

Abstract
Intracerebral hemorrhage (ICH), the most common form of hemorrhagic stroke, accounts for up to 15% of all strokes. Despite maximal surgical intervention and supportive care, ICH is associated with significant morbidity and mortality, in part due to a lack of viable treatment options. Astrogliosis, a key feature of secondary injury that is characterized by glial proliferation, is a poorly-defined process that may produce both beneficial and detrimental outcomes after brain injury. Using a pre-clinical murine model of collagenase-induced ICH, we demonstrate a delayed upregulation of survivin, a key molecule involved in tumor cell proliferation and survival, by 72 h post-ICH. Notably, this increase in survivin expression was prominent in GFAP-positive astrocytes, but absent in neurons. Survivin was not expressed at detectable levels in the striatum of sham-operated mice. The expression of survivin after ICH was temporally and spatially associated with the expression of proliferating cell nuclear antigen (PCNA), an established marker of cellular proliferation. Moreover, the survivin expression was co-localized in proliferating astrocytes as evidenced by triple-label immunohistochemistry. Finally, shRNA-mediated silencing of survivin expression attenuated PCNA expression and reduced cellular proliferation in human glial cells. Together, these data suggest a potentially novel role for survivin in functionally promoting astrocytic proliferation after ICH.
Key words: astrogliosis, Inhibitor of Apoptosis protein family, proliferation, repair, stroke
Introduction
Intracerebral hemorrhage (ICH), the most common form of hemorrhagic stroke, accounts for up to 15% of all strokes and afflicts over 50,000 Americans annually.1 Spontaneous ICH is primarily caused by the rupture of small blood vessels damaged by chronic hypertension or amyloid angiopathy, resulting in the formation of a space-occupying hematoma within the brain parenchyma. Despite neurosurgical intervention and maximal supportive care, ICH induces 50–60% mortality within the first year, and less than 20% of survivors regain functional independence.2,3 Moreover, the incidence of ICH is increasing due to an aging population and changing racial demographics.3 Unfortunately, effective medical treatment options are currently lacking, at least in part due to the poorly defined pathophysiology of ICH.2,3
Astrocytes, a prominent brain cell type, play an essential part in brain homeostasis, apart from providing structural support to neurons. Astrocytes actively promote the formation and maintenance of the blood–brain barrier,4–6 regulate cerebral blood flow,7–9 maintain oxidative and ionic homeostasis,10 and secrete neuroprotective factors.11–15 Astrocytes also respond to brain insults with conserved, phenotypic changes that may exert both beneficial and detrimental effects. This process, called reactive astrogliosis, is characterized by enhanced astrocyte proliferation, glial fibrillary acidic protein (GFAP) expression, cellular hypertrophy, and glial scar formation;16 however, the molecular and cellular mechanisms underlying astroglial cell proliferation, a critical process in reactive astrogliosis, remain poorly defined. Given the potential for astrocytes to influence secondary brain injury and central nervous system (CNS) repair, an improved understanding of astrogliosis may provide novel therapeutic targets after brain injuries, including ICH.
Survivin, a member of the Inhibitor of Apoptosis (IAP) protein family, is ubiquitously present in embryonic and fetal tissues,17 yet its expression is absent or low in most adult human differentiated tissues.18 In contrast to other IAP family members that exert antiapoptotic functions, survivin promotes both cell survival and cellular proliferation.18,19 Tight regulation of these distinct functions occurs, at least in part, via the subcellular distribution of survivin. Along these lines, nuclear localization of survivin correlated with increased cell division, whereas the antiapoptotic actions associated with cytosolic localization.20 Herein, we hypothesized that survivin promotes reactive astrogliosis after ICH.
Methods
Collagenase-induced ICH
Our animal studies were reviewed and approved by the Committee on Animal Use for Research and Education at the Medical College of Georgia, in compliance with National Institutes of Health (NIH) and U.S. Department of Agriculture (USDA) guidelines. Male CD1 mice (8 weeks old; Charles River, Wilmington, MA) were anesthetized with an intraperitoneal injection of ketamine and xylazine and positioned prone in a stereotaxic head frame. A small-animal temperature controller (David Kopf Instruments, Tujunga, CA) was used to maintain the body temperature at 37°±0.50°C. A 0.5-mm burr hole was made 2.2 mm lateral to the bregma, using care not to damage the underlying dura, with a high-speed dental drill. A 26-gauge Hamilton syringe containing 0.04 U of bacterial type IV collagenase (Sigma-Aldrich, St. Louis, MO) in 0.5 μL saline was inserted with stereotaxic guidance 3.0 mm into the left striatum to induce a spontaneous ICH, per our laboratory's earlier work.11 Collagenase, a proteolytic enzyme, induces spontaneous intraparenchymal bleeding by disrupting the basal lamina of cerebral vessels, creating a rupture aneurysm. The syringe plunger was depressed at a slow rate (0.5 μL over 10 min), and the syringe remained in place for 5 min to prevent reflux. After removal of the needle, the burr hole was sealed with bone wax and the incision was surgically stapled. Sham-operated mice received a burr hole and needle placement, but only a (0.5 μL) saline (vehicle) injection was performed. The mice were maintained at 37°C until recovery.
Immunohistochemistry
Deeply anesthetized mice (n=4 mice/group), were transcardially perfused with 0.9% saline (pH 7.4), followed by 4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS; pH 7.4). The brains were post-fixed overnight in 4% paraformaldehyde and cryoprotected in 30% sucrose until the brains were permeated. They were snap-frozen, sectioned at 25 μM using a cryostat, and mounted on glass slides. The sections were incubated at 20°C with 10% normal donkey serum in PBS containing 0.4% Triton X-100 for 1 h, followed by incubation with primary antibodies at 4°C for 24 h in 0.2% Triton X-100 containing PBS (survivin, 1:50; Cell Signaling Technologies, Danvers, MA), GFAP (1:500; Dako Corporation, Carpinteria, CA), proliferating cell nuclear antigen (PCNA, 1:100; Cell Signaling Technologies), and NeuN (1:100; Millipore, Billerica, MA), followed by incubation with Alexa Fluor-tagged secondary antibodies at room temperature for 1 h in 0.2% Triton X-100 containing 0.1 M PBS. Omission of primary antibody served as a negative control. Immunofluorescence was determined using an LSM510 Meta confocal laser microscope (Carl Zeiss, Thornwood, NY), as described previously by our laboratory.21 The numbers of surviving-positive and PCNA-positive cells in a 200×200-μm2 area in the peri-hematomal brain region were counted.
Western blotting
Anesthetized mice were perfused with saline and the brains were removed. Brain slices (2 mm) centered on the injection site, were prepared with the aid of a brain matrix. The hematomal and peri-hematomal region (16–20 mm2) in the striatum from ICH-induced mice or comparable striatal tissue from sham-operated mice (S) was carefully dissected on ice and homogenized for Western blotting. Protein concentrations from the homogenates were measured using a BCA protein assay kit. Proteins (50 μg) were separated using a 4–20% gradient gel and transferred onto a PVDF membrane. The membrane was then blocked with 5% non-fat dry milk in Tris-buffered saline containing Tween-20 (TBS-T), and then incubated with appropriate primary antibody followed by fluorescent-tagged secondary antibody. The blots were visualized using a Li-Cor Odyssey near-infrared imaging system, and densitometry analysis was performed using Quantity One software (Bio-Rad, Foster City, CA), as previously detailed by our group.22–24 β-Actin was used as a loading control for Western blotting and for normalization of the densitometry analysis.
Lentiviral-mediated protein knockdown
Human U87MG glial cells (American Type Culture Collection, Manassas, VA) was cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and antibiotics in a 37°C humidified incubator. For survivin knockdown, control shRNA or survivin shRNA particles (Santa Cruz Biotechnology, Santa Cruz, CA) were used per the manufacturer's recommended protocols. Briefly, U87MG cells were transduced with lentiviral shRNA particles at multiplicity of infection (MOI) of 1 in the presence of 5 μg/mL polybrene. Medium was replaced 18 h later with growth media. At 72 h post-infection, 2 μg/mL puromycin, the minimum concentration sufficient to kill non-transduced U87MG cells (data not shown), was added to the culture media to select stably transduced cells. Puromycin selection continued for a week with partial media replenishment every 48 h. Western blotting was utilized to confirm protein knockdown. U87MG cells stably transduced with control shRNA served as control. For cellular proliferation studies, U87MG cells stably expressing either control shRNA or survivin shRNA were plated at 2×105 cells/well in a 6-well plate and cultured in growth medium. Total cell counts were assessed 1 week after plating with the aid of a hemocytometer.
Statistical analysis
The data are presented as mean±standard error of the mean (SEM), and were analyzed using analysis of variance (ANOVA) or the Student's t-test as appropriate, and as detailed in the figure legends. A p value < 0.05 was considered to be statistically significant.
Results
Astrocyte-specific survivin expression after ICH
To establish whether survivin expression is modulated in the peri-hematoma region following ICH, a murine collagenase model of ICH was utilized. Survivin was expressed at undetectable or low levels in sham-operated mice at 1 day post-ICH (Fig. 1), as assessed by Western blotting. In contrast, a significant upregulation of survivin was noted within the striatum (directly adjacent to the hematoma) by day 3 and day 5 after injury (Fig. 1). This increase was followed by a reduction in survivin expression by day 7 after injury (Fig. 1). Figure 2 depicts representative coronal brain sections from sham and ICH mice to demonstrate the temporal pattern of hematoma development and resolution after injury. The maximal expression of survivin directly correlated with the pattern of spontaneous clot resolution.
FIG. 1.

Survivin expression following intracerebral hemorrhage (ICH). (A) Temporal pattern of survivin expression after ICH, as assessed by Western blotting. Tissue was collected from the hematomal and peri-hematomal striatum at 1, 3, 5, and 7 days post-ICH. Striatal tissue collected from sham-operated mice (S) served as a baseline control. Representative blots were normalized to β-actin to control for equal protein loading. (B) Densitometric analysis of Western blotting data. Quantification of survivin expression was normalized to β-actin. Data were analyzed using two-way analysis of variance with Bonferroni post-tests (n=3–7/group, **p<0.01, ***p<0.001 versus shams).
FIG. 2.

Temporal pattern of hematoma resolution following intracerebral hemorrhage (ICH). Coronal brain slices (2 mm) were obtained after sham or ICH and digital photographs were captured.
We next sought to determine the cellular localization of the increased expression of survivin using immunohistochemistry at 3 days post-ICH. As was observed with Western blotting (Fig. 1), basal survivin expression was undetectable in sham-operated mice (Fig. 3A and B, top panels). In contrast, the nuclear neuronal-specific marker NeuN exhibited a punctate, homogenous appearance throughout the uninjured striatum. By 72 h post-ICH, a considerable reduction in NeuN immunoreactivity and a diffuse appearance were observed within the peri-hematomal region (Fig. 3A). This pattern of staining is consistent with widespread neuronal injury. In accord with the results from Western blotting, survivin was significantly upregulated in the injured striatum adjacent to the hematoma. Notably, survivin did not co-localize with NeuN. Conversely, a remarkable level of survivin expression was observed in GFAP-positive cells exhibiting classical reactive astrocyte morphology (Fig. 3B) in the injured striatum, compared to sham-operated mice.
FIG. 3.

Cellular localization of survivin after intracerebral hemorrhage (ICH). (A and B) Dual-label fluorescence immunohistochemistry was performed for survivin and the neuronal nuclear marker NeuN, or the astrocyte-specific marker glial fibrillary acidic protein (GFAP), in sham-operated mice or at 3 days post-ICH (scale bar=20 μm).
Increased astrocytic proliferation following ICH
Reactive gliosis is characterized by increased cellular proliferation. Thus we next determined whether the increase in GFAP immunoreactivity was associated with increased cellular proliferation after ICH. Consistent with this possibility, expression of the proliferative marker PCNA was increased within the peri-hematoma region beginning at day 3 post-ICH, compared to sham mice with levels remaining elevated through day 7 (Fig. 4A and C). This pattern of expression temporally mirrored the expression pattern of the reactive astrocyte marker GFAP, suggesting the increase in PCNA may occur within reactive glial cells (Fig. 4A and B). Immunohistochemistry revealed increased expression of PCNA in GFAP-positive astrocytes, supporting the notion that ICH induces delayed astrocytic proliferation and reactivity (Fig. 4D).
FIG. 4.

Astrocyte proliferation following intracerebral hemorrhage (ICH). (A) Representative Western blot illustrating the temporal pattern of expression of the reactive astrocyte marker glial fibrillary acidic protein (GFAP), and proliferating cell nuclear antigen (PCNA), a proliferative marker in sham-operated mice (S), or at 1, 3, 5, or 7 days post-ICH. Data were normalized to β-actin to control for equal protein loading. Right side panels depict the densitometric analysis of Western blotting data. Quantification of (B) GFAP expression (n=3/group), or (C) PCNA expression (n=3–9/group) was normalized to β-actin. Data were analyzed using one-way analysis of variance followed by the Student-Newman-Keuls post-hoc test (*p<0.05, ***p<0.001 versus sham animals). (D) Dual-label fluorescence immunohistochemistry was performed for GFAP and PCNA in sham-operated mice or at 3 days post-ICH (scale bar=20 μm).
Survivin inhibition attenuates glial cell proliferation
We next investigated whether the induction of survivin in reactive astrocytes functionally promoted glial cell proliferation after ICH. Dual immunohistochemistry revealed an overlap between survivin and PCNA-positive cells (Fig. 5). Notably, 36% of cells expressing survivin were also immunoreactive for PCNA, suggesting that survivin may contribute to astrocytic proliferation after ICH. Moreover, the triple-label immunohistochemical analysis revealed a remarkable co-localization of survivin in proliferating astrocytes (Fig. 6).To further define the role of survivin in the astrocyte proliferation we inhibited survivin expression in glial cells. Consistent with astrocytes under physiological conditions in vivo, primary astrocyte cultures are quiescent and do not express detectable protein levels of survivin (data not shown). In contrast, the human U87MG glial cell line expresses survivin and exhibits a high proliferation rate. Stable transduction of a survivin shRNA in U87MG (Fig. 7A and B) resulted in abnormally large and flattened cells with decreased cellular proliferation, as assessed by attenuated PCNA expression (Fig. 7A and C), and by a reduction in cell numbers (Fig. 7D). Together, these findings suggest that increased survivin expression may promote the proliferative phenotype in reactive astrocytes.
FIG. 5.
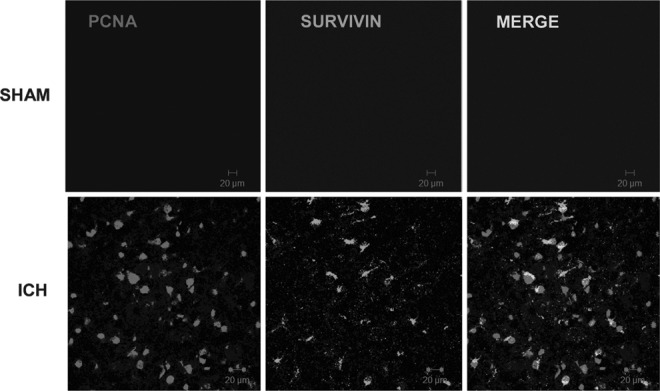
Relationship between survivin expression and cellular proliferation. Dual-label fluorescence immunohistochemistry was performed for proliferating cell nuclear antigen (PCNA), a cellular proliferation marker, and survivin, in sham-operated mice or at 3 days post-intracerebral hemorrhage (ICH). Images were obtained in peri-hematomal brain tissue after ICH or in the comparable brain region of sham-operated mice (scale bar=20 μm).
FIG. 6.

Relationship between survivin expression and astrocyte proliferation. Triple-label fluorescence immunohistochemistry was performed for proliferating cell nuclear antigen (PCNA), and survivin, and glial fibrillary acidic protein (GFAP), in sham-operated mice or at 3 days post-ICH. The arrows and box indicate co-localization of survivin, PCNA, and GFAP. Images were obtained in the peri-hematomal brain tissue after ICH, or in the comparable brain region of sham-operated mice (scale bar=20 μm). The bottom-most ICH panels show magnified images (scale bar=10 μm) of co-localization of survivin, PCNA, and GFAP.
FIG. 7.

Survivin promotes glial proliferation. Stable expression of survivin shRNA represses proliferating cell nuclear antigen (PCNA) expression. (A) Survivin knockdown and PCNA expression were demonstrated by Western blots. Data were normalized to β-actin to control for equal protein loading. Experiments are representative of three independent trials. The right panels depict β-actin-normalized densitometric quantification of (B) survivin and (C) PCNA expression. The data were analyzed using Student's t-test (**p<0.01, *p<0.05 versus control shRNA transduced cells). (D) Stable expression of survivin shRNA represses cellular proliferation in human U87MG glial cells, compared to cells expressing control shRNA. The effect of survivin knockdown on proliferation was analyzed over a 1-week period. Experiments are representative of two independent trials. The data were analyzed using Student's t-test (**p<0.01, *p<0.05 versus control shRNA transduced cells).
Discussion
ICH is a devastating neurological injury that is widely regarded as the least treatable form of stroke. ICH patients frequently exhibit acute neurological deterioration over the first hours and days following presentation, due to the hematoma formation and expansion. Although controversial, current treatment of ICH patients involves hematoma evacuation and supportive measures to reduce patient mortality; however, therapeutic options to enhance recovery are lacking, at least in part, due to a poor mechanistic understanding of the brain response to injury. As such, a large percentage of ICH survivors exhibit long-term neurological deficits and rarely regain full functional independence. Herein we identify for the first time astrocytic survivin as a potential therapeutic target to modulate the gliotic response after ICH, which could lead to novel medical treatment modalities after brain injury.
ICH induces a complex set of molecular and cellular changes within the brain. Following vessel rupture, blood accumulates within the parenchyma to form a space-occupying hematoma that continues to expand to promote neurological demise.25 Hemolysis attenuates the mass effect by reducing hematoma volume; however, this process also generates hemoglobin oxidation products which persist in the brain parenchyma for weeks after ICH, and may result in cell death, vasogenic edema, and poor outcomes.26,27 Based on the established role of secondary brain injury in influencing patient outcomes, pre-clinical research has largely focused on the identification of novel neuroprotective molecules. Unfortunately, to date no therapies have been successfully translated to clinical use in humans, suggesting the need for alternative approaches to improve long-term patient outcomes following ICH.
The neurovascular unit (NVU) is comprised of neurons, astrocytes, and blood vessels, which are anatomically in juxtaposition. Astrocytes, the predominant cell type within the NVU, maintain neurovascular function under physiological conditions; however, controversy remains about whether reactive astrocytes are beneficial or detrimental after brain injury.28 Astrocytes display increased cellular hypertrophy and enhanced expression of inflammatory cytokines after an injury, which stimulate an immune reaction to clear cellular debris and permit axonal regeneration and vascular remodeling.28 Reactive astrocytes also exhibit profound genetic changes that include increased expression of GFAP, S100β, and vimentin, as well as extracellular matrix components that function to form a glial scar, restricting secondary tissue injury from uninjured brain regions.29,30 As serum levels of both GFAP and S100β directly correlated with hematoma volume in patients, reactive astrocytes may exert an important influence on the brain's response to hemorrhage.31–34
In the present report, elevated expression of GFAP and the morphological appearance of reactive astrocytes were prominently detected within the peri-hematoma tissue beginning at 3 days post-ICH. Notably, the phenotypic manifestation of reactive gliosis temporally and spatially paralleled the induction of PCNA, an established marker of cellular proliferation, suggesting the presence of actively-proliferating astrocytes. Although the functional significance of the gliotic changes was not addressed in the present study, it is interesting to note that spontaneous hematoma resolution and neurological improvement were both initiated at 3 days post-ICH,11 coinciding with the development of the glial response observed herein.
We next sought to define the molecular basis for the increased astrocytic proliferation seen after ICH. Herein we observed a strong upregulation of the IAP family member survivin, primarily within GFAP-positive astrocytes, beginning at 3 days post-ICH, correlating with the expression of the proliferative marker PCNA. Interestingly, a similarly delayed induction in survivin expression was noted preferentially within astrocytes following traumatic brain injury,35 or after neuronal excitotoxicity.36 Although the functional significance of survivin after brain injury remains unexplored, it is notable that survivin co-localized with cleaved caspase-3, a marker of apoptotic cells, in surviving glial cells following excitotoxicity in the immature rat brain.36 Together, these data suggest that the increase in survivin expression may represent a conserved cellular pathway after brain injury, and imply a possible anti-apoptotic role in astrocytes.
The biological functions of IAP proteins such as survivin are determined by the conserved presence of one to three zinc-binding motifs called baculoviral inhibitor of apoptosis repeat (BIR) domains.37 Type I BIR domains are essential for caspase inhibition and promote cellular survival. In contrast, type II BIR domains bind caspases, and act on the cell cycle to modulate cellular proliferation.38 Of the known IAP family members, cIAP-1, cIAP-2, XIAP, and NAIP contain three type I BIR domains, whereas livin, APOLLON/BRUCE, ts-IAP/ML-IAP, and survivin contain a single BIR domain or type II domains.39 In this study, survivin expression was correlated with the induction of the proliferative marker PCNA. This finding is consistent with reports in glioblastoma tumors showing that survivin promotes both cellular growth and survival.40–42 Unfortunately, surviving-knockout mice cannot survive, so we were unable to determine whether survivin inhibition reverses the development of a reactive gliotic phenotype after ICH. However, stable genetic knockdown of survivin expression in human U87MG glial cells significantly attenuated PCNA expression and reduced the rate of cellular proliferation. These data suggest that the induction of survivin may functionally promote astrocytic proliferation and the development of reactive gliosis after brain injury.
The functional significance of reactive astrogliosis remains highly controversial following brain injury, with both beneficial and detrimental effects reported.28,43 Thus it remains unclear whether gliosis should be therapeutically enhanced or inhibited to improve outcomes. Herein we identified the delayed upregulation of survivin in GFAP-positive astrocytes by 3 days post-ICH. This induction spatially and temporally correlated with the expression of PCNA, a marker of cellular proliferation, suggesting that survivin may represent a conserved and novel therapeutic target to modify the glial response to hemorrhagic brain injuries. Future work by our laboratory will continue to explore the functional significance of survivin in astrogliosis and neurological outcomes after ICH.
Acknowledgments
This work was supported by grants from the National Institute of Health (R01NS065172, R21NS075774) to KMD.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Ribo M. Grotta J.C. Latest advances in intracerebral hemorrhage. Curr. Neurol. Neurosci. Rep. 2006;6:17–22. doi: 10.1007/s11910-996-0004-0. [DOI] [PubMed] [Google Scholar]
- 2.Broderick J.P. Adams H.P., Jr. Barsan W. Feinberg W. Feldmann E. Grotta J. Kase C. Krieger D. Mayberg M. Tilley B. Zabramski J.M. Zuccarello M. Guidelines for the management of spontaneous intracerebral hemorrhage: A statement for healthcare professionals from a special writing group of the Stroke Council, American Heart Association. Stroke. 1999;30:905–915. doi: 10.1161/01.str.30.4.905. [DOI] [PubMed] [Google Scholar]
- 3.Qureshi A.I. Tuhrim S. Broderick J.P. Batjer H.H. Hondo H. Hanley D.F. Spontaneous intracerebral hemorrhage. N. Engl. J. Med. 2001;344:1450–1460. doi: 10.1056/NEJM200105103441907. [DOI] [PubMed] [Google Scholar]
- 4.Abbott N.J. Revest P.A. Romero I.A. Astrocyte-endothelial interaction: physiology and pathology. Neuropathol. Appl. Neurobiol. 1992;18:424–433. doi: 10.1111/j.1365-2990.1992.tb00808.x. [DOI] [PubMed] [Google Scholar]
- 5.Hayashi Y. Nomura M. Yamagishi S. Harada S. Yamashita J. Yamamoto H. Induction of various blood-brain barrier properties in non-neural endothelial cells by close apposition to co-cultured astrocytes. Glia. 1997;19:13–26. [PubMed] [Google Scholar]
- 6.Janzer R.C. Raff M.C. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature. 1987;325:253–257. doi: 10.1038/325253a0. [DOI] [PubMed] [Google Scholar]
- 7.Howard J.L. Cipolle M.D. Anderson M. Sabella V. Shollenberger D. Li P.M. Pasquale M.D. Outcome after decompressive craniectomy for the treatment of severe traumatic brain injury. J. Trauma. 2008;65:380–385. doi: 10.1097/TA.0b013e31817c50d4. [DOI] [PubMed] [Google Scholar]
- 8.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat. Rev. Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- 9.Takano T. Tian G.F. Peng W. Lou N. Libionka W. Han X. Nedergaard M. Astrocyte-mediated control of cerebral blood flow. Nat. Neurosci. 2006;9:260–267. doi: 10.1038/nn1623. [DOI] [PubMed] [Google Scholar]
- 10.Wilson J.X. Antioxidant defense of the brain: a role for astrocytes. Can. J. Physiol. Pharmacol. 1997;75:1149–1163. [PubMed] [Google Scholar]
- 11.King M.D. McCracken D.J. Wade F.M. Meiler S.E. Alleyne C.H. Dhandapani K.M. Attenuation of hematoma size and neurological injury with curcumin following intracerebral hemorrhage in mice. J. Neurosurg. 2011;115:116–123. doi: 10.3171/2011.2.JNS10784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahesh V.B. Dhandapani K.M. Brann D.W. Role of astrocytes in reproduction and neuroprotection. Mol. Cell Endocrinol. 2006;246:1–9. doi: 10.1016/j.mce.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 13.Pfrieger F.W. Barres B.A. Synaptic efficacy enhanced by glial cells in vitro. Science. 1997;277:1684–1687. doi: 10.1126/science.277.5332.1684. [DOI] [PubMed] [Google Scholar]
- 14.Ullian E.M. Christopherson K.S. Barres B.A. Role for glia in synaptogenesis. Glia. 2004;47:209–216. doi: 10.1002/glia.20082. [DOI] [PubMed] [Google Scholar]
- 15.Ullian E.M. Sapperstein S.K. Christopherson K.S. Barres B.A. Control of synapse number by glia. Science. 2001;291:657–661. doi: 10.1126/science.291.5504.657. [DOI] [PubMed] [Google Scholar]
- 16.Ridet J.L. Malhotra S.K. Privat A. Gage F.H. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 1997;20:570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- 17.Adida C. Crotty P.L. McGrath J. Berrebi D. Diebold J. Altieri D.C. Developmentally regulated expression of the novel cancer anti-apoptosis gene survivin in human and mouse differentiation. Am. J. Pathol. 1998;152:43–49. [PMC free article] [PubMed] [Google Scholar]
- 18.Li F. Ambrosini G. Chu E.Y. Plescia J. Tognin S. Marchisio P.C. Altieri D.C. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396:580–584. doi: 10.1038/25141. [DOI] [PubMed] [Google Scholar]
- 19.Altieri D.C. Marchisio P.C. Survivin apoptosis: an interloper between cell death and cell proliferation in cancer. Lab. Invest. 1999;79:1327–1333. [PubMed] [Google Scholar]
- 20.Moon W.S. Tarnawski A.S. Nuclear translocation of survivin in hepatocellular carcinoma: a key to cancer cell growth? Hum. Pathol. 2003;34:1119–1126. doi: 10.1053/j.humpath.2003.07.016. [DOI] [PubMed] [Google Scholar]
- 21.Laird M.D. Sukumari-Ramesh S. Swift A.E. Meiler S.E. Vender J.R. Dhandapani K.M. Curcumin attenuates cerebral edema following traumatic brain injury in mice: a possible role for aquaporin-4? J. Neurochem. 2010;113:637–648. doi: 10.1111/j.1471-4159.2010.06630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sukumari-Ramesh S. Laird M.D. Singh N. Vender J.R. Alleyne C.H., Jr. Dhandapani K.M. Astrocyte-derived glutathione attenuates hemin-induced apoptosis in cerebral microvascular cells. Glia. 2010;58:1858–1870. doi: 10.1002/glia.21055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sukumari-Ramesh S. Singh N. Jensen M.A. Dhandapani K.M. Vender J.R. Anacardic acid induces caspase-independent apoptosis and radiosensitizes pituitary adenoma cells. J. Neurosurg. 2011;114:1681–1690. doi: 10.3171/2010.12.JNS10588. [DOI] [PubMed] [Google Scholar]
- 24.Wakade C. Sukumari-Ramesh S. Laird M.D. Dhandapani K.M. Vender J.R. Delayed reduction in hippocampal postsynaptic density protein-95 expression temporally correlates with cognitive dysfunction following controlled cortical impact in mice. J. Neurosurg. 2010;113:1195–1201. doi: 10.3171/2010.3.JNS091212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dowlatshahi D. Demchuk A.M. Flaherty M.L. Ali M. Lyden P.L. Smith E.E. Defining hematoma expansion in intracerebral hemorrhage: relationship with patient outcomes. Neurology. 2011;76:1238–1244. doi: 10.1212/WNL.0b013e3182143317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xi G. Keep R.F. Hoff J.T. Erythrocytes and delayed brain edema formation following intracerebral hemorrhage in rats. J. Neurosurg. 1998a;89:991–996. doi: 10.3171/jns.1998.89.6.0991. [DOI] [PubMed] [Google Scholar]
- 27.Xi G. Wagner K.R. Keep R.F. Hua Y. de Courten-Myers G.M. Broderick J.P. Brott T.G. Hoff J.T. Role of blood clot formation on early edema development after experimental intracerebral hemorrhage. Stroke. 1998b;29:2580–2586. doi: 10.1161/01.str.29.12.2580. [DOI] [PubMed] [Google Scholar]
- 28.Laird M.D. Vender J.R. Dhandapani K.M. Opposing roles for reactive astrocytes following traumatic brain injury. Neurosignals. 2008;16:154–164. doi: 10.1159/000111560. [DOI] [PubMed] [Google Scholar]
- 29.Busch S.A. Silver J. The role of extracellular matrix in CNS regeneration. Curr. Opin. Neurobiol. 2007;17:120–127. doi: 10.1016/j.conb.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 30.Rolls A. Shechter R. Schwartz M. The bright side of the glial scar in CNS repair. Nat. Rev. Neurosci. 2009;10:235–241. doi: 10.1038/nrn2591. [DOI] [PubMed] [Google Scholar]
- 31.Delgado P. Alvarez Sabin J. Santamarina E. Molina C.A. Quintana M. Rosell A. Montaner J. Plasma S100B level after acute spontaneous intracerebral hemorrhage. Stroke. 2006;37:2837–2839. doi: 10.1161/01.STR.0000245085.58807.ad. [DOI] [PubMed] [Google Scholar]
- 32.Foerch C. Curdt I. Yan B. Dvorak F. Hermans M. Berkefeld J. Raabe A. Neumann-Haefelin T. Steinmetz H. Sitzer M. Serum glial fibrillary acidic protein as a biomarker for intracerebral haemorrhage in patients with acute stroke. J. Neurol. Neurosurg. Psychiatry. 2006;77:181–184. doi: 10.1136/jnnp.2005.074823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.James M.L. Blessing R. Phillips-Bute B.G. Bennett E. Laskowitz D.T. S100B and brain natriuretic peptide predict functional neurological outcome after intracerebral haemorrhage. Biomarkers. 2009;14:388–394. doi: 10.1080/13547500903015784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Volterra A. Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat. Rev. Neurosci. 2005;6:626–640. doi: 10.1038/nrn1722. [DOI] [PubMed] [Google Scholar]
- 35.Johnson E.A. Svetlov S.I. Pike B.R. Tolentino P.J. Shaw G. Wang K.K. Hayes R.L. Pineda J.A. Cell-specific upregulation of survivin after experimental traumatic brain injury in rats. J. Neurotrauma. 2004;21:1183–1195. doi: 10.1089/neu.2004.21.1183. [DOI] [PubMed] [Google Scholar]
- 36.Villapol S. Acarin L. Faiz M. Castellano B. Gonzalez B. Survivin and heat shock protein 25/27 colocalize with cleaved caspase-3 in surviving reactive astrocytes following excitotoxicity to the immature brain. Neuroscience. 2008;153:108–119. doi: 10.1016/j.neuroscience.2008.01.054. [DOI] [PubMed] [Google Scholar]
- 37.Dubrez-Daloz L. Dupoux A. Cartier J. IAPs: more than just inhibitors of apoptosis proteins. Cell Cycle. 2008;7:1036–1046. doi: 10.4161/cc.7.8.5783. [DOI] [PubMed] [Google Scholar]
- 38.Miller L.K. An exegesis of IAPs: salvation and surprises from BIR motifs. Trends Cell Biol. 1999;9:323–328. doi: 10.1016/s0962-8924(99)01609-8. [DOI] [PubMed] [Google Scholar]
- 39.Clem R.J. Hardwick J.M. Miller L.K. Anti-apoptotic genes of baculoviruses. Cell Death Differ. 1996;3:9–16. [PubMed] [Google Scholar]
- 40.Blum R. Jacob-Hirsch J. Rechavi G. Kloog Y. Suppression of survivin expression in glioblastoma cells by the Ras inhibitor farnesylthiosalicylic acid promotes caspase-dependent apoptosis. Mol. Cancer Ther. 2006;5:2337–2347. doi: 10.1158/1535-7163.MCT-06-0193. [DOI] [PubMed] [Google Scholar]
- 41.Chakravarti A. Zhai G.G. Zhang M. Malhotra R. Latham D.E. Delaney M.A. Robe P. Nestler U. Song Q. Loeffler J. Survivin enhances radiation resistance in primary human glioblastoma cells via caspase-independent mechanisms. Oncogene. 2004;23:7494–7506. doi: 10.1038/sj.onc.1208049. [DOI] [PubMed] [Google Scholar]
- 42.Das A. Tan W.L. Teo J. Smith D.R. Expression of survivin in primary glioblastomas. J. Cancer Res. Clin. Oncol. 2002;128:302–306. doi: 10.1007/s00432-002-0343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sofroniew M.V. Reactive astrocytes in neural repair and protection. Neuroscientist. 2005;11:400–407. doi: 10.1177/1073858405278321. [DOI] [PubMed] [Google Scholar]