

Abstract
High-fat feeding or CNS leptin overexpression in chow-fed rats results in a region-specific cellular leptin resistance in medial basal hypothalamic regions and the ventral tegmental area (VTA). The present investigation examined the effects of targeted chronic leptin overexpression in the VTA as compared with the medial basal hypothalamus on long-term body weight homeostasis. The study also examined if this targeted intervention conserves regional leptin sensitivity or results in localized leptin resistance. Cellular leptin resistance was assessed by leptin-stimulated phosphorylation of signal transducers and activators of transcription 3 (STAT3). Tyrosine hydroxylase was measured in hypothalamus and VTA along with brown adipose tissue uncoupling protein 1. Leptin overexpression in VTA tempered HF-induced obesity, but to a slightly lesser extent than that with leptin overexpression in the hypothalamus. Moreover, the overexpression of leptin in the VTA stimulated cellular STAT3 phosphorylation in several regions of the medial basal hypothalamus, whereas verexpression in the hypothalamus did not activate STAT3 signaling in the VTA. This unidirectional trans-stimulation did not appear to involve migration of either the vector or the gene product. Long-term leptin overexpression in either the medial basal hypothalamus or VTA caused desensitization of leptin signaling in the treated region and cross-desensitization of leptin signaling in the untreated region. These results demonstrate a role of leptin receptors in the VTA in long-term body weight regulation, but the trans-activation of the hypothalamus following VTA leptin stimulation suggests that an integrative response involving both brain regions may account for the observed physiological outcomes.
1. Introduction
The adipocyte-derived hormone, leptin, regulates appetite and energy expenditure through its action in the hypothalamus and other brain sites (Li, 2011). The arcuate nucleus (ARC) of the hypothalamus within the forebrain and the ventral tegmental area (VTA) within the midbrain are two regions responsive to leptin stimulation (Cota et al., 2006). Whereas leptin action in hypothalamic regions has long been the subject of investigation, identification of the physiological function of leptin in the VTA midbrain reward circuitry has been more recent. Knockdown of leptin receptors in the midbrain demonstrated a role for leptin in reward-based feeding without discernable effect on body weight (Davis et al., 2011), which questions the importance of leptin function in the VTA in long-term body weight homeostasis. However, direct leptin injection into the VTA induces short-term decreases in food consumption and body weight (Bruijnzeel et al., 2011). The long-term consequences of direct leptin stimulation of the VTA are unknown.
Augmentation of leptin in the central nervous system (CNS) via chronic overexpression of leptin induces impressive anorexia and weight loss (Scarpace et al., 2002a, 2002b). However, these leptin physiological responses appear to be biphasic. There is an initial leptin sensitive phase during which rapid reductions in food intake and body weight occur (Scarpace et al., 2002a, 2002b) and a second desensitizing phase, in which the responses to leptin wane over time. Eventually, the food intake and body weight of the leptin-treated animals revert to those of controls. Notably, rats at this point fail to respond to exogenously administered leptin and display exacerbated diet-induced obesity (DIO) upon exposure to a high-fat (HF) diet (Scarpace et al., 2005). This second period is considered a leptin-induced leptin resistant phase and characterized by a diminished leptin receptor signaling to centrally administered leptin (Scarpace and Zhang, 2009). This signaling impairment is often referred to as cellular leptin resistance, and the loss of leptin-mediated STAT3 phosphorylation serves as one marker of this resistance (Scarpace and Zhang, 2009).
We recently reported that either HF-feeding or chronic leptin overexpression in the CNS in chow-fed rats induces cellular leptin resistance in multiple regions within the hypothalamus as well as in the VTA (Matheny et al., 2011). Leptin impinges upon a distributed neural network to regulate energy balance. Our previous studies involving central leptin gene delivery (leptin overexpression) into the third ventricle produce leptin elevation in the CNS (Scarpace et al., 2002b). Therefore, the observed leptin physiological effects or the occurrence of leptin-induced leptin resistance are likely an integration of leptin activity in many, if not all brain regions bearing functional leptin receptors. It is not known currently if leptin function in an individual CNS site such as the ARC or VTA is dissociable from that of other regions and how or if leptin resistance develops when leptin overexpression is targeted to a specific region rather than the entire CNS.
The purpose of the present investigation is to determine the role of chronic leptin overexpression targeted to the VTA as compared with leptin overexpression targeted to the medial basal hypothalamus (MBH) on long-term body weight homeostasis, and if this targeted intervention conserves regional leptin sensitivity or results in localized leptin resistance. To this end, we overexpressed leptin in the VTA or MBH for either 9 or 54 days via delivery of a recombinant adeno-associated viral (rAAV) vector encoding rat leptin. Subsequently, we assessed signaling by analysis of STAT3 phosphorylation in multiple brain regions, including the VTA, arcuate nucleus (ARC), lateral hypothalamus (LH), ventromedial hypothalamus (VMH), and dorsomedial hypothalamus (DMH) as well as the fate of the expressed transgene message and the protein product.
2. Materials and methods
2.1. Experimental animals
Three-month-old male F344 × Brown Norway (F344×BN) rats were obtained from Harlan Sprague-Dawley (Indianapolis, IN). Upon arrival, rats were examined and remained in quarantine for one week. Animals were cared for in accordance with the principles of the Guide to the Care and Use of Experimental Animals and protocols were approved by the University of Florida Institutional Animal Care and Use Committee. Rats were housed individually with a 12:12 h light-dark cycle (07:00 to 19:00 hr). During the experimental period, rats were fed either a standard rodent chow (17%kcal from fat, no sucrose, 3.3kcal/g, diet 2018, Harlan Teklad; Madison, WI) or a HF diet (60% kcal from fat, 7% kcal from sucrose, 5.24 kcal/g, D12492, Research Diets, New Brunswick, NJ).
2.2. Experimental design
This study consists of two experiments. In both experiments, rats were administered either recombinant adeno-associated virus (rAAV)-leptin or control vector by injection into the VTA or MBH for either 54 days (Experiment 1, N=8-14/group) or 9 days (Experiment 2, N=8/group). Rats were allowed free access to food and water, ad libitum, and food consumption and body weight were recorded daily to weekly. In Experiment 1, prior to death, at day 54, those treated with control vector were further divided into two groups and administered by intracerebroventricular (i.c.v.) injection either artificial cerebral spinal fluid (ACSF, N=6) or leptin (1ug, N=8), whereas those treated with rAAV-leptin were administered leptin (1ug) to determine leptin signaling in various brain regions.
Whole body adiposity was assessed on selected days by time domain nuclear magnetic resonance (TD-NMR) using a Minispec lean fat analyzer (Bruker Optics, Inc., The Woodlands, TX). Validation of TDNMR methodology has been provided (Tinsley et al., 2004).
