

Abstract
Rheumatoid arthritis (RA) is a polygenic autoimmune disease primarily affecting the synovial joints. Numerous animal models show similarities to RA in humans; some of them not only mimic the clinical phenotypes but also demonstrate the involvement of homologous genomic regions in RA. This paper compares corresponding non-MHC genomic regions identified in rodent and human genome-wide association studies (GWAS). To date, over 30 non-MHC RA-associated loci have been identified in humans, and over 100 arthritis-associated loci have been identified in rodent models of RA. The genomic regions associated with the disease are designated by the name(s) of the gene having the most frequent and consistent RA-associated SNPs or a function suggesting their involvement in inflammatory or autoimmune processes. Animal studies on rats and mice preferentially have used single sequence length polymorphism (SSLP) markers to identify disease-associated qualitative and quantitative trait loci (QTLs) in the genome of F2 hybrids of arthritis-susceptible and arthritis-resistant rodent strains. Mouse GWAS appear to be far ahead of rat studies, and significantly more mouse QTLs correspond to human RA risk alleles.
1. Introduction
Rheumatoid arthritis (RA) is a polygenic systemic autoimmune disease that mainly affects the synovial joints, causing chronic inflammation and profound tissue destruction in affected patients. The pathological features of RA include leukocyte infiltration of the synovial tissue (mainly T cells and macrophages), autoantibody production (e.g., against immunoglobulins, citrullinated peptides, or tissue-restricted antigens), the accumulation of inflammatory cells (mainly neutrophils) in the joint fluid, the proliferation of synovial fibroblasts, and the formation of pannus; collectively, these features result in the destruction of articular cartilage and bone erosion. The identification of genetic alterations and variations in RA (involving either the major histocompatibility complex (MHC) or non-MHC genes) and an understanding of their functional consequences may impact the diagnosis, therapy, and prevention of RA [1], an autoimmune disease that affects approximately 1% of the human population. No other autoimmune disease appears in so many different clinical forms or is characterised by such heterogeneous and diverse clinical symptoms and laboratory tests. As a consequence, there are many experimental animal models attempting to mimic the multiple clinical symptoms of RA.
Animal studies may help to fill the gaps in human genome-wide association studies (GWAS) by allowing for gene mapping and functional studies, which cannot be performed in human patients and may yield greater insights into the mechanisms of autoimmune T and B cell responses in RA [2–4]. While the various animal models are tremendously helpful for investigating certain aspects of the human disease, none of these models recreates the full spectrum of diseases collectively called RA. Notably, thousands of investigators and pharmaceutical companies use animal models of RA, perhaps without understanding the differences among the different subtypes of this disease and the corresponding animal models [2–5]. Based upon the clinical, immunological, and genetic components, the most appropriate animal models for RA seem to be (i) those that use genetically controlled systemic autoimmune joint diseases, (ii) those in which the MHC (class II molecules) plays a crucial role, (iii) those in which both T and B cells are involved, and (iv) those that apply (auto)antigenic molecules of cartilage or joint tissues for provoking (“targeting”) synovial joint inflammation.
Among the animal models of RA that fulfil the above listed criteria from a genetic point of view and that are characterised by the presence of the most valuable biomarkers, such as rheumatoid factor (RF) and anticitrullinated peptide antibodies (anti-CCP or ACPA), the closest genetic, and clinical models of RA appear to be cartilage proteoglycan (PG) aggrecan-induced arthritis (PGIA) [6, 7] and cartilage type II collagen- (CII-) induced arthritis (CIA) [3, 8–11].
2. Progresses and Limitations of Human and Animal GWAS
In addition to certain MHC (or human leukocyte antigen (HLA) in humans) class-II alleles on human chromosome 6 that are most commonly (over 40%) associated with a genetic risk for RA [1, 12–16], currently there are 31 non-MHC RA risk alleles that have been confirmed by GWAS and meta-analyses [17, 18]. Many of these risk alleles are weak and are frequently “specific” for different ethnic groups or subpopulations, but there are at least 25 strong RA risk alleles within 23 non-MHC loci in the human genome that control disease susceptibility or severity [19]. These human RA risk alleles were identified and confirmed using hundreds of thousands of single nucleotide polymorphisms (SNPs) and designated by the name of the gene in which the SNP occurred most frequently. However, except for very few cases, none of the genetic risk loci identified to date represent the disease-causing or disease-promoting gene, in which mutations have occurred. SNPs, similar to postal ZIP codes, define only certain regions where a number of genes or noncoding elements (streets in the analogy) are located, but they do not define exact addresses. These risk loci or alleles defined by various numbers and frequencies of SNPs indicate only a chromosome region (carrying dozens to hundreds of genes) expected to have one or a few functionally defective genes involved in the pathomechanism of RA [20]. In fact, these RA-associated SNP risk alleles may indicate a risk for RA or a number of other autoimmune diseases [1, 19, 21–29], or their combination may be used as “predictive” markers for effective therapy selection. Due to extreme heterogeneity in the human population, the highly motivated and exciting early-stage studies have led to the current frustration, and only confirmatory or treatment-related meta-analysis studies have been published during the past couple of years.
In contrast to human studies using heterogeneous populations, there is a chance to use the combination of various arthritis-susceptible and arthritis-resistant inbred strains for GWAS and to identify disease-associated QTLs. There are over a hundred non-MHC genetic risk alleles identified in the rat and mouse models of RA. However, a surprisingly small number of these rodent QTLs (especially in rat arthritis models) correspond to the RA risk alleles or corresponding area in the syntenic human genomic area. Many of these rodent QTLs are listed as new discoveries and were never coordinated as the human studies were, and thus, they are frequently represented by duplicate or triplicate names when described by different research groups. Another limitation of these animal studies is that the different QTLs may represent different, probably over a dozen, phenotypes (e.g., onset, susceptibility, severity, tissue destruction, etc.) in combination with the presence or level of various biomarkers, such as autoantibodies or cytokines either in sera or in vitro stimulated spleen or lymph node cultures. The PCR-based method (single sequence length polymorphism, SSLP) used for the identification of QTLs in either mice or rats is a different technique from SNP microarray-based screening of the human genome, but the principal of the final linkage analysis is based on the same concept. Therefore, as it happened in human SNP-based studies where different sizes and types of arrays, populations, clinical phenotypes, disease durations, environmental factors, and responsiveness to treatment types create a heterogeneous picture of risk alleles, similar heterogeneity in genotype, phenotype, and biomarker distribution exists in animal studies.
3. Significance of Animal Models of RA
Human genetic studies are expected to be fast but fairly less reliable because either the function of the SNP-identified gene or intergenic region is unknown or the consequence of the mutation found in a gene (e.g., transcription factor binding site) is very rarely known in humans. Animal studies are slow and laborious, but using appropriate genetic combinations (selected combinations of intercrosses and GWAS of F2 hybrids, congenic/subcongenic, and interval-specific congenic (IVSC) processes, and genomic sequences of the target inbred region) they can find disease-promoting genes, even with a relatively weak disease-modulating effect. Moreover, animal models allow us to investigate the role of a single gene and the mechanisms of the disease, allowing development of more effective and appropriate treatments. These animal studies, however, are valuable only if they focus on the disease-affecting/causing gene(s) in humans. Human genetics often arrives at a dead end because the disease-affecting genes are unknown [20]. Furthermore, due to the enormous heterogeneity of the human population, it is not feasible to sequence large genomic areas of thousands of people before careful selection of a relatively homogeneous subpopulation of RA patients. This selection requires extensive bioinformatics analysis comparing hundreds or thousands of disease-associated SNPs and RA patients to identify homogeneous (identical, or close to identical) SNP combinations and allele frequency for the selected RA-associated locus in affected patients. In a recent study, we compared a few hundred seropositive RA patients (all carrying the PTPN22 risk allele) but found only a dozen patients with the same SNP combinations. We expect that after high-throughput sequencing, there may be only a few (2–4) RA patients who show high genomic similarity within a small genomic region using bioinformatics analysis, but the appropriate programs and appropriate functional tests are not available at the moment.
Although there are limitations surrounding both human and animal genome-wide screening studies, in the future, the two lines of research may support similar findings and be consolidated to provide additional insight. There are a few animal models of RA that have identified highly significant disease-associated loci. Induced autoimmune models of RA usually represent an accelerated form of RA. For example, both CIA and PGIA are known to involve MHC class-II-restricted antigen presentation and generation of T cells and autoantibodies that cross-react with self (mouse) antigens such as mouse CII or PG [3, 6, 8, 10, 30, 31]. In addition to MHC, which controls at least 40–50% of the genomic susceptibility to RA, both models require an arthritis-prone non-MHC genetic background. Nonobese diabetic (NOD) mice are resistant to both CIA and PGIA. However, when KRN T cell receptor (TCR) transgenic mice were intercrossed with NOD mice, it resulted in the K/BxN model, which develops spontaneous arthritis. The KRN TCR is specific for the bovine pancreas ribonuclease and apparently cross-reacts with glucose-6-phosphate isomerase (GPI) [32–34]. However, the spontaneous K/BxN model is irrelevant for genomic studies. It has no MHC linkage, a ubiquitous (auto)antigenic component exists (which is present in all mammalian cells [35]), and anti-GPI antibodies can rarely be detected in RA patients [36–38]. The sera of these spontaneously arthritic mice can transfer arthritis to any strain of mice (serum-transfer arthritis); thus, the genetic components of either the K/BxN or serum-transfer arthritis models are vague and unclear. However, a genome-wide screening of serum-transfer-induced arthritis in heterogeneous stock (HS) mice resulted in very interesting results [39]. QTLs identified on six chromosomes matched two human RA risk alleles (TRAF1/C5 and PADI4 loci), of which the Traf1/Hc locus on mouse chromosome 2 (mChr2) is a dominant QTL in both CIA (mCia2 and mCia4) and PGIA (Pgia2) (Table 1).
Table 1.
Arthritis-associated QTLs of mice corresponding to human SNP-based GWAS.
| mChr | Markers | Region (Mbp) | Locus name (mouse) | Peak marker name | Peak marker position |
Mouse gene And its position (Mbp) |
References (mouse) |
Corresponding human Chr position (Mbp) | Human locus name |
||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | D1Mit188– D1Mit122 |
3.9–40.5 | mCia14 | D1Mit244 | 37.6 | Aff3 | (38.2) | [9, 40, 92, 93] | 2q11 | (100.2) | AFF3 |
| 1 | D1Mit426– D1Mit209 |
90.4–193.3 |
mCia9
Pgia1 |
D1Mit166 | 178.3 | Fcgr2b | (170.9) | [57, 92, 93] | 1q23 | (172.9) | FCGR2A |
| Pia(Chr1) | D1Mit36 | 171.1 | |||||||||
| 2 | D2Mit79– D2Mit237 |
21.9–40.8 |
mCia2
mCia4 |
D2Mit238 | 33.9 | Traf1 /Hc | (34.8) | [2, 40–44] | 9q33 | (123.6) | TRAF1/ C5 |
|
Pgia2
HS |
D2Mit81 | 33.4 | |||||||||
| 3 | D3Mit75– D3Mit284 |
100.5–103.6 |
mCia21
mCia22 |
D2Mit75 D2Mit284 |
100.5 103.6 |
Cd2
Ptpn22 |
(100.5)
(103.6) |
[45, 46] |
1p23
1p13 |
(114.3)
(117.3) |
CD22
PTPN22 |
| 3 | D3Mit141.1–D3Mit323 | 93.0–152.4 |
Pgia26
(+3 subloci) |
D3Mit158 | 109.1 |
Cd2
Ptpn22 |
(100.5)
(103.6) |
[47, 47, 48, 57, 92, 93] |
1p13
1p23 |
(114.3)
(117.3) |
CD22
PTPN22 |
| Pia-Chr3 | D3Mit100 | 96.8 | |||||||||
| 5 | D5Mit388– D5Mit256 |
33.8–63.3 |
Pgia16
mCia13 |
D5Mit233 | 53.0 | Rbjp | (53.9) | [9, 44, 49] | 4p15 | (25.8) | RBJP |
| 6 | D6Mit86– D6Mit318 |
4.4–65.1 | Pgia19 | D6Mit267 | 29.2 | Irf5 | (29.4) | [40, 50, 93] | 7q32 | (128.2) | IRF5 |
| 10 | D10Mit206– D10Mit130 |
13.8–65.9 |
Pgia6
Pgia6b |
D10Mit124 | 20.9 |
Tnfaip3
Prdm1 |
(18.7)
(44.1) |
[9, 40, 43, 92, 93] |
6q23
6q21 |
(138.2)
(106.5) |
TNFAIP3
PRDM1 |
| 10 | D10Mit12– D10Mit269 |
98.9–128.4 |
mCia8
(“locus 5”) AIL(Chr10) |
D10Mit102 D10Mit261 |
120.3 85.0 |
Kif5a
Pip4k2c |
(126.7)
(126.6) |
[40, 50, 58, 58, 94] |
12q13
12q13 |
(57.9)
(57.9) |
KIF5A
PIP4K2C |
| 13 | D13Mit258– D13Mit78 |
95.6–119.6 |
Pgia15
mCia19 |
D13Mit51 D13Mit53 |
105.3 113.1 |
Ankrd55
IL6st |
(113.0)
(113.2) |
[9, 44, 51, 92, 93] |
5q11
5q11 |
(55.4)
(55.2) |
ANKRD55
IL6ST |
| 15 | D15Mit121– D15Mit242 |
57.9–90.2 | Pgia9 | D15Mit28 | 74.4 |
IL2b
Bik |
(78.3)
(83.4) |
[9, 40, 44, 62, 92, 93] |
22q12
8p3 |
(37.5)
(11.3) |
IL2RB
BIK |
| 15 | D15Mit279– D15Mit192 |
74.0–92.7 | mCia35–mCia37 | D15Mit159 | 87.3 |
IL2b
Bik |
(78.3)
(83.4) |
[53] |
22q12
8p3 |
(37.5)
(11.3) |
IL2RB
BIK |
| 18 | D18Mit51– D18Mit80 |
61.3–77.0 | Pgia11 | D18Mit81 | 66.7 | Ptpn2 | (67.8) | [9, 40, 43, 93] | 18p11.3–p11.2 | (12.8) | PTPN2 |
SKG mice develop arthritis due to a spontaneous mutation in the SH2 domain of Zap70 [40]. Because the Zap70-mutation causes defective TCR signalling, it has been postulated that autoreactive T cells escape thymic deletion and accumulate in the periphery of SKG mice [40]. Altered thymic selection in SKG mice leads to the survival of otherwise negatively selected T cell clones that then spontaneously differentiate into Th17 cells in the periphery and attack the joints. In contrast, interleukin 1 (IL-1) receptor antagonist protein (IRAP) knock-out mice develop spontaneous arthritis due to increased production of proinflammatory cytokines (IL-1β, IL-6, IL-17, and tumour necrosis factor-alpha, TNFα) and autoantibodies in the absence of negative regulation of IL-1 signalling [41, 42]. In addition, human TNFα-expressing transgenic mice develop spontaneous chronic erosive arthritis due to their continuous production of TNFα [43]. This arthritis appears to be a highly simplified proinflammatory cytokine-induced arthritis; thus, it is similar to the serum transfer-induced arthritis (using anti-GPI antibody-containing sera from arthritic K/BxN mice) [44] and the collagen monoclonal antibody cocktail or LPS-induced arthritis (CAIA) [45–47].
All of these models, directly or indirectly, have contributed insights into the complex mechanisms behind RA and have facilitated the development of current therapeutics and biologics. It is important to note that all the previously mentioned experimental animal models of arthritis develop at a relatively young age (beginning at ~4–6-weeks), except PGIA [48], and that arthritis develops in SKG and IRAP-deficient mice only in the BALB/c genetic background [40–42]. This arthritis-prone BALB/c genetic background has also been shown to predispose mice to PGIA [7], human G1 domain-induced arthritis (GIA) [49], link protein [50] or human cartilage HC-gp39 protein [51]. The incidence of spontaneous arthritis in retired, breeder, wild-type BALB/c females is estimated at 0.5–1.0% (TTG, unpublished data), which is close to the ratio observed in the human population. Additionally, BALB/c mice carrying the HLA-DR4 transgene [52] or expressing a PG (5/4E8 epitope)-specific TCR [53, 54] develop arthritis spontaneously but only at an advanced age [55]. Although there are a number of other animal models of RA, we have listed only those that may have conceptual relevance to this paper. However, except for a relatively few studies [39, 56–58], GWAS in mice has almost exclusively been performed in PGIA and CIA; thus, we compare QTLs identified mostly in these two models with human GWAS and their subsequent meta-analyses (Table 1). Therefore, we summarise only those genomic regions (QTLs) of animal studies that correspond to the human chromosome region where risk alleles were identified in RA, and thus, may help to accelerate human studies. Interval-specific congenic (IVSC) mice representing human RA-associated regions present a high potential for sequencing homogeneous genomic regions, and any genes with potentially pathogenic variants (either in exons, introns or intergenic regions and in disease-promoting or disease-suppressive areas) may guide future human studies in terms of selecting appropriate patient populations for more detailed genetic and epigenetic analysis.
4. Tissue-Restricted (Cartilage) Antigens Can Provoke Arthritis in Genetically Susceptible Mice and May Contribute to the Severity of RA
Cartilage is one of the few immune-privileged tissues in the body in that it is essentially avascular and therefore not subjected to close “internal” immunological surveillance [59]. An incomplete central tolerance is most likely the dominant component of this special immune condition, a tolerance that can be breached when transgenes are expressed in cartilage and the cartilage-specific overexpression is “leaky,” especially in the embryo. Several lines of evidence support this hypothesis. For example, cartilage link protein [60] or otherwise arthritogenic human G1 domain (unpublished data) expression in mice, driven by the rat type II collagen promoter and enhancer, may be detected in cartilage tissue, but the transcript and protein could also be detected in other embryonic tissues. Additionally, when cartilage PG (or CII) is degraded by various matrix metalloproteinases, the newly generated neoepitopes may provoke an autoimmune reaction [61]. Further evidence is provided by posttranslational events (e.g., citrullination), as molecules unrelated to cartilage (e.g., filaggrin [62–64]) are first citrullinated far before the onset of joint inflammation. Subsequently, additional molecules (e.g., fibrinogen, vimentin, type II collagen, PG aggrecan, α-enolase, and a few virus proteins) also undergo posttranslational modifications (citrullination), and the cumulative effect of (auto)immune reactions may breach the immune tolerance in genetically susceptible human individuals.
Although immunity to the cartilage PG aggrecan has been less extensively studied than immunity to type II collagen (CII), cartilage PG is also considered to be a causal factor in rheumatoid joint diseases [65–67]. Either humoral or cellular immunity, or both, to human cartilage PGs have been detected in patients with RA [65–79], and the two most recent studies reported that the citrullinated version of a dominant arthritogenic (5/4E8) peptide of human cartilage PG [80, 81] induced substantial cytokine (IL-17, IL-22, IL-6, TNFα, IFNγ) production by T cells from the majority of RA patients [78, 79]. T cells from the same RA patients responded poorly to the native (noncitrullinated) peptide in both studies, and T cells from healthy subjects did not respond [78] or responded only to the citrullinated peptide by producing IL-6 [79]. Although the majority of RA patients tested were positive for anti-citrullinated cyclic peptide (anti-CCP) antibodies (ACPA), T-cell response to the citrullinated PG peptide was also noted in some ACPA patients [78, 79].
5. Overlapping Genomic Loci of RA and Autoimmune Mouse Models of RA
In this paper, we collected results from GWAS in mice and rats (over 100 QTLs) and compared the QTL localisations to those identified in human studies (over 30 RA-associated loci). It is technically impossible and scientifically unnecessary to cite all these studies; rather, we tried to select those that represent syntenic regions in humans and mice (and rats if available). We cite the most appropriate publications in Table 1 or in the text rather than indicating SNP codes (rsXXXX). The levels of significant association between the same SNP and RA is variable in different papers, and for the novelty of a new meta-analysis, investigators may preferentially use a SNP in close proximity to those that have already been published. In brief, we selected data from RA risk allele groups that also have syntenic regions in rodent studies and show one of a few on-going animal/human studies (mouse Chr3 versus human Chr1) in which the combined information may be not only quantitative but also qualitative (Figures 1 and 2). In other words, two chromosome regions (Figure 1) have not only SSLPs (andSNPs) in the “candidate” target regions but also functional defects in the protein encoded by the mutated gene that may either suppress or promote the onset and severity of arthritis. Thus, these particular mouse studies aid in the discovery of functional defects in disease-associated genes in humans with RA.
Figure 1.
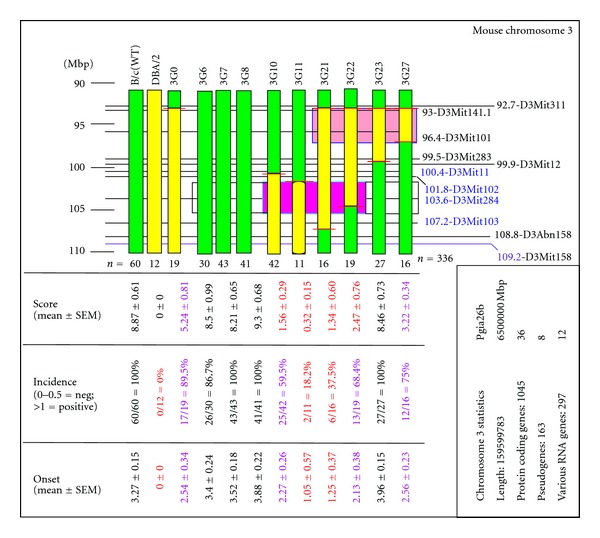
Summary of the genotypes and corresponding clinical phenotypes of parent stains and Pgia26 (3G0) and Pgia26 subloci that were identified in IVSC lines with overlapping chromosome intervals. The original mChr3 region (3G0: 90.4–156.5 Mbp in size) was reduced and separated into several subloci in 27 interval-specific subcongenic (IVSC) lines (3G1-3G27). For simplicity, only a 16.5 Mbp region is shown. Green columns represent BALB/c, and yellow columns represent the DBA/2 chromosome regions. Horizontal black lines with numbers at the right side (and with marker names) are shown. The short red lines crossing the IVSC chromosome region indicate the position between the two markers, where the DBA/2 allele continued as BALB/c [84]. The blue-framed red rectangular area indicates the position of the Pgia26d locus (between 101.4 and 107.2 Mbp); in the worst case, this region may include the entire flanking region between 99.9 and 108.8 Mbp where the disease-promoting gene(s) in BALB/c mice is located (or reciprocally, the suppressive genes in DBA/2). This area contains the most prominent Ptpn22 (protein tyrosine phosphatase non-receptor-22) identified in human GWAS with SNPs, an allele that is associated with many autoimmune diseases. The mutation affecting R620W amino acid appears to affect both peripheral and central B-cell tolerance [120]. Under the worst scenario, this region contains 128 protein-coding genes, 19 miRNAs, 13 pseudogenes, and 9 non-protein-coding transcripts (http://www.ensembl.org/Mus_musculus/Info/Index). Other Pgia26 subloci (with large scales) are presented in Figure 2 with the corresponding human, rat, and mouse RA risk alleles. Another disease-suppressive region (inherited from the DBA/2 strain), between 92.7 Mbp and 96.4/99.9 Mbp position (framed), is currently under sequencing and examination.
Figure 2.
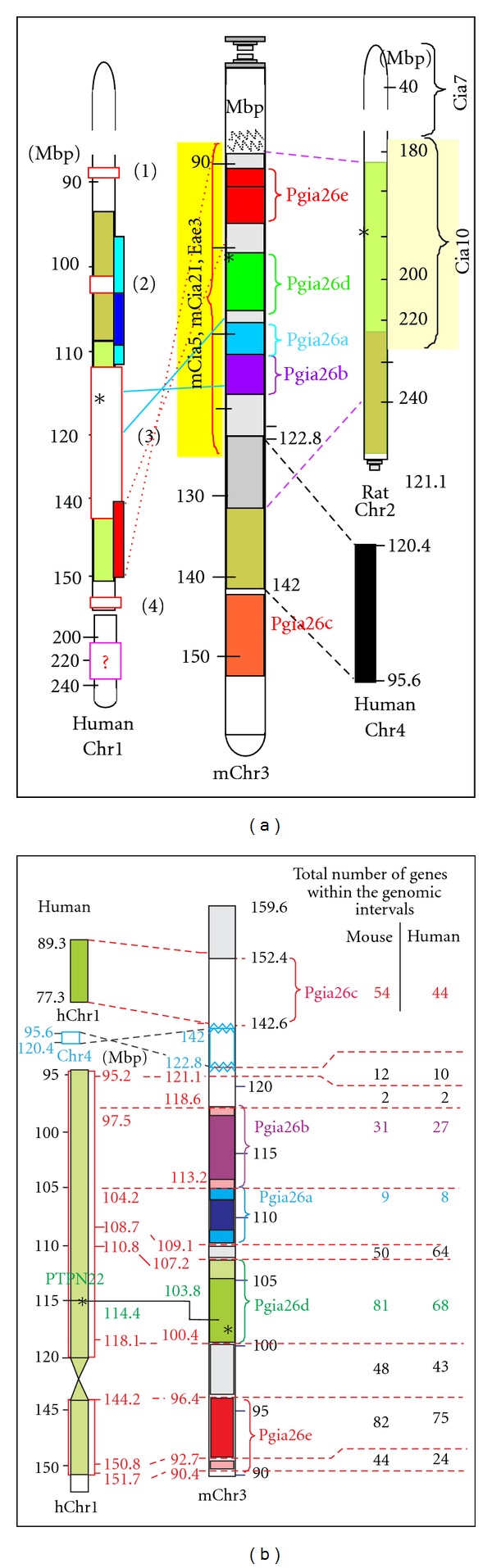
Mouse chromosome 3 (Chr3) with Pgia26 subloci identified in IVSC mice (Figure 1) and corresponding human and rat chromosome regions with their corresponding risk alleles. Panel (a) summarises the location of five Pgia26 subloci with corresponding mouse mCia5 and mCia21 (collagen-induced arthritis) [3, 108, 121], mouse Eae3 (experimental allergic encephalomyelitis) [122, 123] (between mChr1:84.3–126 Mbp), and the corresponding rat Chr2 region of rat Cia10 [124, 125]. The left side of the panel identifies risk alleles on human Chr1 [126], with red-framed boxes and numbers in parentheses indicate the following regions: (1) between 87–89 Mbp [127], (2) 105.4 Mbp [128], (3) 113–142 (including the PTPN22 gene at 114.4) Mbp positions [129–131], and (4) the FCGR family between 158 and 159 Mbp [132, 133]. Panel (b) displays the syntenic risk alleles of human Chr1 and mouse Chr3 (Pgia26a-e) with the number of genes localized in the different chromosome regions.
As mentioned, over 100 rodent QTLs have been described to date, but relatively few are syntenic with any of the 30 human RA risk alleles. In our laboratory, over 5,000 inbred wild-type parents, approximately 500 F1 hybrids (all negative for PGIA, data not shown) and 3,200 F2 hybrids of six different genetic intercrosses were genotyped using a total of 240 SSLP markers. The goal was to identify genetic alterations responsible for individual and overlapping qualitative (binary) QTLs that are linked to PGIA or CIA in the mouse genome and then compare the results with loci identified in human autoimmune diseases, preferably RA. Many of the risk alleles in RA overlap with a number of risk alleles of other autoimmune diseases [19, 21–27, 29], and a number of Pgia and Cia loci [10, 82–87] overlap with chromosomal regions identified in GWAS studies of RA patients [17, 19, 88–91]. CIA was considered as a model of seronegative RA, whereas PGIA, which has both rheumatoid factors and ACPA [7, 49], was considered a seropositive RA model. The overall hypothesis was that genes associated with a QTL in one or more genetic combinations of murine autoimmune arthritis should correspond to genes involved in RA. (A total of 26 loci out of 31 confirmed non-MHC loci were screened for corresponding mouse QTLs. Only those that were found in comparative studies of mouse genome-wide association (GWAS) studies (n = 17) are listed under the “human locus name.” These mouse GWAS studies include over a dozen intercrosses screened in different laboratories. Occasionally, the same (mouse) Cia locus-number appears on different chromosomes in different publications, thus the references corresponding to the appropriate mouse Cia (mCia) loci are listed here. QTL of Pgia (n = 9) and mCia (n = 2) identified in our laboratory are italic and bold faced. Each human locus is listed by the gene-name and chromosome location using the “standard” name of the given RA risk allele; the corresponding mouse region/gene is indicated by the same gene name and location in the mouse genome given by the mega-base pair (Mbp) position (bold-faced). Tissue samples (tails and kidney) of each F2 hybrid mouse are catalogued and stored at −80°C. Many of the F2 hybrids were retested with additional, new markers in confirmatory studies (9 Pgia and 2 mCia loci). The average marker density in these confirmatory studies was 8.2 Mbp. Some of these reference markers shifted slightly after confirmatory studies using high density marker screening. Two QTLs on mouse chromosomes 3 and 15 have overlapping regions; therefore, they are listed in the Table 1 twice due to the information from different studies.)
Although there are a number of weaknesses for both human and animal GWAS, they may supplement and support each other. During the past 15 years, we and others have identified 29 Pgia and 40 Cia loci in different genetic combinations of F2 hybrid mice [3, 10, 11, 57, 58, 82, 86, 95–99] and a couple of corresponding QTLs in rats [100–105]. With a strong confirmation in the literature, we selected QTLs from all (published) mouse genomic studies [10, 56–58, 82–84, 92–96, 98, 106–112] that also correspond to one of the major risk loci of RA confirmed in a number of meta-analyses [19, 29, 90, 113–117]. Table 1 summarises the risk alleles selected that have corresponding genomic regions from human and mouse GWAS. Only QTLs that correspond to at least one major RA-associated locus in the human genome are listed; these QTLs were found on mouse chromosomes 1 (2x), 2, 3 (2x), 5, 6, 10 (2x), 13, 15 (2x), and 18 (a total of 13 QTLs). The list was organised in order of mouse chromosomes. At least one, and possibly two or three, QTLs from various animal studies covered the syntenic chromosome region of human RA-associated loci. Standard abbreviations of genes were used as they are listed in gene bank databases (e.g., http://www.informatics.jax.org/; http://www.ensembl.org/Mus_musculus/Info/Index or http://genome.ucsc.edu/cgi-bin/hgGateway), and many of their known functions are described in publications available from PubMed (http://www.ncbi.nlm.nih.gov/pubmed). Thus, we did not list the full names or discuss the function(s) of these genes used to identify RA susceptibility loci or the “most frequent” associated SNPs of meta-analyses. These “marker-specific” genes were usually located near the unknown genes that might carry the disease-causing genomic defect. For example, SNPs of two of the strongest RA risk alleles, TRAF1/C5 and TNFAIP3/OLIG3, are in the intergenic regions, making it difficult to establish causality of these regions at this moment [20, 88, 118]. Although both TRAF1 and TNFAIP3 are “preferential” gene candidates based on their function in TNF signalling, known to be important in RA [119], none of the genes having SNPs or genomic mutations evidently affect their function.
In the next section, we show an example of how we can integrate information from the human and mouse studies. This method may be one of the potential ways to identify causal variants that map to human RA-associated chromosome regions.
6. Benefits of MHC-Matched Susceptible and Resistant Mouse Strains: IVSC Strains Targeting Human RA Risk Alleles
To eliminate or reduce the dominant effect of MHC in cases where the association of a QTL with an arthritis phenotype has been sufficiently confirmed, one of the most successful alternative approaches is to use MHC-matched arthritis-susceptible and arthritis-resistant strains to establish congenic and subcongenic lines. Either a disease-promoting chromosome region can be “inserted” into a resistant strain, or reciprocally, the same region containing a disease-suppressive allele can be inserted into a fully susceptible genetic background. Either direction is acceptable, but from a practical point of view and based on many congenic experiments during the past decade, the latter solution appears to be more manageable. First, F1 males are selected, for example, from the intercross of a PGIA-susceptible BALB/c female and a resistant DBA/2 male (both MHC H2d) carrying the DBA/2 genomic region of interest. These F1 males are backcrossed several times with wild-type BALB/c females, and the offspring are genotyped for each litter until the N1-NX generations have sufficient numbers of recombination events (and, if possible, overlapping areas) (Figure 1). These Nx males are intercrossed with wild-type BALB/c females, and the resulting heterozygous Nx−1 males and Nx−1 females are intercrossed to establish a homozygous IVSC strain(s).
During the ongoing backcrossing process, fewer and fewer previously heterozygous loci need to be tested by PCR. If a gender effect is expected, it is necessary to replace the Y chromosome with a single reciprocal backcross, but it is both practical and sufficient to do this replacement near the final step.
Subsequently, the chromosome intervals from the resistant strain of a relatively (and usually) large QTL (several cM or Mbp in size) need to be tested for clinical phenotypes. For example (Figure 1), the “Chr3G0” (“3G0”) subcongenic line contains an overlapping region ~66 Mbp in size that significantly affected all clinical phenotypes when compared to either susceptible BALB/c or resistant DBA/2 parental strains [84], a finding that needs to be further confirmed by in vitro tests (i.e., measuring biomarkers). In this case, males from the congenic 3G0 strain can be used to reduce the chromosome interval with new recombination events with matings into inbred BALB/c females. On the other hand, only the critical interval of mChr3 with high-density markers needs to be genotyped because the entire genome was previously genotyped for BALB/c (during the selection of 3G0 congenic line). Then, mice with the most appropriate recombination products are used as founders for fine mapping of chromosome intervals generating IVSC strains. Conceptually, the same backcrossing to the susceptible BALB/c strain and genotyping approach, as described above, are used for the selection of new congenic strains. However, investigators need to (i) focus on the new recombination events within selected chromosome interval using high marker density within the region of interest (e.g., Pgia26) and (ii) genotype both males and females. Depending on the volume of backcrossing (i.e., the number of breeding pairs and offspring) and the shortest chromosome interval achievable after a few generations, we are able to select a number of heterozygous males and females with identical recombination events at different positions (if possible with overlapping regions as shown in Figure 1: e.g., Chr3G0-Chr3G27) to establish homozygous IVSC strains for in vivo and in vitro tests.
To save time, it is practical to genotype both males and females for all new recombination products within the chromosome interval of interest, a locus that corresponds to the selected human RA risk allele. As shown in Figures 2(a) and 2(b) and Table 1, the PTPN22/CD2 human risk locus most likely represents a complex trait on mChr3 (syntenic with hChr1) containing both disease-suppressive and disease-promoting alleles [84]. Distinct regions, alone or in combination, may result in clinically similar phenotypes (Figure 1), while the IVSC-associated biomarkers may show significant differences. Thus, a relatively small IVSC chromosome region may be separated for different genotypes representing similar clinical phenotypes (Figure 1, only the centromeric region of the mapped mChr3 is shown). However, while clinical phenotypes are comparable, fundamentally different genes in nearby chromosome regions may control disease susceptibility, onset and severity. Needless to say, fine mapping of chromosome regions and selecting narrow genomic regions with high probability for successful genomic high-throughput sequencing might be difficult, if not impossible, to complete using RA patients from the heterogeneous human population. Further, this highly specific and laborious animal study is valuable only if it represents human relevance, that is, if the corresponding region where the human risk allele was localised had already been identified.
Figure 2 shows simplified schematics comparing the previously outlined IVSC approach (Pgia26 on mChr3) in combination with mouse (mCia) and rat CIA loci syntenic with the RA loci identified on human Chr1. Colours, numbers of genes, locations of syntenic genomic loci, and their flanking regions are indicated in Figure 1 and legend. With the advent of genome sequencing techniques, SureSelect Target Enrichment kit (Agilent, San Diego, CA, USA), library amplification and Illumina parallel sequencing methods made it realistic to oversequence 10–30 Mbp of homogeneous genomic regions from inbred IVSC strains and compare sequences with parent strains (susceptible versus resistant). It is also a reasonable approach to confirm the function of arthritis-susceptible or arthritis-resistant murine strains with transgenic methodologies. Today, the real challenge in human genetics is to find and select appropriate human patients with nearly identical genomic region(s) for high-throughput genomic sequencing due to the extreme heterogeneity of the human population. While SNP analyses using thousands of samples can give an extremely high statistical power, the same approach (SNP selection for genomic sequencing) is unsuccessful in the selection of human samples [20].
However, there are promising directions based on the combination of human-mouse GWAS. Selected homozygous regions of IVSC mice sequenced first with high-throughput sequencing method and affected genes and/or intergenic (relatively small) regions are genome-sequenced from selected humans with appropriate primers. In fact, a certain number of mutations/SNPs of the syntenic regions (identified in IVSC mouse and confirmed using conventional Sanger sequencing of human genomic DNA) may guide the selection of human RA patients for high-throughput sequencing of the region of interest (Figure 2). Alternatively, for example, if miRNA-related sequences are expected, the high-throughput sequencing of RNAs isolated by cross-linking immunoprecipitation (HITS-CLIP) with antibodies against the RNA-binding protein Argonaut (Ago HITS-CLIP) [134–138] may offer another solution.
7. Overall Summary and Perspectives
Overall, mouse studies, especially with congenic strains, appear to be a fundamental resource for the identification of candidate gene(s) in RA. During the past 15 years or so, almost concurrent with the first human genomic studies in RA, a number of rodent (mouse and rat) GWAS studies have been performed. At approximately the same time, both the human and mouse genome sequencing studies were completed and, simultaneously, unlimited numbers of new markers became available for both species. The number of human studies expanded; tens of thousands of RA patients, along with controls, were genotyped; new and more reliable SNP arrays became available; more risk alleles became identified in RA and in almost all autoimmune diseases. However, after extensive progress in GWAS, the direction of RA research moved towards confirmatory studies of previously tested patients, examinations of different ethnic groups or comparisons of the therapeutic effects of different biologics. Briefly, human studies turned to mainly in silico and meta-analysis studies rather than aimed towards finding causative and functional (aetiological) reasons. The previously identified genomic regions were confirmed using a high marker density, but the large chromosome regions with tens of Mbps in size still remained unmanageable. Only a very few SNPs causing missense mutations proved to be associated with disease, and usually only in a narrow selection of the patient population. However, the number of risk alleles increased, and previously identified marker positions were confirmed.
Unfortunately, animal studies also slowed down, although due to completely different reasons from human studies. Increasing the number of new combinations of disease-susceptible and disease-resistant inbred strains revealed more and more QTLs, but not a disease-causing gene. Recognising the limitations as well as the potential of both human and mouse GWAS, approximately 10 years ago, a number of congenic strains carrying the most promising traits representing the strongest clinical phenotypes were established. These strains carry overlapping traits identified in different animal models and syntenic with genomic regions identified as RA risk alleles. In other words, at the time when the human GWAS explored the most critical RA risk alleles, congenic backcrossing had selected inbred IVSC strains with syntenic regions to the major human risk alleles. We selected two QTLs for more detailed analysis: Pgia26/Cia5/mCia21/Eae3 on mChr3 and rat Cia10, corresponding to the PTPN22/CD2 allele on human Chr1 (Figure 2); Pgia2/Cia2/Cia3 on mChr2 (corresponding to the TRAF1/C5 allele on hChr9). Then, we generated IVSC strains (Figures 1 and 2, Pgia26 is shown). All other congenic and subcongenic strains were cryopreserved. The two major/dominant mouse QTLs were separated into narrow subtraits and simultaneously tested for arthritis susceptibility, for disease onset and severity, and for over 15 biomarkers that might have some potential relevance for RA [84]. Simultaneously, some of the IVSC genomic regions representing homogeneous regions of disease-susceptible and disease-resistant IVSC mice (and the corresponding parent genomic regions) were sequenced, and a few mutated genes were identified (with “known” or completely unknown function). Occasionally, these genes had not been previously associated with arthritis, but all of them had localised in close proximity to a gene used to name the human RA risk alleles. The analyses of these genes and a targeted selection of appropriate human genomic DNA samples used for high-throughput sequencing are currently in progress in a number of laboratories. The approaches and concepts outlined in this paper (especially in Sections 4 and 5) are not the only possible avenues for the identification of the RA (or other autoimmune disease)-related defects in the genome. However, these approaches may allow us to merge currently available results of human GWAS with findings of GWAS and IVSC studies in mice. Nonetheless, to confirm the role of these genes in RA, researchers must identify not only the genomic identity but also the corresponding functional defects in mice analogous with those present in patients with RA. Unfortunately, mechanistic and functional studies, manipulation of the genome, and pretesting of new therapeutic approaches cannot be applied in human patients, which underlines the relevance of and necessity for laborious genetic studies in animal models.
Conflict of Interests
The authors declare no conflict of interests.
Authors' Contribution
T. Besenyei and A. Kadar wrote the first draft of the paper, and then all other authors wrote and corrected the final version of the paper, which was submitted by the corresponding author (Z. Szekanecz).
Acknowledgments
This work was supported by research Grants ETT 315/2009 from the Medical Research Council of Hungary (Z. Szekanecz) and by the TÁMOP 4.2.1/B-09/1/KONV-2010-0007 project. It was cofinanced by the European Union and the European Social Fund (Z. Szekanecz), the NIH (AR040310, AR045652, and AR059356), the Grainger Foundation (Lake Forest, IL, USA) (T. T. Glant and K. Mikecz) and the J. O. Galante Endowment Chair (Rush University Medical Center, Chicago, IL, USA) (T. T. Glant).
Abbreviations:
- Anti-CCP:
Anti citrullinated peptide antibody
- ACPA:
Anti-citrullinated protein antibody
- CII:
Type II collagen
- CAIA:
Collagen antibody-induced arthritis
- CIA:
Type II collagen-induced arthritis
- Chr:
Chromosome
- cM:
Centi-Morgan
- GIA:
Human cartilage PG's G1 domain-induced arthritis
- GPI:
Glucose-6-phosphate isomerase
- GWAS:
Genome-wide association studies
- HLA:
Human leukocyte antigen
- HS:
Heterogeneous stock
- IFNγ:
Interferon gamma
- IL:
Interleukin
- IVSC:
Interval-specific congenic
- mCia:
QTL of mouse CIA
- Mbp:
Mega-base pair
- MHC:
Major-histocompatibility complex
- QTLs:
Quantitative trait loci
- PG:
Cartilage proteoglycan aggrecan
- PGIA:
PG aggrecan-induced arthritis
- Pgia:
QTL of PGIA
- Pia:
QTL of pristane-induced arthritis
- RA:
Rheumatoid arthritis
- RF:
Rheumatoid factor
- SSLP:
Single sequence length polymorphism
- TCR:
T cell receptor
- TNFα:
Tumor necrosis factor alpha.
References
- 1.Kurko J, Besenyei T, Laki J, et al. Genetics of rheumatoid arthritis. doi: 10.1007/s12016-012-8346-7. Clinical Reviews in Allergy & Immunology. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berg WBVD. Lessons from animal models of arthritis over the past decade. Arthritis Research and Therapy. 2009;11(5, article 250) doi: 10.1186/ar2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahlqvist E, Hultqvist M, Holmdahl R. The value of animal models in predicting genetic susceptibility to complex diseases such as rheumatoid arthritis. Arthritis Research and Therapy. 2009;11(3, article 226) doi: 10.1186/ar2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bevaart L, Vervoordeldonk MJ, Tak PP. Evaluation of therapeutic targets in animal models of arthritis: how does it relate to rheumatoid arthritis? Arthritis and Rheumatism. 2010;62(8):2192–2205. doi: 10.1002/art.27503. [DOI] [PubMed] [Google Scholar]
- 5.Cai C, La Cava A. Mimicking self-antigens with synthetic peptides in systemic autoimmune rheumatic diseases. Current Clinical Pharmacology. 2009;4(2):142–147. doi: 10.2174/157488409788184936. [DOI] [PubMed] [Google Scholar]
- 6.Glant TT, Mikecz K, Arzoumanian A, Poole AR. Proteoglycan-induced arthritis in BALB/c mice. Clinical features and histopathology. Arthritis and Rheumatism. 1987;30(2):201–212. doi: 10.1002/art.1780300211. [DOI] [PubMed] [Google Scholar]
- 7.Mikecz K, Glant TT, Poole AR. Immunity to cartilage proteoglycans in BALB/c mice with progressive polyarthritis and ankylosing spondylitis induced by injection of human cartilage proteoglycan. Arthritis and Rheumatism. 1987;30(3):306–318. doi: 10.1002/art.1780300310. [DOI] [PubMed] [Google Scholar]
- 8.Trentham DE, Townes AS, Kang AH. Autoimmunity to type II collagen: an experimental model of arthritis. Journal of Experimental Medicine. 1977;146(3):857–868. doi: 10.1084/jem.146.3.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Courtenay JS, Dallman MJ, Dayan AD. Immunisation against heterologous type II collagen induces arthritis in mice. Nature. 1980;283(5748):666–668. doi: 10.1038/283666a0. [DOI] [PubMed] [Google Scholar]
- 10.Glant TT, Finnegan A, Mikecz K. Proteoglycan-induced arthritis: immune regulation, cellular mechanisms, and genetics. Critical Reviews in Immunology. 2003;23(3):199–250. doi: 10.1615/critrevimmunol.v23.i3.20. [DOI] [PubMed] [Google Scholar]
- 11.Popovic M, Ahlqvist E, Rockenbauer E, Bockermann R, Holmdahl R. Identification of new loci controlling collagen-induced arthritis in mouse using a partial advanced intercross and congenic strains. Scandinavian Journal of Immunology. 2008;68(4):405–413. doi: 10.1111/j.1365-3083.2008.02151.x. [DOI] [PubMed] [Google Scholar]
- 12.Zanelli E, Breedveld FC, De Vries RRP. HLA class II association with rheumatoid arthritis: facts and interpretations. Human Immunology. 2000;61(12):1254–1261. doi: 10.1016/s0198-8859(00)00185-3. [DOI] [PubMed] [Google Scholar]
- 13.Weyand CM, Goronzy JJ. Association of MHC and rheumatoid arthritis HLA polymorphisms in phenotypic variants of rheumatoid arthritis. Arthritis Research. 2000;2(3):212–216. doi: 10.1186/ar90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Plenge RM. Rheumatoid arthritis genetics: 2009 update. Current Rheumatology Reports. 2009;11(5):351–356. doi: 10.1007/s11926-009-0050-0. [DOI] [PubMed] [Google Scholar]
- 15.Bax M, Van Heemst J, Huizinga TWJ, Toes REM. Genetics of rheumatoid arthritis: what have we learned? Immunogenetics. 2011;63(8):459–466. doi: 10.1007/s00251-011-0528-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perricone C, Ceccarelli F, Valesini G. An overview on the genetic of rheumatoid arthritis: a never-ending story. Autoimmunity Reviews. 2011;10(10):599–608. doi: 10.1016/j.autrev.2011.04.021. [DOI] [PubMed] [Google Scholar]
- 17.Craddock N, Hurles ME, Cardin N, et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464(7289):713–720. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cong F, Spencer S, Côté JF, et al. Cytoskeletal protein PSTPIP1 directs the PEST-type protein tyrosine phosphatase to the c-Abl kinase to mediate Abl dephosphorylation. Molecular Cell. 2000;6(6):1413–1423. doi: 10.1016/s1097-2765(00)00138-6. [DOI] [PubMed] [Google Scholar]
- 19.Stahl EA, Raychaudhuri S, Remmers EF, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nature Genetics. 2010;42(6):508–514. doi: 10.1038/ng.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Vries R. Genetics of rheumatoid arthritis: time for a change! Current Opinion in Rheumatology. 2011;23(3):227–232. doi: 10.1097/BOR.0b013e3283457524. [DOI] [PubMed] [Google Scholar]
- 21.Velaga MR, Wilson V, Jennings CE, et al. The codon 620 tryptophan allele of the Lymphoid Tyrosine Phosphatase (LYP) gene is a major determinant of Graves’ disease. Journal of Clinical Endocrinology and Metabolism. 2004;89(11):5862–5865. doi: 10.1210/jc.2004-1108. [DOI] [PubMed] [Google Scholar]
- 22.Smyth D, Cooper JD, Collins JE, et al. Replication of an association between the lymphoid tyrosine phosphatase locus (LYP/PTPN22) with type 1 diabetes, and evidence for its role as a general autoimmunity locus. Diabetes. 2004;53(11):3020–3023. doi: 10.2337/diabetes.53.11.3020. [DOI] [PubMed] [Google Scholar]
- 23.Criswell LA, Pfeiffer KA, Lum RF, et al. Analysis of families in the multiple autoimmune disease genetics consortium (MADGC) collection: the PTPN22 620W allele associates with multiple autoimmune phenotypes. American Journal of Human Genetics. 2005;76(4):561–571. doi: 10.1086/429096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hinks A, Barton A, John S, et al. Association between the PTPN22 gene and rheumatoid arthritis and juvenile idiopathic arthritis in a UK population: further support that PTPN22 is an autoimmunity gene. Arthritis and Rheumatism. 2005;52(6):1694–1699. doi: 10.1002/art.21049. [DOI] [PubMed] [Google Scholar]
- 25.Viken MK, Amundsen SS, Kvien TK, et al. Association analysis of the 1858C > T polymorphism in the PTPN22 gene in juvenile idiopathic arthritis and other autoimmune diseases. Genes and Immunity. 2005;6(3):271–273. doi: 10.1038/sj.gene.6364178. [DOI] [PubMed] [Google Scholar]
- 26.De Jager PL, Sawcer S, Waliszewska A, et al. Evaluating the role of the 620W allele of protein tyrosine phosphatase PTPN22 in Crohn’s disease and multiple sclerosis. European Journal of Human Genetics. 2006;14(3):317–321. doi: 10.1038/sj.ejhg.5201548. [DOI] [PubMed] [Google Scholar]
- 27.Vandiedonck C, Capdevielle C, Giraud M, et al. Association of the PTPN22∗R620W polymorphism with autoimmune myasthenia gravis. Annals of Neurology. 2006;59(2):404–407. doi: 10.1002/ana.20751. [DOI] [PubMed] [Google Scholar]
- 28.Centola M, Szekanecz Z, Kiss E, et al. Gene expression profiles of systemic lupus erythematosus and rheumatoid arthritis. Expert Review of Clinical Immunology. 2007;3(5):797–806. doi: 10.1586/1744666X.3.5.797. [DOI] [PubMed] [Google Scholar]
- 29.Coenen MJH, Gregersen PK. Rheumatoid arthritis: a view of the current genetic landscape. Genes and Immunity. 2009;10(2):101–111. doi: 10.1038/gene.2008.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brand DD, Kang AH, Rosloniec EF. Immunopathogenesis of collagen arthritis. Springer Seminars in Immunopathology. 2003;25(1):3–18. doi: 10.1007/s00281-003-0127-1. [DOI] [PubMed] [Google Scholar]
- 31.Hanyecz A, Berlo SE, Szántó S, Broeren CPM, Mikecz K, Glant TT. Achievement of a synergistic adjuvant effect on arthritis induction by activation of innate immunity and forcing the immune response toward the Th1 phenotype. Arthritis and Rheumatism. 2004;50(5):1665–1676. doi: 10.1002/art.20180. [DOI] [PubMed] [Google Scholar]
- 32.Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87(5):811–822. doi: 10.1016/s0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- 33.Korganow AS, Hong J, Mangialaio S, et al. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity. 1999;10(4):451–461. doi: 10.1016/s1074-7613(00)80045-x. [DOI] [PubMed] [Google Scholar]
- 34.Mangialaio S, Ji H, Korganow AS, et al. The arthritogenic T cell receptor and its ligand in a model of spontaneous arthritis. Arthritis and Rheumatism. 1999;42(12):2517–2523. doi: 10.1002/1529-0131(199912)42:12<2517::AID-ANR3>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 35.Matsumoto I, Maccioni M, Lee DM, et al. How antibodies to a ubiquitous cytoplasmic enzyme may provoke joint-specific autoimmune disease. Nature Immunology. 2002;3(4):360–365. doi: 10.1038/ni772. [DOI] [PubMed] [Google Scholar]
- 36.Kassahn D, Kolb C, Solomon S, et al. Few human autoimmune sera detect GPI (multiple letters) [2] Nature Immunology. 2002;3(5):411–413. doi: 10.1038/ni0502-411b. [DOI] [PubMed] [Google Scholar]
- 37.Kassahn D, Kolb C, Solomon S, et al. Few human autoimmune sera detect GPI (multiple letters) Nature Immunology. 2002;3(5):411–413. doi: 10.1038/ni0502-411b. [DOI] [PubMed] [Google Scholar]
- 38.Matsumoto I, Lee DM, Goldbach-Mansky R, et al. Low prevalence of antibodies to glucose-6-phosphate isomerase in patients with rheumatoid arthritis and a spectrum of other chronic autoimmune disorders. Arthritis and Rheumatism. 2003;48(4):944–954. doi: 10.1002/art.10898. [DOI] [PubMed] [Google Scholar]
- 39.Johnsen AK, Valdar W, Golden L, et al. A genome-wide and species-wide dissection of the genetics of arthritis severity in hetrogeneous stock mice. Arthritis and Rheumatism. 2011;63(9):2630–2640. doi: 10.1002/art.30425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakaguchi N, Takahashi T, Hata H, et al. Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature. 2003;426(6965):454–460. doi: 10.1038/nature02119. [DOI] [PubMed] [Google Scholar]
- 41.Horai R, Saijo S, Tanioka H, et al. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin I receptor antagonist-deficient mice. Journal of Experimental Medicine. 2000;191(2):313–320. doi: 10.1084/jem.191.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakae S, Saijo S, Horai R, Sudo K, Mori S, Iwakura Y. IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(10):5986–5990. doi: 10.1073/pnas.1035999100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keffer J, Probert L, Cazlaris H, et al. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO Journal. 1991;10(13):4025–4031. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Solomon S, Rajasekaran N, Jeisy-Walder E, Snapper SB, Illges H. A crucial role for macrophages in the pathology of K/B × N serum-induced arthritis. European Journal of Immunology. 2005;35(10):3064–3073. doi: 10.1002/eji.200526167. [DOI] [PubMed] [Google Scholar]
- 45.Stuart JM, Dixon FJ. Serum transfer of collagen-induced arthritis in mice. Journal of Experimental Medicine. 1983;158(2):378–392. doi: 10.1084/jem.158.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Englert ME, Ferguson KM, Suarez CR. Passive transfer of collagen arthritis: heterogeneity of anti-collagen IgG. Cellular Immunology. 1986;101(2):373–379. doi: 10.1016/0008-8749(86)90150-4. [DOI] [PubMed] [Google Scholar]
- 47.Holmdahl R, Klareskog L, Rubin K. T lymphocytes in collagen II-induced arthritis in mice. Characterization of arthritogenic collagen II-specific T-cell lines and clones. Scandinavian Journal of Immunology. 1985;22(3):295–306. doi: 10.1111/j.1365-3083.1985.tb01884.x. [DOI] [PubMed] [Google Scholar]
- 48.Tarjanyi O, Boldizsar F, Nemeth P, Mikecz K, Glant TT. Age-related changes in arthritis susceptibility and severity in a murine model of rheumatoid arthritis. Immunity and Ageing. 2009;6, article 8 doi: 10.1186/1742-4933-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Glant TT, Radacs M, Nagyeri G, et al. Proteoglycan-induced arthritis and recombinant human proteoglycan aggrecan G1 domain-induced arthritis in BALB/c mice resembling two subtypes of rheumatoid arthritis. Arthritis and Rheumatism. 2011;63(5):1312–1321. doi: 10.1002/art.30261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y, Guerassimov A, Leroux JY, et al. Induction of arthritis in BALB/c mice by cartilage link protein: involvement of distinct regions recognized by T and B lymphocytes. American Journal of Pathology. 1998;153(4):1283–1291. doi: 10.1016/S0002-9440(10)65673-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Verheijden GFM, Rijnders AWM, Bos E, et al. Human cartilage glycoprotein-39 as a candidate autoantigen in rheumatoid arthritis. Arthritis and Rheumatism. 1997;40(6):1115–1125. doi: 10.1002/art.1780400616. [DOI] [PubMed] [Google Scholar]
- 52.Bárdos T, Zhang J, Mikecz K, David CS, Glant TT. Mice lacking endogenous major histocompatibility complex class II develop arthritis resembling psoriatic arthritis at an advanced age. Arthritis and Rheumatism. 2002;46(9):2465–2475. doi: 10.1002/art.10637. [DOI] [PubMed] [Google Scholar]
- 53.Berlo SE, Van Kooten PJ, Ten Brink CB, et al. Naive transgenic T cells expressing cartilage proteoglycan-specific TCR induce arthritis upon in vivo activation. Journal of Autoimmunity. 2005;25(3):172–180. doi: 10.1016/j.jaut.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 54.Berlo SE, Guichelaar T, Ten Brink CB, et al. Increased arthritis susceptibility in cartilage proteoglycan-specific T cell receptor-transgenic mice. Arthritis and Rheumatism. 2006;54(8):2423–2433. doi: 10.1002/art.22013. [DOI] [PubMed] [Google Scholar]
- 55.Boldizsar F, Kis-Toth K, Tarjanyi O, et al. Impaired activation-induced cell death promotes spontaneous arthritis in antigen (cartilage proteoglycan)-specific T cell receptor-transgenic mice. Arthritis and Rheumatism. 2010;62(10):2984–2994. doi: 10.1002/art.27614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jensen JR, Peters LC, Borrego A, et al. Involvement of antibody production quantitative trait loci in the susceptibility to pristane-induced arthritis in the mouse. Genes and Immunity. 2006;7(1):44–50. doi: 10.1038/sj.gene.6364271. [DOI] [PubMed] [Google Scholar]
- 57.Yu X, Bauer K, Koczan D, Thiesen HJ, Ibrahim SM. Combining global genome and transcriptome approaches to identify the candidate genes of small-effect quantitative trait loci in collagen-induced arthritis. Arthritis Research and Therapy. 2007;9, article R3 doi: 10.1186/ar2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu X, Teng H, Marques A, Ashgari F, Ibrahim SM. High resolution mapping of Cia3: a common arthritis quantitative trait loci in different species. Journal of Immunology. 2009;182(5):3016–3023. doi: 10.4049/jimmunol.0803005. [DOI] [PubMed] [Google Scholar]
- 59.Poole AR. Cartilage in health and disease. In: McCarty DJ, Koopman WJ, editors. Arthritis and Allied Conditions. A Textbook of Rheumatology. Philadelphia, Pa, USA: Lea & Febiger; 1993. pp. 279–333. [Google Scholar]
- 60.Czipri M, Otto JM, Cs-Szabó G, et al. Genetic rescue of chondrodysplasia and the perinatal lethal effect of cartilage link protein deficiency. Journal of Biological Chemistry. 2003;278(40):39214–39223. doi: 10.1074/jbc.M303329200. [DOI] [PubMed] [Google Scholar]
- 61.Singer II, Kawka DW, Bayne EK, et al. VDIPEN, a metalloproteinase-generated neoepitope, is induced and immunolocalized in articular cartilage during inflammatory arthritis. Journal of Clinical Investigation. 1995;95(5):2178–2186. doi: 10.1172/JCI117907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bizzaro N. Antibodies to citrullinated peptides: a significant step forward in the early diagnosis of rheumatoid arthritis. Clinical Chemistry and Laboratory Medicine. 2007;45(2):150–157. doi: 10.1515/CCLM.2007.027. [DOI] [PubMed] [Google Scholar]
- 63.Simon M, Girbal E, Sebbag M, et al. The cytokeratin filament-aggregating protein filaggrin is the target of the so-called ’antikeratin antibodies,’ autoantibodies specific for rheumatoid arthritis. Journal of Clinical Investigation. 1993;92(3):1387–1393. doi: 10.1172/JCI116713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Girbal-Neuhauser E, Durieux JJ, Arnaud M, et al. The epitopes targeted by the rheumatoid arthritis-associated antifilaggrin autoantibodies are posttranslationally generated on various sites of (pro)filaggrin by deimination of arginine residues. Journal of Immunology. 1999;162(1):585–594. [PubMed] [Google Scholar]
- 65.Poole AR, Glant TT, Mikecz K. Autoimmunity to cartilage collagen and proteoglycan and the development of chronic inflammatory arthritis. In: Glaurer AM, editor. The Control of Tissue Damage. Amsterdam, The Netherlands: Elsevier Publishers (Biomedical Division); 1988. pp. 55–65. [Google Scholar]
- 66.Glant TT, Mikecz K, Thonar EJMA, Kuettner KE. Immune responses to cartilage proteoglycans in inflammatory animal models and human diseases. In: Woessner JF, Howell DS, editors. Cartilage Degradation: Basic and Clinical Aspects. New York, NY, USA: Marcel Dekker; 1992. pp. 435–473. [Google Scholar]
- 67.Glant TT, Fulop C, Mikecz K, Buzas E, Molnar G, Erhardt P. Proteoglycan-specific autoreactive antibodies and T-lymphocytes in experimental arthritis and human rheumatoid joint diseases. Biochemical Society Transactions. 1990;18(5):796–799. doi: 10.1042/bst0180796. [DOI] [PubMed] [Google Scholar]
- 68.Herman JH, Wiltse DW, Dennis MV. Immunopathologic significance of cartilage antigenic components in rheumatoid arthritis. Arthritis and Rheumatism. 1973;16(3):287–297. doi: 10.1002/art.1780160302. [DOI] [PubMed] [Google Scholar]
- 69.Glant T, Csongor J, Szucs T. Immunopathologic role of proteoglycan antigens in rheumatoid joint disease. Scandinavian Journal of Immunology. 1980;11(3):247–252. doi: 10.1111/j.1365-3083.1980.tb00232.x. [DOI] [PubMed] [Google Scholar]
- 70.Golds EE, Stephen IBM, Esdaile JM. Lymphocyte transformation to connective tissue antigens in adult and juvenile rheumatoid arthritis, osteoarthritis, ankylosing spondylitis, systemic lupus erythematosus, and a nonarthritic control population. Cellular Immunology. 1983;82(1):196–209. doi: 10.1016/0008-8749(83)90153-3. [DOI] [PubMed] [Google Scholar]
- 71.Karopoulos C, Rowley MJ, Ilic MZ, Handley CJ. Presence of antibodies to native G1 domain of aggrecan core protein in synovial fluids from patients with various joint diseases. Arthritis and Rheumatism. 1996;39(12):1990–1997. doi: 10.1002/art.1780391207. [DOI] [PubMed] [Google Scholar]
- 72.Boots AMH, Verheijden GFM, Schöningh R, et al. Selection of self-reactive peptides within human aggrecan by use of a HLA-DRB1*0401 peptide binding motif. Journal of Autoimmunity. 1997;10(6):569–578. doi: 10.1006/jaut.1997.0164. [DOI] [PubMed] [Google Scholar]
- 73.Guerassimov A, Zhang Y, Banerjee S, et al. Autoimmunity to cartilage link protein in patients with rheumatoid arthritis and ankylosing spondylitis. Journal of Rheumatology. 1998;25(8):1480–1484. [PubMed] [Google Scholar]
- 74.Guerassimov A, Zhang YP, Banerjee S, et al. Cellular immunity to the G1 domain of cartilage proteoglycan aggrecan is enhanced in patients with rheumatoid arthritis but only after removal of keratan sulfate. Arthritis and Rheumatism. 1998;41(6):1019–1025. doi: 10.1002/1529-0131(199806)41:6<1019::AID-ART8>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 75.Li NL, Zhang DQ, Zhou KY, et al. Isolation and characteristics of autoreactive T cells specific to aggrecan G1 domain from rheumatoid arthritis patients. Cell Research. 2000;10(1):39–49. doi: 10.1038/sj.cr.7290034. [DOI] [PubMed] [Google Scholar]
- 76.Zou J, Zhang Y, Thiel A, et al. Predominant cellular immune response to the cartilage autoantigenic G1 aggrecan in ankylosing spondylitis and rheumatoid arthritis. Rheumatology. 2003;42(7):846–855. doi: 10.1093/rheumatology/keg230. [DOI] [PubMed] [Google Scholar]
- 77.De Jong H, Berlo SE, Hombrink P, et al. Cartilage proteoglycan aggrecan epitopes induce proinflammatory autoreactive T-cell responses in rheumatoid arthritis and osteoarthritis. Annals of the Rheumatic Diseases. 2010;69(1):255–262. doi: 10.1136/ard.2008.103978. [DOI] [PubMed] [Google Scholar]
- 78.Von Delwig A, Locke J, Robinson JH, Ng WF. Response of Th17 cells to a citrullinated arthritogenic aggrecan peptide in patients with rheumatoid arthritis. Arthritis and Rheumatism. 2010;62(1):143–149. doi: 10.1002/art.25064. [DOI] [PubMed] [Google Scholar]
- 79.Law SC, Street S, Yu CH, et al. T cell autoreactivity to citrullinated autoantigenic peptides in rheumatoid arthritis patients carrying HLA-DRB1 shared epitope alleles. Arthritis Research and Therapy. 2012;14(3):p. R118. doi: 10.1186/ar3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Buzás EI, Hanyecz A, Murad Y, et al. Differential recognition of altered peptide ligands distinguishes two functionally discordant (arthritogenic and nonarthritogenic) autoreactive T cell hybridoma clones. Journal of Immunology. 2003;171(6):3025–3033. doi: 10.4049/jimmunol.171.6.3025. [DOI] [PubMed] [Google Scholar]
- 81.Buzás EI, Végvári A, Murad YM, Finnegan A, Mikecz K, Glant TT. T-cell recognition of differentially tolerated epitopes of cartilage proteoglycan aggrecan in arthritis. Cellular Immunology. 2005;235(2):98–108. doi: 10.1016/j.cellimm.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 82.Glant TT, Adarichev VA, Nesterovitch AB, et al. Disease-associated qualitative and quantitative trait loci in proteoglycan-induced arthritis and collagen-induced arthritis. American Journal of the Medical Sciences. 2004;327(4):188–195. doi: 10.1097/00000441-200404000-00004. [DOI] [PubMed] [Google Scholar]
- 83.Otto JM, Cs-Szabó G, Gallagher J, et al. Identification of multiple loci linked to inflammation and autoantibody production by a genome scan of a murine model of rheumatoid arthritis. Arthritis and Rheumatism. 1999;42(12):2524–2531. doi: 10.1002/1529-0131(199912)42:12<2524::AID-ANR4>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 84.Glant TT, Adarichev VA, Boldizsar F, et al. Disease-promoting and -protective genomic loci on mouse chromosomes 3 and 19 control the incidence and severity of autoimmune arthritis. Gene Immunity. 2012;13(4):336–345. doi: 10.1038/gene.2012.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Adarichev VA, Bárdos T, Christodoulou S, Phillips MT, Mikecz K, Glant TT. Major histocompatibility complex controls susceptibility and dominant inheritance, but not the severity of the disease in mouse models of rheumatoid arthritis. Immunogenetics. 2002;54(3):184–192. doi: 10.1007/s00251-002-0462-8. [DOI] [PubMed] [Google Scholar]
- 86.Adarichev VA, Valdez JC, Bárdos T, Finnegan A, Mikecz K, Glant TT. Combined autoimmune models of arthritis reveal shared and independent qualitative (binary) and quantitative trait loci. Journal of Immunology. 2003;170(5):2283–2292. doi: 10.4049/jimmunol.170.5.2283. [DOI] [PubMed] [Google Scholar]
- 87.Adarichev VA, Vegvari A, Szabo Z, Kis-Toth K, Mikecz K, Glant TT. Congenic strains displaying similar clinical phenotype of arthritis represent different immunologic models of inflammation. Genes and Immunity. 2008;9(7):591–601. doi: 10.1038/gene.2008.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Plenge RM, Seielstad M, Padyukov L, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis - A genomewide study. The New England Journal of Medicine. 2007;357(12):1199–1209. doi: 10.1056/NEJMoa073491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Etzel CJ, Chen WV, Shepard N, et al. Genome-wide meta-analysis for rheumatoid arthritis. Human Genetics. 2006;119(6):634–641. doi: 10.1007/s00439-006-0171-8. [DOI] [PubMed] [Google Scholar]
- 90.Barton A, Thomson W, Ke X, et al. Re-evaluation of putative rheumatoid arthritis susceptibility genes in the post-genome wide association study era and hypothesis of a key pathway underlying susceptibility. Human Molecular Genetics. 2008;17(15):2274–2279. doi: 10.1093/hmg/ddn128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Eleftherohorinou H, Wright V, Hoggart C, et al. Pathway analysis of GWAS provides new insights into genetic susceptibility to 3 inflammatory diseases. PloS One. 2009;4(11, article e8068) doi: 10.1371/journal.pone.0008068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Glant TT, Szántó S, Vegvari A, et al. Two loci on chromosome 15 control experimentally induced arthritis through the differential regulation of IL-6 and lymphocyte proliferation. Journal of Immunology. 2008;181(2):1307–1314. doi: 10.4049/jimmunol.181.2.1307. [DOI] [PubMed] [Google Scholar]
- 93.Ahlqvist E, Bockermann R, Holmdahl R. Fragmentation of two quantitative trait loci controlling collagen-induced arthritis reveals a new set of interacting subloci. Journal of Immunology. 2007;178(5):3084–3090. doi: 10.4049/jimmunol.178.5.3084. [DOI] [PubMed] [Google Scholar]
- 94.Liljander M, Sällström MA, Anderson S, et al. Identification of collagen-induced arthritis loci in aged multiparous female mice. Arthritis Research and Therapy. 2006;8(2, article R45) doi: 10.1186/ar1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bauer K, Yu X, Wernhoff P, Koczan D, Thiesen HJ, Ibrahim SM. Identification of new quantitative trait loci in mice with collagen-induced arthritis. Arthritis and Rheumatism. 2004;50(11):3721–3728. doi: 10.1002/art.20624. [DOI] [PubMed] [Google Scholar]
- 96.Yu X, Bauer K, Wernhoff P, et al. Fine mapping of collagen-induced arthritis quantitative trait loci in an advanced intercross line. Journal of Immunology. 2006;177(10):7042–7049. doi: 10.4049/jimmunol.177.10.7042. [DOI] [PubMed] [Google Scholar]
- 97.Johannesson M, Olsson LM, