signal (Bachmair, et al., 1986; Bachmair and Varshavsky, 1989; Gonda, et al., 1989; Park, et al., 1992; Suzuki and Varshavsky, 1999; Varshavsky, 1996). The N-terminal amino acid is the primary determinant of this tag. In the two reporters described here, MΔkGFP* and YΔkGFP*, the N-terminal Met (M) is stabilizing whereas Tyr (Y) is destabilizing (Varshavsky, 1996). The second determinant of the N-degron is the presence of one or more lysine residues that are stericly positioned for conjugation to ubiquitin. The number, positioning and context of the lysine residues directly influence the efficiency of ubiquitination by the N-end rule pathway ubiquitin ligase, Ubr1, and consequently degradation by the proteasome (Bachmair and Varshavsky, 1989; Suzuki and Varshavsky, 1999). The Δk sequence fused to GFP* in these reporters is a flexible linker of 23 amino acids with a lysine motif that satisfies this requirement (Bachmair and Varshavsky, 1989; Hackett, et al., 2006; Suzuki and Varshavsky, 1999).
To construct UBI-MΔkGFP* and UBI-YΔkGFP* reporters expressed from either the GAL1 or FUS1 upstream activating sequence (UAS), we inserted different UAS-UBI-[X] cassettes into the KpnI-BamHI cloning site in the [Δk]-GFP* targeting cassette (Fig. 1) (Hackett, et al., 2006). The UAS cassettes carry the UBI4 coding sequence followed by an engineered codon (X= ATG, Met; or X =TAT, Tyr) which specifies the primary determinant of N-degron for end rule pathway recognition. The [Δk] sequence needed to complete the N-degron tag is fused to GFP* in the targeting cassette. This cassette includes the Schizosaccharomyces pombe His5 gene (SpHis5) as a selectable marker and flanking sequences that target the reporters for integration at Chromosome V of S. cerevisiae (Fig. 1). Notably, any UAS of interest can be cloned into the unique BamHI and KpnI or XhoI sites of the UAS cassettes described previously (Hackett, et al., 2006). This feature makes these tools easily adaptable as transcriptional reporters.
Figure 1.
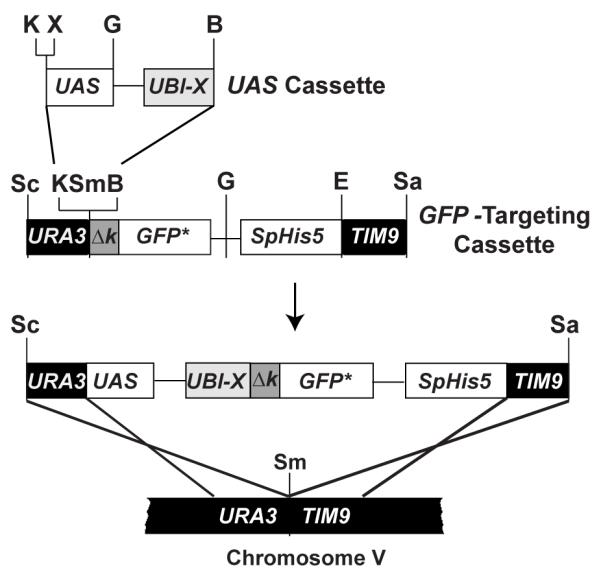
Strategy for N-degron GFP* reporter gene construction and integration into the S. cerevisiae genome. Plasmids with different UAS-UBI-X-ΔkGFP* reporters are constructed by combining UAS and GFP targeting cassettes. The X-Δk element of the reporter gene represents different bipartite N-degron signal sequences (see text). Digestion of the reporter gene plasmid with SacI and SalI restriction endonucleases releases the reporter gene from the plasmid backbone and generates free ends that target the allele to the URA3-TIM9 intergenic region of chromosome V. Unique restriction enzyme sites in the different cassettes are shown (B, BamHI; G, BglII; E, EcoRV; K, KpnI; Sa, SalI; Sc, SacI; Sm, SmaI; X, XhoI.)
Comparison of galactose induction profiles for MΔkGFP* and YΔkGFP* reporter alleles
To estimate the time differential between mRNA synthesis and detection of protein and fluorescence for the reporters, we monitored their galactose induction profiles in strains expressing the PGAL1-UBI-M ΔkGFP* (C699-181) or PGAL1-UBI-YΔkGFP* (C699-183) allele (Fig. 2). Cultures were grown to early log phase in medium with a non-inducing carbon source. Galactose then was added to induce transcription of the reporter gene. Samples of the liquid cultures were removed for either RNA or protein extract preparation before (t=0) and at indicated times after addition of galactose. RNA samples were treated as described in Materials and Methods for quantification of time dependent galactose induction of UBI-MΔkGFP* or UBI-YΔkGFP* mRNA relative to the non-induced ACT1 mRNA by qPCR. Protein extracts were fractionated by SDS-PAGE and transferred to PVDF membranes for quantification of MΔkGFP* or YΔkGFP* proteins relative to a tubulin loading control using western blot methods as described in Materials and Methods. To compare fluorescence induction profiles for MΔkGFP* and YΔkGFP*, cells were taken from the uniniduced (t=0) and induced cultures at indicated times and spread on microscope slides holding a thin layer of agarose medium. Images of different fields were captured for quantification of fluorescence, as detailed in Materials and Methods.
Figure 2.

Galactose induction kinetics for detection of reporter gene mRNA, protein, and fluorescence. (A & B) Plots comparing galactose induction profiles for GFP* mRNA ( ), protein (
), protein ( ), and fluorescence (
), and fluorescence ( ) in strains with the PGAL1-UBI-MΔkGFP* (C699-181) or PGAL1-UBI-YΔkGFP* (C699-183) reporters, as specified. The data points for mRNA and protein are the average values from four and three independent induction time courses, respectively. The data points for fluorescence are the average of the mean fluorescence from four independent induction time courses. (The mean fluorescence value at each time in given time course is determined from quantification of at least 25 cells.) Error bars show 95% confidence limits calculated from the independent time course determinations.
) in strains with the PGAL1-UBI-MΔkGFP* (C699-181) or PGAL1-UBI-YΔkGFP* (C699-183) reporters, as specified. The data points for mRNA and protein are the average values from four and three independent induction time courses, respectively. The data points for fluorescence are the average of the mean fluorescence from four independent induction time courses. (The mean fluorescence value at each time in given time course is determined from quantification of at least 25 cells.) Error bars show 95% confidence limits calculated from the independent time course determinations.
The plots comparing mRNA, fluorescence, and protein induction profiles for MΔkGFP* and YΔkGFP* point out several important features of this system (Fig. 2A & 2B). The inherent difference in the short-lived and stable reporters influences whether basal signal may be amplified or underestimated, respectively. In this example, the short-lived YΔkGFP* reporter protein and fluorescence is undetectable under non-inducing conditions, whereas the stable MΔkGFP* reporter accumulates sufficiently to have a measurable basal signal (compare t=0, Fig. 2A & 2B). It is also evident that GFP* makes the fluorescence detection method more sensitive than the ECL-Plus western blot detection system used here to monitor protein accumulation. This difference in sensitivity is evident for MΔkGFP* at early times when protein abundance is still low (Fig. 2A). It is also evident for YΔkGFP* throughout the profile because absolute protein amounts remain low (Fig. 2B). As a consequence, the protein profile for the short-lived reporter is shifted to the right compared with that for the stable reporter. By contrast, the induction profiles based on relative fluorescence are the same within experimental error for the two reporters. Thus the destabilized and stable reporters function equally well for monitoring induction profiles. Fluorescence induction for the reporters is shifted toward later times compared with mRNA induction profiles. The extent offset is influenced by the threshold for fluorescence detection and the effective maturation time that includes the time for translation of the mRNA, protein folding, and oxidative steps that develop the fluorophore.
Comparison of MΔkGFP* and YΔkGFP* half-lives
To determine the half-lives for the reporter mRNA and protein, cultures of strains expressing either the PGAL1-UBI-MΔkGFP* (C699-181) or PGAL1-UBI-YΔkGFP* (C699-183) allele were grown in galactose medium to induce their steady state expression. Cultures were then switched to glucose medium, which immediately inhibits further transcription (Johnston, et al., 1994). Samples of the cultures were removed before (t=0) and at indicated times after addition of glucose to prepare mRNA and protein extracts and to mount cells for fluorescence microscopy. In contrast to half-life determinations made in the presence of general translation inhibitors, such as cycloheximide, the strategy used here measures rates of degradation under normal growth conditions. Quantification of amounts of reporter mRNA, protein, and fluorescence was done as described for the galactose induction profiles (above). Plots comparing the exponential decay of these species for MΔkGFP* and YΔkGFP* are shown in Figs. 3A & 3B, respectively. The half-lives extrapolated from these data are summarized in Table 3. The half-life of the UBI-X- ΔkGFP* mRNA (~ 7 min) was the same for both alleles, but is somewhat longer than for the reference GAL1 mRNA (~4 min). The half-life of the stable MdkGFP* protein is approximately 10-times longer than for the destabilized YdkGFP* protein whether calculated from data collected by western-blot or fluorescence methods. Because the YdkGFP* species is undetectable 10 min after inhibition of transcription, the time available for making measurements is constrained to the period where mRNA has not yet been depleted from the cells. In this situation the measured half-life reflects the dynamics between the translation and degradation.
Figure 3.

Degradation kinetics of reporter gene mRNA, protein, and fluorescence. (A & B) Plots show the exponential decay of GFP* mRNA ( ) and protein quantified by Western blot (
) and protein quantified by Western blot ( ) or fluorescence (
) or fluorescence ( ) in strains with the GAL1-UBI-MΔkGFP* (C699-181) or GAL1-UBI-YΔkGFP* (C699-183) reporters, as specified. Amounts of each species are relative to steady state amounts before inhibition of transcription (t=0) by the addition of glucose to the cultures. Data points shown in each plot are the average of N independent experiments as specified in Table 3. Error bars show 95% confidence limits.
) in strains with the GAL1-UBI-MΔkGFP* (C699-181) or GAL1-UBI-YΔkGFP* (C699-183) reporters, as specified. Amounts of each species are relative to steady state amounts before inhibition of transcription (t=0) by the addition of glucose to the cultures. Data points shown in each plot are the average of N independent experiments as specified in Table 3. Error bars show 95% confidence limits.
Table 3.
Half-life of GAL1,10-UBI-X-GFP Reporters.
| Strain | Allele | Species | Detection Method |
Half-life ± StDev (min) |
N |
|---|---|---|---|---|---|
| C699-181 | GAL1,10-UBIMdkGFP | mRNA | qPCR | 7 | 1 |
| Protein | Western | 71 ± 6 | 4 | ||
| Protein | Fluorescence | 84 ± 7 | 3 | ||
| C699-183 | GAL1,10-UBIYdkGFP | mRNA | qPCR | 7 ± 1 | 3 |
| Protein | Western | 8 ± 2 | 4 | ||
| Protein | Fluorescence | 7 ± 3 | 3 | ||
| C699-183 C699-184 |
GAL1 | mRNA | qPCR | 4 ± 0.4 | 4 |
Assessment of MdkGFP* and YdkGFP* as reporters of transient gene expression
Yeast respond to mating pheromone by inducing a transient transcription program (Roberts, et al., 2000). FUS1 encodes a membrane protein that is localized to the tips of mating projections and is required for cell fusion between mating partners. Its expression is strongly induced by pheromone and serves as a standard indicator for signal dependent transcription in yeast. Therefore, the FUS1 UAS was exploited to examine fluorescent proteins as reporters of transient transcription. Mating pheromone α-factor was added to cultures of strains expressing either the PFUS1-UBI-MΔkGFP* (C699-198) or PFUS1-UBI-YΔkGFP* (C699-199) allele. Samples of the cultures were removed before (t=0) and at indicated times after pheromone addition to prepare mRNA for quantification by qPCR and to mount cells for microscopy for fluorescence determination as before.
As expected, FUS1 mRNA is rapidly and transiently induced with a peak occurring between 0-30 min (Fig. 4A). Because of our limited early sampling times, we use the 15 min time point as the apparent time at which maximal mRNA expression occurs. FUS1 mRNA abundance then declines and stabilizes at ~50-60% of the maximal amount. The GFP* mRNA profiles expressed from the 45 bp FUS1 UAS for both UBI-MΔkGFP* and UBI-YΔkGFP* are indistinguishable from each other and the endogenous FUS1 (Fig. 4A). The pheromone induction kinetics of GFP* fluorescence is similar for stable (MΔkGFP*) and destabilized (YΔkGFP*) reporters, yet only the destabilized GFP* reveals attenuation that is characteristic of pheromone induced mRNAs (Fig. 4B). The mRNA and fluorescence profiles for the YΔkGFP* are similar in that both reveal attenuation of expression that declines to 50-60% of the maximum. However, because of the delay in fluorescence detection compared with mRNA, other details are different. Notably, the mRNA profile has a sharp peak at 15 min and the fluorescence profile has a delayed and broad peak spanning 45-90 min.
Figure 4.

Comparison of pheromone induction profiles for FUS1 and reporter gene expression. (A) Plot comparing pheromone induction profiles for FUS1 ( ) and GFP* (
) and GFP* ( ) mRNA in PFUS1-UBI-MΔkGFP* (C699-198) and PFUS1-UBI-YΔkGFP* (C699-199) strains. Data points are the average from six independent induction time courses (three with strain C699-198 and three with strain C699-199). Error bars show 95% confidence limits. (B) Plot comparing the pheromone induction profile for GFP* fluorescence in the PFUS1-UBI-MΔkGFP* (C699-198) (
) mRNA in PFUS1-UBI-MΔkGFP* (C699-198) and PFUS1-UBI-YΔkGFP* (C699-199) strains. Data points are the average from six independent induction time courses (three with strain C699-198 and three with strain C699-199). Error bars show 95% confidence limits. (B) Plot comparing the pheromone induction profile for GFP* fluorescence in the PFUS1-UBI-MΔkGFP* (C699-198) ( ) and PFUS1-UBI-YΔkGFP* (C699-199)
) and PFUS1-UBI-YΔkGFP* (C699-199)  reporter strains. More than 25 or 50 individual cells were scored for each time point in a single time course with strain C699-198 or C699-199, respectively. Each data point is the average from four independent time courses. Error bars show 95% confidence limits. (C) Model prediction of the pheromone induced FUS1 mRNA profile using YΔkGFP* fluorescence measurements as input. Plot compares the model mean (
reporter strains. More than 25 or 50 individual cells were scored for each time point in a single time course with strain C699-198 or C699-199, respectively. Each data point is the average from four independent time courses. Error bars show 95% confidence limits. (C) Model prediction of the pheromone induced FUS1 mRNA profile using YΔkGFP* fluorescence measurements as input. Plot compares the model mean ( ) and range (min, max
) and range (min, max  ) to the empirically determined reporter GFP* mRNA profile
) to the empirically determined reporter GFP* mRNA profile  from (A).
from (A).
Here we show that despite the delay in detection for fluorescence compared with mRNA, the induction profile of the destabilized GFP* is indicative of the corresponding mRNA profile. Previously some of us reported on a way to back predict the mRNA from a shorted-lived cyan fluorescent protein reporter (Wang, et al., 2008). Here we apply that same strategy to the destabilized YΔkGFP* reporter under control of the FUS1 UAS. Figure 4C shows the predicted mRNA profile (solid black line) compared to the measured FUS1 UAS dependent GFP* mRNA profile (cirlces). The uncertainty in the PFUS1-UBI-YΔkGFP* pheromone induction profile corresponds to a range of values that the fluorescence could take. We used the maximum and minimum range, set by the uncertainty in fluorescence measurements, to estimate the maximum and minimum range of mRNA levels that could explain our data (Fig. 4C, grey lines). The mRNA profile was modeled as a sum of exponentials so that the resulting dynamical equation for the GFP* could be solved analytically and the resulting solution could be fit to the data using a nonlinear least squares method (see methods section for details). Figure 4C demonstrates the success of back-predicting the mRNA profile from the fluorescence.
Discussion
Theoretical and computational analyses of mathematical models designed to investigate biological responses to various environmental stimuli are providing deeper and often non-intuitive understanding of the underlying regulatory systems. These studies often rely on measurements of changes in gene expression in individual living cells as they respond to stimuli in real time. Fluorescent proteins are obvious tools for generating the data used to evaluate and validate model predictions. To be faithful as reporters of temporal transcription programs, it is important that the fluorescent protein has fast maturation and turnover kinetics. These two qualities make the window in which they are detected more closely match that of transcript profiles.
We previously provided proof of principle that flexibility in fluorescent protein reporter stabilities could be realized by exploiting the N-end rule pathway for programmable protein degradation (Hackett, et al., 2006). This pathway targets proteins to the proteasome for their destruction with efficiencies that are determined by the identity of a bipartite “N-degron” signal (Bachmair, et al., 1986; Bachmair and Varshavsky, 1989; Gonda, et al., 1989; Park, et al., 1992; Varshavsky, 1996). This first generation of N-degron reporters was based on cyan fluorescent protein (CFP). CFP has a slow functional maturation time (~ 2 hr), which encompasses the time it takes for translation, protein folding, and fluorophore development. This slow maturation and the low fluorescence intensity of CFP severely limited the utility of this first generation of N-degron fluorescent reporters.
The MΔkGFP* (stable) and YΔkGFP* (destabilized) reporters described here are derivatives of a fast maturing and thermostable version of GFP (F64L, S65T, V163A). GFP* has a far better fluorescence intensity than CFP at the optimal growth temperature (30°C) for S. cerevisiae. This attribute improves detection sensitivity, which is especially advantageous for the destabilized reporter. It is noteworthy that fluorescence of the GFP* reporters proved to be more sensitive for detection of the reporter gene product than did the commonly used ECL-Plus western blot system. The brightness and faster maturation of GFP* compared with CFP also allows the destabilized YΔkGFP* reporter to track induction kinetics equally well as the more abundant stable MΔkGFP* reporter. In addition, the destabilized reporter revealed the transient profile characteristic of pheromone induced transcription that was otherwise masked by the accumulation of the longer-lived protein. These characteristics of YΔkGFP* fluorescence make this reporter especially well-suited for tracking the temporal dynamics of transcription. In support of this claim, we applied a mathematical approach that successfully back-predicted the pheromone induced mRNA profile from the fluorescence data of the YΔkGFP* reporter. The approach we used is similar to that of Wang et al. (Wang, et al., 2008) except that the rapid maturation of GFP* required only information about the half-life of the reporter and allowed us to use a quasi-equilibrium approach to reduce the number of equations and parameters needed for fitting. Because the N-end rule pathway is evolutionarily conserved, the utility of this strategy for generating families of short-lived fluorescent reporter proteins and application of the mathematical approach to predict mRNA profiles should be generally applicable (Gonda, et al., 1989).
There has also been great interest in determining the origins and consequences of variability in gene expression observed from isogenic cell populations. This variability arises from two general sources: “intrinsic noise”, which is due to the inherent random nature of the biochemical processes necessary for expression of a particular gene, and “extrinsic noise”, which affects all genes (Elowitz, et al., 2002). Many of these investigations relied on stable fluorescent proteins as reporters of transcriptional activity (Blake, et al., 2003; Colman-Lerner, et al., 2005; Elowitz, et al., 2002; McCullagh, et al., 2010; Raser and O’Shea, 2004). Therefore, the fluorescent reporters are expressed at levels greater than many endogenous proteins, which have mRNA and protein half-lives of 15 min or shorter. When either the maturation or the turnover rate of the reporter is slow compared with the endogenous proteins, the reporter acts as a low pass filter and obscures intrinsic fluctuations that contribute to variability in gene expression.
The programmable features of the N-degron tagging approach expand the range of half-lives that is possible with fluorescent reporter proteins. This flexibility, in principle, allows investigators to match the decay rates of the reporters to the proteins in the system that they are designed to track and thereby provide a more accurate picture of the dynamic properties of the system under investigation.
Acknowledgements
We thank Drs. L.R. Schenkman and J.R. Pringle (Stanford University, Palo Alto CA) for the plasmid pYEPGFP*-BUD8. We also thank Drs. J. Kelley and M. Pena for critical reading of the manuscript. This work was supported by NIH Grants GM 079271 and GM 084071.
References
- Amberg DC, Burke DJ, Strathern JN. Methods in Yeast Genetics. Cold Spring Harbor Lab. Press: Cold Spring Harbor; New York: 2005. [Google Scholar]
- Andersen JB, Sternberg C, Poulsen LK, Bjorn SP, Givskov M, Molin S. New unstable variants of green fluorescent protein for studies of transient gene expression in bacteria. Appl Environ Microbiol. 1998;64:2240–6. doi: 10.1128/aem.64.6.2240-2246.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmair A, Finley D, Varshavsky A. In vivo half-life of a protein is a function of its amino-terminal residue. Science. 1986;234:179–86. doi: 10.1126/science.3018930. [DOI] [PubMed] [Google Scholar]
- Bachmair A, Varshavsky A. The degradation signal in a short-lived protein. Cell. 1989;56:1019–32. doi: 10.1016/0092-8674(89)90635-1. [DOI] [PubMed] [Google Scholar]
- Blake WJ, M KA, Cantor CR, Collins JJ. Noise in eukaryotic gene expression. Nature. 2003;422:633–7. doi: 10.1038/nature01546. [DOI] [PubMed] [Google Scholar]
- Collart MA, Oliviero S. Preparation of yeast RNA. In: Ausubel Frederick M., et al., editors. Current protocols in molecular biology. 2001. Chapter 13, Unit13 12. [DOI] [PubMed] [Google Scholar]
- Colman-Lerner A, Gordon A, Serra E, Chin T, Resnekov O, Endy D, Pesce CG, Brent R. Regulated cell-to-cell variation in a cell-fate decision system. Nature. 2005;437:699–706. doi: 10.1038/nature03998. [DOI] [PubMed] [Google Scholar]
- Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP) Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic gene expression in a single cell. Science. 2002;297:1183–6. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH, Willems AR, Woods RA. Studies on the transformation of intact yeast cells by the LiAc/SS- DNA/PEG procedure. Yeast. 1995;11:355–60. doi: 10.1002/yea.320110408. [DOI] [PubMed] [Google Scholar]
- Gonda DK, Bachmair A, Wunning I, Tobias JW, Lane WS, Varshavsky A. Universality and structure of the N-end rule. J Biol Chem. 1989;264:16700–12. [PubMed] [Google Scholar]
- Hackett EA, Esch RK, Maleri S, Errede B. A family of destabilized cyan fluorescent proteins as transcriptional reporters in S. cerevisiae. Yeast. 2006;23:333–49. doi: 10.1002/yea.1358. [DOI] [PubMed] [Google Scholar]
- Harkins HA, Page N, Schenkman LR, De Virgilio C, Shaw S, Bussey H, Pringle JR. Bud8p and Bud9p, proteins that may mark the sites for bipolar budding in yeast. Molecular biology of the cell. 2001;12:2497–518. doi: 10.1091/mbc.12.8.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim R, Cubitt AB, Tsien RY. Improved green fluorescence. Nature. 1995;373:663–4. doi: 10.1038/373663b0. [DOI] [PubMed] [Google Scholar]
- Iizuka R, Yamagishi-Shirasaki M, Funatsu T. Kinetic study of de novo chromophore maturation of fluorescent proteins. Analytical biochemistry. 2011;414:173–8. doi: 10.1016/j.ab.2011.03.036. [DOI] [PubMed] [Google Scholar]
- Johnston M, Flick JS, Pexton T. Multiple mechanisms provide rapid and stringent glucose repression of GAL gene expression in Saccharomyces cerevisiae. Mol Cell Biol. 1994;14:3834–41. doi: 10.1128/mcb.14.6.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhao X, Fang Y, Jiang X, Duong T, Fan C, Huang CC, Kain SR. Generation of destabilized green fluorescent protein as a transcription reporter. J Biol Chem. 1998;273:34970–5. doi: 10.1074/jbc.273.52.34970. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–61. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Mateus C, Avery SV. Destabilized green fluorescent protein for monitoring dynamic changes in yeast gene expression with flow cytometry. Yeast. 2000;16:1313–23. doi: 10.1002/1097-0061(200010)16:14<1313::AID-YEA626>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Mattison CP, Spencer SS, Kresge KA, Lee J, Ota IM. Differential regulation of the cell wall integrity mitogen-activated protein kinase pathway in budding yeast by the protein tyrosine phosphatases Ptp2 and Ptp3. Mol Cell Biol. 1999;19:7651–60. doi: 10.1128/mcb.19.11.7651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullagh E, Seshan A, El-Samad H, Madhani HD. Coordinate control of gene expression noise and interchromosomal interactions in a MAP kinase pathway. Nature Cell Biology. 2010;12:954–62. doi: 10.1038/ncb2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki A, Nagai T, Mizuno H. Mechanisms of protein fluorophore formation and engineering. Curr Opin Chem Biol. 2003;7:557–62. doi: 10.1016/s1367-5931(03)00097-8. [DOI] [PubMed] [Google Scholar]
- Park EC, Finley D, Szostak JW. A strategy for the generation of conditional mutations by protein destabilization. Proc Natl Acad Sci U S A. 1992;89:1249–52. doi: 10.1073/pnas.89.4.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raser JM, O’Shea EK. Control of stochasticity in eukaryotic gene expression. Science. 2004;304:1811–4. doi: 10.1126/science.1098641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts CJ, Nelson B, Marton MJ, Stoughton R, Meyer MR, Bennett HA, He YD, Dai H, Walker WL, Hughes TR, Tyers M, Boone C, Friend SH. Signaling and circuitry of multiple MAPK pathways revealed by a matrix of global gene expression profiles. Science. 2000;287:873–80. doi: 10.1126/science.287.5454.873. [DOI] [PubMed] [Google Scholar]
- Rothstein RJ. One-step gene disruption in yeast. Methods Enzymol. 1983;101:202–11. doi: 10.1016/0076-6879(83)01015-0. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Lab. Press; Plainview NY: 1989. [Google Scholar]
- Straight AF, Sedat JW, Murray AW. Time-lapse microscopy reveals unique roles for kinesins during anaphase in budding yeast. The Journal of cell biology. 1998;143:687–94. doi: 10.1083/jcb.143.3.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Varshavsky A. Degradation signals in the lysine-asparagine sequence space. Embo J. 1999;18:6017–26. doi: 10.1093/emboj/18.21.6017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A. The N-end rule: functions, mysteries, uses. Proc Natl Acad Sci U S A. 1996;93:12142–9. doi: 10.1073/pnas.93.22.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Errede B, Elston TC. Mathematical analysis and quantification of fluorescent proteins as transcriptional reporters. Biophysical journal. 2008;94:2017–26. doi: 10.1529/biophysj.107.122200. [DOI] [PMC free article] [PubMed] [Google Scholar]