2.3. rAAV-vector administration
A single dose (5 ×1012 viral genomes/ml) of control vector encoding green fluorescent protein (GFP, 1 μl) was unilaterally delivered by injection; in half of the control animals, the injections were directed into hypothalamus, targeting the MBH, and in the other half, the injections targeted the VTA. In parallel, half the experimental animals received an equivalent dose of rAAV-leptin (1 μl) delivered by injection into the MBH (n=8-9) and the other half into the VTA (n=8-9). The coordinates for the injection targeting the MBH were 2mm posterior to Bregma, 0.4mm lateral from the midsagittal suture and 9.8mm ventral from the surface of the skull. The coordinates for the injection targeting the VTA were 5.3mm posterior to Bregma, 0.8mm lateral from the midsagittal suture and 8.5mm ventral from the surface of the skull. Stereotaxic coordinates for injection into the brain were determined using the Rat Brain Atlas (Paxinos and Watson, 2005). Coordinates were verified and refined by use of a water soluble blue dye and visualization in brain sections using low-power scopy. Additional verification of the accuracy of the vector delivery was performed by injections of rAAV-GFP into either the MBH or the VTA followed by fluorescence imaging of fixed brain sections.
2.4. Leptin administration
A single dose of leptin (1 μg) was injected into the third cerebral ventricle as previously described (Scarpace et al., 2007). The coordinates for injection are 1.3mm anterior to Bregma, 9.4mm ventral from the skull surface, at an angle of 20 degrees anterior to posterior. Rats were killed one hour later to assess leptin-mediated STAT3 signaling.
2.5. Tissue harvest and preparation
Rats were killed by thoracotomy under 150-mg/kg pentobarbital anesthetic. Subsequently, 40 ml of cold saline were perfused through the circulatory system. The perirenal and retroperitoneal white adipose depots (PWAT and RTWAT, respectively) and interscapular brown adipose tissue (BAT) were each excised and their individual weights recorded.
Additionally, 2 mm coronal sections containing the regions of the VTA and MBH were sliced from fresh brains using a meter controlled tissue slicer (Stoelting Co, Wood Dale, II.) and a punch of the respective regions were taken as subsequently described. Aligning a straight edge razor blade with the optic tract (−1.5 mm posterior bregma), a 2 mm caudal coronal section was cut. Hypothalamic tissue punches (ARC, VMH, and DMH) were removed under a low power scope that provides identification of landmarks found in each 2mm tissue slice. For the ARC, the 1mm punch was placed just lateral to the 3rd ventricle and approximately ½ of one millimeter from the ventral most border of the brain slice. The punches were then moved progressively dorsal on the border of the 3rd ventricle, providing those for VMH and DMH. Similarly, aligning a straight edge razor blade with the caudal end of the hypothalamus (−5 mm posterior to bregma), a coronal section was cut 2 mm posterior. For the 2mm section containing the VTA, a 1mm diameter punch was centered ventral to the red nucleus, parvicellular (RPC) and medial to the border of the substantia nigra (SN). All punches were taken bilaterally. Brain punches and BAT were briefly sonicated in 40 ul 10 mM Tris, pH 6.8, 2% SDS for Western analysis (Experiment 1) or in 10 mM Tris, pH 6.8 for leptin peptide assessment (experiment 2). The remaining lysate was spiked with 22% SDS to a final concentration of 2% SDS for subsequent Western analysis. Protein was determined by DC Bradford (Bio-Rad, Hercules, CA).
2.6 Leptin transgene expression and protein production
In a separate set of rats, RNA was extracted from VTA and ARC punches using Tri reagent (Sigma, St. Louis, MO). Leptin transgene expression in various brain regions was determined by RT-PCR using sense (TGACACCAAAACCCTCATCA) and antisence primers (TGAGCTATCTGCAGCACGTT) as described previously (Scarpace et al., 2002b).
Leptin peptide levels in VTA and ARC brain regions were assessed in tissue punch lysates by ELISA. (Crystal Chem Inc, Downers Grove, IL.)
Serum leptin levels were determined by radioimmunoassay (Millipore, Billerica, MA).
2.7. Western analysis of STAT3 and TH
Protein homogenate (20μg, P-STAT; 0.5 μg, TH) was separated on a SDS-PAGE gel and electro-transferred to nitrocellulose membranes (Scarpace et al., 2001). Immunoreactivity was assessed with antibodies specific to phospho-tyrosine 705 of STAT3, and reprobed with antibodies specific to STAT3 regardless of phosphorylation state (Cell Signaling, Danvers, MA). Immunoreactivity to P-STAT3 was compared with that for beta-tubulin (Abcam, Cambridge, MA). For TH, Immunoreactivity was assessed with antibodies specific to TH (Pel-Freeze, Rogers, AR).
2.8. Imaging analysis of MBH and VTA
Separate rats received rAAV-GFP injection in either the MBH or VTA. Four weeks after virus injections, animals were killed and perfused with 100 ml of isotonic saline, followed by 200 ml of ice-cold 4% paraformaldehyde in 0.1 M phosphate buffer (PB), pH 7.4. Brains were fixed with 4% paraformaldehyde for 4–5 h with gentle shaking at 4°C, and cryo-preserved in 20% sucrose overnight at 4°C. Brains were frozen in 2-methylbutane at −55°C, sect ioned on a cryostat at 40 μm, and thaw mounted on Superfrost Plus glass slides (Fisher Scientific, Pittsburgh, PA). The slides were stored at −20°C until use. For imaging, the SuperfrostPlus sli des were secured onto standard glass plates, air-dried, and a cover slip placed with Vectashield mounting medium (Vector, Burlingame, CA). The fluorescence was recorded by a motorized Nikon scope (Tokyo, Japan) equipped with a Nikon DS digital camera. Subsequently, MBH sections were counterstained for NeuN and immunofluorescence recorded, whereas VAT sections were stained with an antibody to TH and immunofluorescence detected.
2.9. Statistical analysis
Data were analyzed by one-way ANOVA with repeated measures when appropriate or by two-way ANOVA with repeated measures. A post-hoc test (Bonferroni) was applied to determine individual differences between means. A p-value of less that 0.05 was considered significant.
3. Results
3.1. Body weight and food consumption over 54 days
Prior to surgery, rats consumed an average of 58.2 ± 7 kcal/day laboratory chow, and there was the expected surgery-related decrease in food intake (Fig 1, top, days 1-4). For clarity, the rats administered control vector either into the hypothalamus or into the VTA were combined into a single control group (rAAV-control). At day 4 after administration of the vectors, at a time when the anorexic response to rAAV-leptin is first expected to surface, a HF diet was introduced. An immediate hyperphagia occurred in the control rats, and this effect was partially attenuated in the MBH and VTA leptin treated groups (Fig 1, top). This initial hyperphagia was normalized by day 7 or 8 with leptin treatment in the MBH or VTA, respectively. In contrast, the normalization was not attained until day 15 in the control group (Fig 1, top). Between days 5 and 15, caloric intake was diminished by 32% and 22%, respectively in the MBH- and VTA-treated rats. Food consumption appeared to be reduced in the MBH compared with the VTA treated animals, but this trend was not significant. The leptin response waned over time and between days 16 and 31, there was no longer a difference in food intake between groups. We noted in the past that chronic leptin treatment results in a leptin-induced leptin resistance that manifests itself by an exacerbated hyperphagic response to HF exposure (Scarpace et al., 2005). This was also the case in the present experiment; cumulative caloric consumption was approximately 20% greater in both MBH and VTA treated rats commencing at day 35 through the end of the study (1368 ± 37 kcal, control; 1654 ±± 41, MBH; 1546 ± 44, VTA; p<0.001).
Figure 1.

Top: Daily food consumption in kcal following administration of control vector (closed circles) or rAAV-leptin to MBH (open circles) or VTA (closed squares). For clarity, the rats administered either control vector into the hypothalamus or into the VTA were combined into a single control group (rAAV-control). The rAAV-leptin or control vectors were administered at day 0 in rats maintained on a chow diet. The HF diet was introduced at day 4, and cumulative caloric intake was significantly different with rAAV-leptin treatment (MBH or VTA) from day 5 through day 14 (P<0.001, one-way ANOVA with Bonferroni post-hoc analysis). After this period food consumption was not difference between the three groups for the remainder of the study.
Bottom: Change in body weight in rats following administration of control vector (closed circles) or rAAV-leptin to MBH (open circles) or VTA (closed squares). Delta body weight was significantly different with leptin treatment. (P<0.0001, two-way ANOVA with repeated measures) with significance obtained at day 5 for MBH and day 8 for VTA (P<0.001 by Bonferroni post-hoc analysis). Delta body was significantly different between MBH and VTA treated groups between days 5 and 14 (P <0.01, Bonferroni post-hoc analysis). Values represent the mean ± SE of 8 rats per group.
The change in body weight paralleled the food consumption. The introduction of the HF diet induced a rapid increase in body weight in control rats that was significantly attenuated by leptin treatment in VTA starting at day 8 or by leptin treatment in the MBH starting at day 5 and continued through day 50 (Fig. 1, bottom). Notably, the weight gain was suppressed to a significantly greater extent with leptin treatment in the MBH compared with leptin treatment in the VTA from days 5 through day 14 (Fig. 1, bottom). After the apparent onset of leptin-induced leptin resistance, beginning around day 20 or so, the rate of body weight gain was greater in both the rAAV-leptin treated groups as evidenced by the progressive merging of the delta body weights (Fig. 1, bottom). At the end of the study, body weight change (Fig 1, bottom) and absolute body weights were no longer different between the control and rAAV-leptin treated groups (424 ± 7g, control; 411 ± 9g, MBH; 402 ± 8g, VTA, p=0.18).
3.2. Adiposity levels
Adiposity levels were determined by time-domain NMR on conscious rats at day 14 and prior to death at day 54. In addition, the weights of three adipose depots, PWAT, RTWAT, and EWAT were determined at death. Total adiposity at day 14 was diminished by 17% in the VTA treated and 23% in the MBH treated rats compared with control. There was a corresponding ∼ 3% decrease in percent body fat in either treatment group (Table 1). In contrast, percent lean mass was unchanged with leptin treatment, indicating that adiposity and body weight diminished in parallel (Table 1). At the end of the study, there were no longer differences in total body fat or percent body fat among groups, providing evidence of leptin resistance with leptin overexpression in either the VTA or MBH (Table 1). Combined tissue weights of three adiposity tissues, PWAT, RTWAT and EWAT at death also confirmed no statistical differences in adiposity at death (Table 1). Serum leptin levels, another marker of adiposity, were not assessed due to the administration of leptin prior to death.
Table 1.
Body weight, adiposity, and lean mass following chronic leptin overexpression.
| rAAV-Control | rAAV-Leptin into VTA | rAAV-Leptin into MBH | ||||
|---|---|---|---|---|---|---|
| Day 14 | Day 55 | Day 14 | Day 55 | Day 14 | Day 55 | |
| BW, g | 352± 2a | 427 ± 7 | 327± 8c | 408 ± 8 | 313± 6b | 419 ± 10 |
| Adiposity, g | 86 ± 1a | 117 ± 2 | 71 ± 3b | 110 ± 32 | 66 ± 2b | 118 ± 4 |
| Adiposity, % | 24.5 ± 0.3a | 27.3 ± 0.2 | 21.9 ± 0.5b | 27.0 ± 0.4 | 21.1 ± 0.3b | 27.8 ± 0.4 |
| Lean Mass, % | 60.0 ± 0.4 | 58.5 ± 0.2 | 61.1 ± 0.4 | 58.8 ± 0.3 | 61.1 ± 0.5 | 58.0 ± 0.4 |
| WAT, g | 20.5 ± 1.1 | 17.1 ± 1.1 | 20.5 ± 1.6 | |||
Data represent the mean ± SE of 8-9 rats per group.
Adiposity and Lean Mass were determined by TD-NMR at day 14 and prior to death at day 55.
WAT represents the sum of PWAT, RTWAT, and EWAT at death.
P<0.001 for difference with treatment at day 14 by one-way ANOVA. There were no significant changes across treatments at day 55.
P<0.01 for difference from corresponding control group by post-hoc analysis.
P<0.05 for difference from corresponding control group by post-hoc analysis.
3.3. Regional signaling in response to acute leptin stimulation
Leptin signaling following acute injection of 1μg of leptin into the third ventricle was examined at day 54 in the rats with leptin overexpression and corresponding controls. This acute dose of leptin corresponded to a supra-maximal dose of leptin based on a previously determined full dose-response curve (Scarpace et al., 2001). Administration of leptin into the third ventricle activates STAT3 phosphorylation in multiple brain regions including areas in the hypothalamus and VTA (Matheny et al., 2011). As expected, the control animals demonstrated a nearly five-fold increase in phosphorylated STAT3 (P-STAT3) in the ARC with acute leptin administration as compared with ACSF administration (Fig 2, top, first two bars). This level of stimulation was consistent with what we reported previously (Matheny et al., 2011). However, in rats with chronic leptin overexpression in the ARC, acute leptin stimulation elevated P-STAT3 only two-fold compared with ACSF administration (Fig 2, top, third bar), consistent with the presence of cellular leptin resistance. Surprisingly, leptin overexpression in the VTA also desensitized the P-STAT3 response to acute leptin stimulation in the ARC (Fig. 2, top, fourth bar).
Figure 2.

STAT3 phosphorylation following a single i.c.v. injection of ACSF (open bars) or 1 μg of leptin in rats administered control vector (sold bars), rAAV-leptin into the MBH (hatched bars) or rAAV-leptin into the VTA (stripped bars) 54 days earlier. STAT3 phosphorylation was assessed 1 hour later in the ARC (Top) or VTA (Bottom). Western images of P-STAT3 are below figure.
Values represent the mean ± SE of 6-8 rats per group. The value of ACSF injected control for each individual tissue is arbitrarily set to 100 with SE adjusted proportionally with remaining groups normalized to the level in ACSF injected control. P< 0.001 for difference with leptin injection in P-STAT3 levels in ARC or VTA by one-way ANOVA. Top: *P<0.001 for difference between leptin injection and ACSF among AAV-Control by Bonferroni post-hoc analysis; **P<0.05 for difference between leptin injection and ACSF and P<0.001 for difference between rAAV-control and either rAAV-leptin to MBH or rAAV-leptin to VTA among leptin injected by Bonferroni post-hoc analysis. Bottom: *P<0.001 for difference between leptin injection and ACSF among AAV-Control by Bonferroni post-hoc analysis.
Similar findings were observed when leptin signaling was examined in the VTA. In control animals receiving leptin, there was the expected 2-fold increase in P-STAT3 compared with ACSF administration (Fig 2, bottom, first two bars). Moreover, leptin overexpression in the VTA diminished the P-STAT3 response to the acute VTA leptin stimulation (Fig 2, bottom, fourth bar). In addition, chronic leptin overexpression in the ARC also desensitized leptin signaling in the VTA (Fig. 2, bottom, third bar). Because levels of total STAT3 increase with chronic leptin overexpression (Scarpace et al., 2002b, Scarpace et al., 2007), changes in P-STAT3 were normalized to immunoreactivity of beta-tublin. Protein levels of beta-tublin were unchanged across treatments, and patterns of P-STAT3 signaling remained the same after the normalization (data not shown).
3.4. Short-term leptin overexpression
The unexpected cross-desensitization of leptin signaling with leptin overexpression in the MBH and VTA prompted us to examine if rAAV-leptin delivered to the MBH or VTA activated leptin receptors in a region-specific or trans-regional manner. To this end, leptin was overexpressed in the MBH and VTA, and the experiment was terminated at day 9, a time during the leptin-sensitive phase and when the anorexic and weight reducing responses to rAAV-leptin gene delivery were near maximum. Because the rats were maintained on standard chow in this experiment, the responses were quantitatively different than Experiment 1. As with experiment 1, the rats administered control vector either into the hypothalamus or into the VTA were combined into a single control group (rAAV-control). There was the expected decrease in both food consumption and body weight with leptin overexpression in either the VTA or MBH compared with controls (Fig. 3). Adiposity levels assessed by time-domain NMR as well as serum leptin and white adipose depot weights were all significantly reduced in the leptin-treated animals consistent with a leptin active state at time of at death (Table 2).
Figure 3.

Body weight and following administration of control vector (closed circles) or rAAV-leptin to MBH (open circles) or VTA (closed squares) in chow-fed rats. Body weight significantly diverged from controls starting at day 8 in MBH treated group (P<0.05) and by day 9 in the VTA treated group (P<0.05) by two-way ANOVA with repeated measures and Bonferroni post-hoc analysis. Values represent the mean ± SE of 7-8 rats per group. For data presentation, the rats administered either control vector into the hypothalamus or into the VTA were combined into a single control group (rAAV-control).
Inset: Cumulative food consumption from days 4 to 9. *P<0.001 for difference with leptin treatment by one-way ANOVA with Bonferroni post-hoc analysis.
Table 2.
Body weight, adiposity and serum leptin levels with short-term leptin overexpression.
| rAAV-Control | rAAV-Leptin into VTA | rAAV-Leptin into MBH | |
|---|---|---|---|
| BW, g | 332 ± 4a | 304 ± 8c | 294 ± 6b |
| Adiposity, g | 83.6 ± 1.5a | 65.0 ± 2.3b | 61.6 ± 3.0b |
| WAT, g | 6.9 ± 0.3a | 3.7 ± 0.3b | 3.4 ± 1.1b |
| Serum Leptin, ng/ml | 3.3 ± 0.5a | 1.4 ± 0.4b | 1.3 ± 0.3c |
Data represent the mean ± SE of 7-8 rats per group.
Adiposity was determined by TD-NMR prior to death at day 9.
WAT represents the sum of PWAT, RTWAT, and EWAT at death.
P<0.001 for difference with treatment at day 9 by one-way ANOVA.
P<0.01 for difference from control group by post-hoc analysis.
P<0.05 for difference from control group by post-hoc analysis.
STAT3 phosphorylation was examined in various hypothalamic regions and the VTA. As opposed to Experiment 1, where leptin signaling was assessed following acute administration of leptin, in this experiment, no exogenous drug was administered, thus the levels of P-STAT3 reflected those stimulated by vector-mediated leptin overexpression in VTA and MBH relative to the basal non-stimulated P-STAT3 in control vector-treated animals. With respect to the VTA region, leptin overexpression in the VTA stimulated P-STAT3 by greater than 3-fold over basal levels in control rats, whereas leptin overexpression in MBH did not affect the VTA P-STAT3 signal (Fig 4, top). In contrast, examination of P-STAT3 in the ARC revealed that leptin overexpression in either the MBH or the VTA resulted in elevated P-STAT3 levels in the ARC (Fig 4, bottom). Further analysis revealed that the VTA-mediated simulation of STAT3 phosphorylation involved other hypothalamic sites besides the ARC. In fact, elevated P-STAT3 was found in all three examined sites ranging from greater than 2-fold in the DMH to 4-fold in the VMH and nearly 5-fold in the LH (Fig 5).
Figure 4.

STAT3 phosphorylation in the VTA (Top) or ARC (Bottom) following treatment for 9 days with control vector (open bars), rAAV-leptin into the VTA (solid bars) or rAAV-leptin into the ARC (hatched bars). STAT3 phosphorylation represents that stimulated by leptin gene delivery. No further exogenous leptin was administered. Western images of P-STAT3 are below figure. C: AAV-Control; A: AAV-Leptin to MBH; V: AAV-Leptin to VTA.
Values represent the mean ± SE of 7-8 rats per group. The value of rAAV-Control for each individual tissue is arbitrarily set to 100 with SE adjusted proportionally with remaining groups normalized to the level in rAAV-Control. P< 0.001 for difference with rAAV-leptin treatment for leptin signaling in VTA (top) and ARC (bottom) by one-way ANOVA; *P<0.01 for difference between rAAV-Leptin to VTA and rAAV-Control by Bonferroni post-hoc analysis; **P<0.001 for difference between either rAAV-leptin into the MBH or VTA and rAAV-Control by Bonferroni post-hoc analysis.
Figure 5.

STAT3 phosphorylation in the DMH, LH or VMH following treatment for 9 days with control vector (open bars), rAAV-leptin into the VTA (solid bars) or rAAV-leptin into the MBH (hatched bars). STAT3 phosphorylation represents that stimulated by leptin gene delivery. No further exogenous leptin was administered.
Values represent the mean ± SE of 7-8 rats per group. The value of rAAV-Control for each individual tissue is arbitrarily set to 100 with SE adjusted proportionally with remaining groups normalized to the level in rAAV-Control. P< 0.001 for difference with rAAV-leptin treatment for leptin signaling in DMH, LH, or VMH by one-way ANOVA; *P<0.001 for difference between rAAV-Leptin treated and corresponding rAAV-Control by Bonferroni post-hoc analysis.
3.5. GFP and Leptin overexpression
Potential viral spread following delivery of the control vector encoding GFP was determined by examining native GFP fluorescence in selected coronal sections in conjunction with immunofluorescence of TH or NeuN. The GFP native fluorescence was observed only in the injected ipsilateral VTA (Fig. 6, top) or predominantly in the injected side of the MBH with minor leakage into the contralateral side (Fig. 6, bottom). Intense GFP expression was predominant in a paramedian field in the rostral VTA involving also the medial extensions of VTA sub-nuclei parabrachial pigmented area (PBP). GFP fluorescence was also seen in the adjacent SN area (Fig 6, top). There was prevailing GFP fluorescence in the lateral and medial arcuate hypothalamus with obvious intrusion into the adjacent VMH area but only minor spread beyond that (Fig 6, bottom). No GFP fluorescence was visible in hypothalamic regions post VTA control vector delivery or in the VTA/SN area post control vector delivery in the MBH. Taken together, site-directed rAAV-GFP delivery targeted to either the VTA or MBH achieved high level of GFP expression in both the targeted regions as well as indicated neighboring sub-regions with no detectable trans-regional virus transduction.
Figure 6.
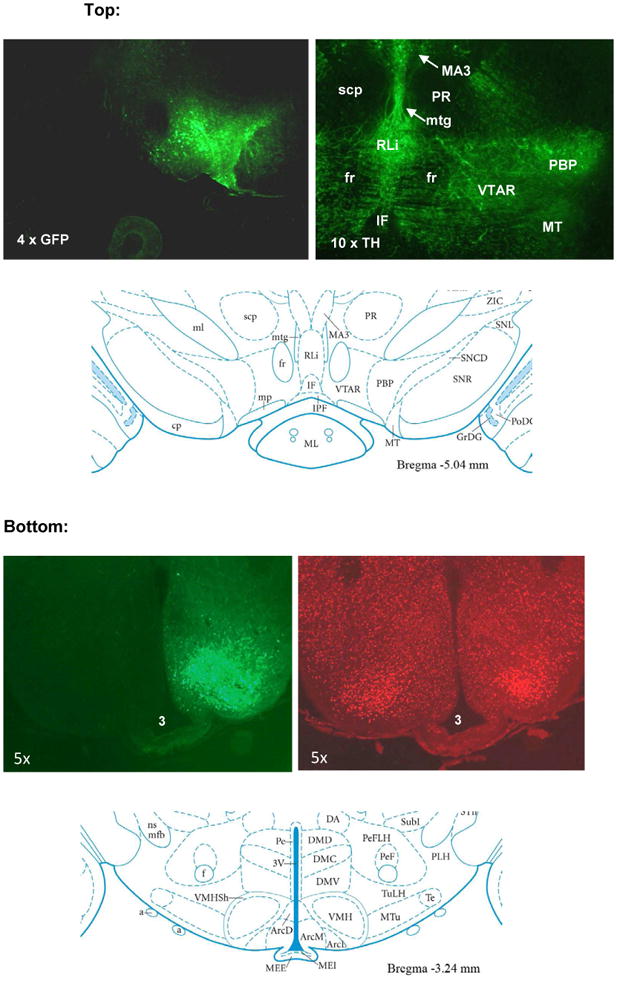
The control rAAV-GFP vector was injected unilaterally (ipsilaterally) into the VTA (top) or MBH (bottom) and coronal sections of VTA and MBH analyzed one month post vector delivery.
Top: Native GFP fluorescence (upper left) and TH immunofluorescence (upper right). TH immunofluorescence identifying dopamine neurons indicate the VTA nuclei (VTAR and PBP) in relation to nearby anatomical landmarks such as IF, fr, RLi, and MT. For reference see schematic on bottom panel (reproduced from Paxinos and Watson, 2005). VTAR: rostral VTA; PBP: parabrach pigment; IF: interfascicular; fr: fac retroflexus; RLi: rost linear raphe; MT: med terminal nu.
Bottom: Native GFP fluorescence (upper left) and counterstaining for NeuN (upper right) in the MBH. Counterstaining with NeuN visualizes neurons within the ARC, VMH and DMH hypothalamic nuclei in relation to the 3rd ventricle (3V) and implicates a localized rAAV-GFP neuron transduction in the basal medial hypothalamic area with high concentration in the VMH region. The schematic on the bottom lower panel indicates hypothalamic nuclei in relation to the 3rd ventricle (reproduced from Paxinos and Watson, 2005).
To determine if the increase in P-STAT3 in the several hypothalamic brain regions by rAAV-leptin gene delivery to the VTA is the result of rAAV-leptin virus migration into the hypothalamus and consequential on-site leptin expression, leptin transgene expression was examined by RT-PCR analysis in the VTA and ARC 54 days after rAAV-leptin administration to the VTA. As expected, site-directed gene delivery to the VTA produced an increase in leptin mRNA levels in the VTA that was absent in the rats treated with control vector (Fig. 7, top). More importantly, there was no detectable leptin mRNA in the corresponding ARC tissue in either control or rAAV-leptin VTA injected rats (Fig. 7, bottom).
Figure 7.
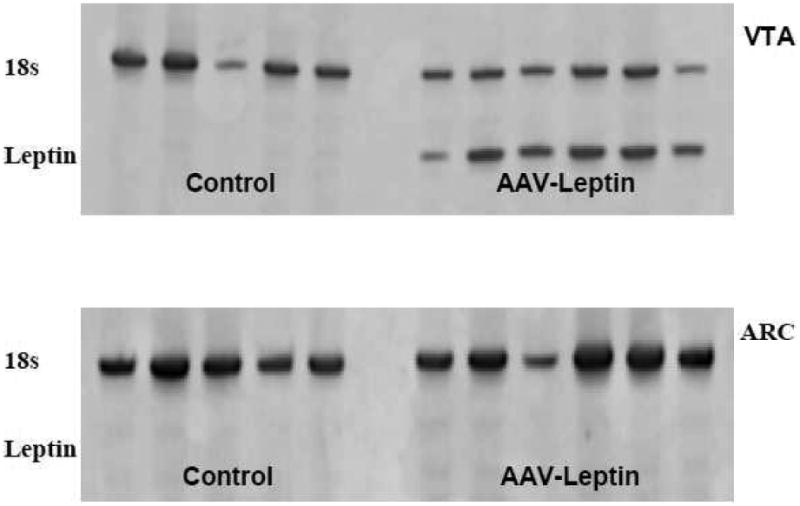
Leptin transgene expression was identified by RT-PCR in VTA or ARC tissue at day 54 following rAAV-leptin injection into the VTA. Top: Leptin RNA (lower band) was identified in the VTA of all rats that were injected with rAAV-leptin into the VTA but not in rAAV-Control injected rats. Bottom: No leptin mRNA was detected in corresponding ARC punches in either rAAV-Control or those injected with rAAV-leptin in the VTA. Upper band represents 18S rRNA as an internal standard.
The gene construct for leptin includes a secretory sequence. Thus, even though the vector-mediated leptin expression in the rAAV-leptin VTA-treated rats was not found in the hypothalamus, it is possible that the peptide product was secreted into the surrounding tissue and diffused to the hypothalamus, or traveled to the hypothalamus via medial forebrain bundle (MFB), or by an unknown facilitated transport pathway. To examine these possibilities, leptin peptide was measured in the ARC and VTA tissue homogenates 9 days after rAAV-leptin delivery into either the MBH or VTA. The leptin product was identified only in the brain region in which the rAAV-leptin was administered and not in either the GFP injected rats nor in the region not directly injected with rAAV-leptin (Table 3).
Table 3.
Leptin peptide content in ARC and VTA punches with short-term leptin overexpression.
| rAAV-Control (ng/punch) | rAAV-Leptin into VTA (ng/punch) | rAAV-Leptin into MBH (ng/punch) | |
|---|---|---|---|
| ARC | 0.26 ± 0.04 | 0.25 ± 0.04 | 6.78 ± 3.32a |
| VTA | 0.29 ± 0.03 | 10.58 ± 1.75b | 0.31± 0.06 |
ARC and VTA punch represents a cylinder of 2mm in length and 1mm in diameter.
Values represent the mean ± SE of 6-8 rats per group.
P<0.01 for difference with leptin injection by one-way ANOVA. P<0.05 for difference between rAAV-Leptin treated and rAAV-Control by Bonferroni post-hoc analysis.
P<0.001 for difference with leptin injection by one-way ANOVA. P<0.05 for difference between rAAV-Leptin treated and rAAV-Control by Bonferroni post-hoc analysis.
3.6. BAT UCP1 with short and long-term leptin overexpression
Centrally administered leptin is known to augment UCP1 in BAT (Scarpace, et al., 1997). We examined the relative efficacy of short or long-term leptin overexpression in the MBH versus VTA to elevate BAT UPC1. Surprisingly, leptin overexpression in the VTA was as efficacious as overexpression in MBH in inducing BAT UCP1 by 30-40% with either short or long-term overexpression (Table 4).
Table 4.
BAT UCP1 levels with short-term and long-term leptin overexpression.
| Leptin Overexpression | rAAV-Control | rAAV-Leptin into VTA | rAAV-Leptin into MBH |
|---|---|---|---|
| 9 days | 100 ± 2.3 | 137 ± 9.3a | 137 ± 3.6a |
| 55 days | 100 ± 6.6b | 130 ± 8.5b | 129 ± 8.2b |
Data represent the mean ± SE of 7-8 rats per group.
The value of rAAV-Control for each time point is arbitrarily set to 100 with SE adjusted proportionally with remaining groups normalized to the level in rAAV-Control.
P<0.0002 for difference with treatment at day 9 by one-way ANOVA. P<0.001 for difference from control group by post-hoc analysis.
P<0.05 for difference with treatment at day 55 by one-way ANOVA. P<0.05 for difference from control group by post-hoc analysis.
3.7. Tyrosine hydroxylase protein levels with short term leptin overexpression
Tyrosine hydroxylase (TH) protein levels were examined in the ARC and VTA 9 days after leptin gene delivery into the MBH or VTA. Leptin overexpression in the VTA resulted in a 63% decrease in TH protein in the VTA compared with control without a concomitant change in the ARC (Fig 8, top). In contrast, TH protein was unchanged in either the VTA or ARC with leptin overexpression in the MBH (Fig. 8, bottom).
Figure 8.

TH protein in the VTA (Top) or ARC (Bottom) following treatment for 9 days with control vector (open bars), rAAV-leptin into the VTA (solid bars) or rAAV-leptin into the MBH (hatched bars). Values represent the mean ± SE of 7-8 rats per group. The value of rAAV-Control for each individual tissue is arbitrarily set to 100 with SE adjusted proportionally with remaining groups normalized to the level in rAAV-Control. Top: P< 0.02 for difference with rAAV-leptin treatment by one-way ANOVA; *P<0.05 for difference between rAAV-Leptin to VTA and rAAV-Control by Bonferroni post-hoc analysis.
4. Discussion
This study identifies a role for leptin action in the VTA in the long-term regulation of body weight, especially with respect to combating diet-induced obesity. Previous evidence linked leptin receptor activity in the VTA to modulation of HF and sucrose consumption with no discernable effect on body weight (Hommel et al., 2006, Davis et al., 2011). Direct leptin injection into the VTA induced short-term decreases in food consumption and body weight (Bruijnzeel et al., 2011). These data underscore functional significance of leptin action in the VTA, but stopped short of establishing a role in long-term body weight homeostasis. The current study reports the chronic effects of leptin elevation in the VTA versus those in the MBH on body weight control under the HF feeding paradigm.
We overexpessed leptin in two brain areas, the medial basal hypothalamus (targeting the ARC), a region recognized for its importance in energy homeostasis, and the VTA, a region involved in reward and motivation (Cota et al., 2006). Whereas leptin overexpression in the MBH tempered HF-induced weight gain, leptin overexpression in the VTA demonstrated a slightly lower yet comparable efficacy. Within either treatment group, leptin resistance developed over time, as evidenced by increased rate of weight gain and reversal of the reduced adiposity apparent at day 14. By day 55, body weight and food consumption were similar across groups. This leptin-induced leptin resistance with regard to physiological responses mimicked what was previously reported with rAAV-leptin delivered into the third ventricle (Scarpace et al., 2002a, Scarpace et al., 2005). Such delivery results in gene expression in cells located along the wall of the ventricle and limited surrounding tissue (Dhillon et al., 2001). The gene construct for leptin includes a secretory sequence, thus this compound is secreted into the third ventricle (Scarpace et al., 2002b) and likely reaching target sites throughout the brain. By use of this i.c.v. delivery method, we previously demonstrated that long-term leptin overexpression resulted in cellular leptin resistance (diminished leptin-mediated STAT3 phosphorylation) in multiple regions within hypothalamus as well as in the VTA in the absence of HF influence (Matheny et al., 2011). This report extends those findings by demonstrating that long-term leptin overexpression targeted to the medial basal hypothalamus or to the VTA in the presence of a HF diet not only induces cellular leptin resistance in the targeted region, but also cross-desensitizes the non-injected brain area.
The new findings suggest that either the viruses containing the transgene or the gene product traversed outside the targeted region or that the signal to desensitize leptin action was communicated to the non-injected region. To test these possibilities, leptin was overexpessed in the medial basal hypothalamus or VTA for a period of 9 days, a time when the physiological responses to rAAV-leptin delivery were at near maximum (present report and Scarpace, et al., 2002b). Previous work has established that leptin overexpression in the CNS produces a submaximal stimulation of STAT3 phosphorylation (2-3 fold over basal) as compared to the level of P-STAT3 following an acute injection of leptin (5-8 fold over basal) (Scarpace, et al., 2001; Scarpace, et al., 2007). In the current study, 9-day leptin overexpression increased P-STAT3 by 3-fold in the hypothalamus, a level consistent with submaximal stimulation. Unexpectedly, leptin overexpression in the VTA also elevated P-STAT3 in the ARC and other regions in the medial basal hypothalamus. In contrast, leptin overexpression in the MBH did not increase P-STAT3 in the VTA.
This unidirectional trans-stimulation was apparently not due to migration of either the vector or the leptin transgene product. We employed GFP as a surrogate marker for viral transduction and transgene expression and performed GFP fluorescence imaging analysis in rats receiving rAAV-GFP vector in either the VTA or MBH. GFP expression was found in the VTA and adjacent SN area post the VTA delivery, while detected mainly in the lateral and medial arcuate hypothalamic nuclei as well as in the VMH post the MBH delivery. Therefore, GFP fluorescence was focused within the injected site and spread into a few indicated neighboring structures with no appearance in the non-injected trans-region. Consist with these findings, increased leptin expression and elevated leptin protein was detected only within the rAAV-leptin injected brain regions and was notably absent in the non-injected trans-regions in the 9-day overexpression experiment. Since neither leptin mRNA nor protein level was measured in the SN area adjacent to the VTA or in the VMH adjacent to the ARC, we cannot rule out the possibility that SN may contribute to the VTA or VMH to the ARC leptin effects.
We have previously measured cerebral spinal fluid (CSF) leptin levels following rAAV-leptin delivery to the third ventricle. Leptin was increased by less than two-fold in the CSF (Control, 0.1 ng/ml vs rAAV-leptin, 0.175ng/ml), well within the physiological range of CSF leptin (Scarpace et al., 2002b). In the present study with overexpression directly targeted to and measured in the tissue, we observed a maximum of 10ng/leptin per tissue punch. The volume of the punch was 3.14 μl, yielding a tissue concentration of ∼ 3,000 ng/ml. The tissue leptin levels were ∼ 100ng/ml in control rats. These data provide two insights: first, local tissue leptin concentration is far greater than CSF leptin levels. How these values relate to each other and the physiological significance of the CSF values remains to be elucidated. Second, our region-specific leptin overexpression produces significant elevation of leptin within the targeted tissue. It is possible that a small amount of leptin diffuses through tissue and is transported either through the brain's ventricular system, the subarachnoid space (Ruiter, et al., 2010), or the MFB neural connections from the VTA to the hypothalamus (Simmons, et al., 1998). This amount of leptin might be sufficient to activate leptin-P-STAT3 signaling, but below the detection limit of our assay. It was recently reported that injection of leptin into the hindbrain triggers STAT3 phosphorylation in hypothalamic neurons (Ruiter, et al 2010). The authors suggested that hindbrain leptin activates neurons in the hypothalamus via neuronal projections, or that leptin reaches leptin-sensitive hypothalamic neurons via the CSF in the subarachnoid space. Collectively, the observations from that and the present study suggest that an integrative response involving multiple brain regions accounts for the observed physiological effects to localized leptin applications. However, the present data cannot determine if the source of the elevated P-STAT3 originates from leptin receptor activation, some other STAT3 coupled receptor, or suppression of inhibitory pathways such as SOCS3 or PTP1B.
Leptin is known to both suppress appetite and augment energy expenditure (Li, 2011). One reliable marker of the latter effect of leptin is an increase in non-shivering thermogenesis through induction in UCP1 protein in BAT (Scarpace, et al., 1997). Although energy expenditure was not directly assessed in this study, BAT UCP1 protein levels were elevated with leptin overexpression in either brain region in both the short- and long-term experiments. Moreover, by visual inspection, the BAT in the short-term study was bright red, suggestive of activated UCP1, whereas the BAT in the long-term study was not, indicative of elevated levels but quiescent UCP1. These findings suggest that leptin overexpression in the hypothalamus or VTA initially activates UCP1, but this effect fades over time, consistent with the development of leptin resistance.
Central leptin induction of UCP1 involves direct sympathetic activation of BAT and can be blocked by severing the intercostal nerve bundle that innervates BAT (Scarpace, et al., 1998). The activation signals are believed to be transmitted via neural projections that originate from the hypothalamus or nucleus of the solitary tract (NTS) (Bartness et al., 2010). Our data hint that leptin may also act in the VTA to promote UCP1. But it is unclear if this action in the VTA requires the involvement of the hypothalamus and/or the NTS to impact BAT UCP1.
Leptin receptor function in the VTA has been linked to reward-based feeding. Leptin tonically inhibits dopaminergic neuron firing rate in the VTA, leading to a decrease in both dopamine release and food intake (Hommel JD, 2006). TH protein level is one marker of dopaminergic neurons (Manfredsson, et al. 2009) and dopamine biosynthesis (Fluharty, et al. 1985). The short-term leptin overexpression in the VTA decreased TH protein, indicating a probable leptin-mediated decrease in dopamine release. In contrast, TH protein was unaffected in the ARC with either leptin overexpression in the VTA or MBH. This observation agrees with a recent notion that leptin action in the ARC may not be coupled to dopamine synthesis within the ARC (Fulton et al., 2000). Moreover, the observation that overexpression of leptin in the MBH does not suppress TH levels in the VTA is consistent with our other observations that leptin action in the MBH does not evoke corresponding responses in the VTA.
Leptin resistance is generally believed to have a causative role in obesity and predisposes rodents to HF-induced obesity (Scarpace and Zhang, 2009). Chronic CNS overexpression of leptin via third ventricle gene delivery in conjunction with chronic HF feeding results in greater weight gain than without leptin treatment (Scarpace et al., 2005). Leptin overexpression directed specifically to the VTA or MBH in the presence of a HF diet resulted in diminished leptin-mediated signaling that was associated with an increased trajectory of body weight gain. Therefore, region-specific leptin elevation failed to preserve regional leptin sensitivity and produced similar physiological and cellular consequences as the CNS leptin overexpression approach (Matheny et al., 2011).
VTA leptin receptor activity influences sugar intake (Hommel et al., 2006, Davis et al., 2011). We demonstrate earlier an association between dietary preference for HF foods and VTA leptin-STAT3 signaling (Scarpace et al., 2010). Both reports support a role of VTA leptin function in curbing the consumption of palatable foods and associated weight gain. We speculate that the onset of any leptin resistance in the VTA would disrupt leptin function, and in turn, that predicts increased vulnerability to HF-induced weight gain. Thus, the elevated leptin levels associated with obesity may induce leptin resistance in the VTA, in addition to the leptin resistance in the hypothalamus, thus, providing an additional mechanism, underlying the increased susceptibility of leptin resistant rats to HF-induced obesity.
There is a dichotomy in our findings with short and long-term leptin overexpression. The short-term experiment revealed a unidirectional trans-stimulation from the VTA to the ARC, whereas the long-term overexpression indicated a bidirectional cross-desensitization. Short-term overexpression of leptin in the VTA activates STAT3 signaling in the ARC, which, over time, may lead to cross-desensitization of the ARC signaling. This phenomenon may involve the MFB neural pathway connecting the VTA to the hypothalamus or other processes discussed earlier. However, the ARC cross-desensitization of VTA is puzzling. The short-term leptin overexpression in the MBH did not activate P-STAT3 in the VTA and did not appear to involve migration of either virus or leptin to the VTA. Since the leptin receptor-bearing POMC/CART and NPY/AgRP neurons in the ARC project to the LH in the hypothalamus (Wynne et. al., 2005) and orexin neurons in the LH project to the VTA (Myers et. al., 2009), the ARC cross-desensitization of the VTA may be relayed via these primary and secondary projections. We may also speculate that chronic leptin production in the MBH, over time, radially diffuses, and ultimately desensitizes leptin action in the VTA.
In summary, leptin functions in the VTA to temper HF-induced obesity, but slightly less effectively than that of leptin overexpression in the MBH during the leptin-responsive phase. Moreover, the action of leptin in the VTA induces cellular STAT3 phosphorylation in the medial basal hypothalamus, which suggests an integrative response involving both brain regions accounts for the observed physiological effects. Long-term leptin overexpression in either the MBH or VTA leads to desensitization of leptin signaling in the treated region and cross-desensitization of leptin signaling in the untreated region. It remains to be determined if leptin acts in the VTA independently from the hypothalamus to modulate body weight.
Highlights.
Leptin overexpression in VTA or hypothalamus tempered HF feeding.
VTA leptin overexpression stimulated STAT3 phosphorylation in hypothalamus.
Overexpression in the hypothalamus did not activate P-STAT3 in the VTA.
Leptin overexpression in VTA induced UCP1 in BAT.
VTA or ARC leptin overexpression cross-desensitized leptin signaling.
Acknowledgments
Supported by the National Institute on Aging Grant AG-26159 and the Medical Research Service of the Department of Veterans Affairs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bartness TJ, Vaughan CH, Song CK. Sympathetic and sensory innervation of brown adipose tissue. Int J Obes (Lond) 2010;34(1):S36–42. doi: 10.1038/ijo.2010.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijnzeel AW, Corrie LW, Rogers JA, Yamada H. Effects of insulin and leptin in the ventral tegmental area and arcuate hypothalamic nucleus on food intake and brain reward function in female rats. Behav Brain Res. 2011;219:254–264. doi: 10.1016/j.bbr.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cota D, Barrera JG, Seeley RJ. Leptin in energy balance and reward: two faces of the same coin? Neuron. 2006;51:678–680. doi: 10.1016/j.neuron.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Davis JF, Choi DL, Schurdak JD, Fitzgerald MF, Clegg DJ, Lipton JW, Figlewicz DP, Benoit SC. Leptin regulates energy balance and motivation through action at distinct neural circuits. Biol Psychiatry. 2011;69:668–674. doi: 10.1016/j.biopsych.2010.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon H, Kalra SP, Prima V, Zolotukhin S, Scarpace PJ, Moldawer LL, Muzyczka N, Kalra PS. Central leptin gene therapy suppresses body weight gain, adiposity and serum insulin without affecting food consumption in normal rats: a long-term study. Regul Pept. 2001;99:69–77. doi: 10.1016/s0167-0115(01)00237-3. [DOI] [PubMed] [Google Scholar]
- Fluharty SJ, Snyder GL, Zigmond MJ, Stricker EM. Tyrosine hydroxylase activity and catecholamine biosynthesis in the adrenal medulla of rats during stress. J Pharmacol Exp Ther. 1985;233:32–38. [PubMed] [Google Scholar]
- Fulton S, Woodside B, Shizgal P. Modulation of brain reward circuitry by leptin. Science. 2000;287:125–128. doi: 10.1126/science.287.5450.125. [DOI] [PubMed] [Google Scholar]
- Hommel JD, Trinko R, Sears RM, Georgescu D, Liu ZW, Gao XB, Thurmon JJ, Marinelli M, DiLeone RJ. Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron. 2006;51:801–810. doi: 10.1016/j.neuron.2006.08.023. [DOI] [PubMed] [Google Scholar]
- Li MD. Leptin and beyond: an odyssey to the central control of body weight. Yale J Biol Med. 2011;84:1–7. [PMC free article] [PubMed] [Google Scholar]
- Manfredsson FP, Tumer N, Erdos B, Landa T, Broxson CS, Sullivan LF, Rising AC, Foust KD, Zhang Y, Muzyczka N, et al. Nigrostriatal rAAV-mediated GDNF overexpression induces robust weight loss in a rat model of age-related obesity. Mol Ther. 2009;17:980–991. doi: 10.1038/mt.2009.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheny M, Shapiro A, Tumer N, Scarpace PJ. Region-Specific Diet-induced and Leptin-Induced Cellular Leptin Resistance Includes the Ventral Tegmental Area in Rats. Neuropharmacology. 2011;60:480–487. doi: 10.1016/j.neuropharm.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MG, Münzberg H, Leinninger GM, Leshan RL. The geometry of leptin action in the brain: more complicated than a simple ARC. Cell Metab. 2009;9:117–23. doi: 10.1016/j.cmet.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Elsevier Academic Press; 2005. [DOI] [PubMed] [Google Scholar]
- Ruiter M, Duffy P, Simasko S, Ritter RC. Increased hypothalamic signal transducer and activator of transcription 3 phosphorylation after hindbrain leptin injection. Endocrinology. 2010;151:1509–19. doi: 10.1210/en.2009-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpace PJ, Matheny M. Leptin induction of UCP1 is dependent on sympathetic innervation. Am J Physiol. 1998;275:E259–264. doi: 10.1152/ajpendo.1998.275.2.E259. [DOI] [PubMed] [Google Scholar]
- Scarpace PJ, Matheny M, Pollock BH, Tümer N. Leptin increases uncoupling protein expression and energy expenditure. Am J Physiol. 1997;273:E226–230. doi: 10.1152/ajpendo.1997.273.1.E226. [DOI] [PubMed] [Google Scholar]
- Scarpace PJ, Matheny M, Tumer N. Hypothalamic leptin resistance is associated with impaired leptin signal transduction in aged obese rats. Neuroscience. 2001;104:1111–1117. doi: 10.1016/s0306-4522(01)00142-7. [DOI] [PubMed] [Google Scholar]
- Scarpace PJ, Matheny M, Tumer N, Cheng KY, Zhang Y. Leptin resistance exacerbates diet-induced obesity and is associated with diminished maximal leptin signalling capacity in rats. Diabetologia. 2005;48:1075–1083. doi: 10.1007/s00125-005-1763-x. [DOI] [PubMed] [Google Scholar]
- Scarpace PJ, Matheny M, Zhang Y. Wheel running eliminates high-fat preference and enhances leptin signaling in the ventral tegmental area. Physiol Behav. 2010;100:173–179. doi: 10.1016/j.physbeh.2010.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpace PJ, Matheny M, Zhang Y, Cheng KY, Tumer N. Leptin antagonist reveals anuncoupling between leptin receptor signal transducer and activator of transcription 3 signaling and metabolic responses with central leptin resistance. J Pharmacol Exp Ther. 2007;320:706–712. doi: 10.1124/jpet.106.112813. [DOI] [PubMed] [Google Scholar]
- Scarpace PJ, Matheny M, Zhang Y, Shek EW, Prima V, Zolotukhin S, Tumer N. Leptin-induced leptin resistance reveals separate roles for the anorexic and thermogenic responses in weight maintenance. Endocrinology. 2002a;143:3026–3035. doi: 10.1210/endo.143.8.8966. [DOI] [PubMed] [Google Scholar]
- Scarpace PJ, Matheny M, Zhang Y, Tumer N, Frase CD, Shek EW, Hong B, Prima V, Zolotukhin S. Central leptin gene delivery evokes persistent leptin signal transduction in young and aged-obese rats but physiological responses become attenuated over time in aged-obese rats. Neuropharmacology. 2002b;42:548–561. doi: 10.1016/s0028-3908(02)00003-5. [DOI] [PubMed] [Google Scholar]
- Scarpace PJ, Zhang Y. Leptin resistance: a prediposing factor for diet-induced obesity. Am J Physiol Regul Integr Comp Physiol. 2009;296:R493–500. doi: 10.1152/ajpregu.90669.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons JM, Ackermann RF, Gallistel CR. Medial forebrain bundle lesions fail to structurally and functionally disconnect the ventral tegmental area from many ipsilateral forebrain nuclei: implications for the neural substrate of brain stimulation reward. J Neurosci. 1998;18:8515–8533. doi: 10.1523/JNEUROSCI.18-20-08515.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinsley FC, Taicher GZ, Heiman ML. Evaluation of a quantitative magnetic resonance method for mouse whole body composition analysis. Obes Res. 2004;12:150–160. doi: 10.1038/oby.2004.20. [DOI] [PubMed] [Google Scholar]
- Wynne K, Stanley S, McGowan B, Bloom S. Appetite control. J Endocrinol. 2005;184:291–318. doi: 10.1677/joe.1.05866. [DOI] [PubMed] [Google Scholar]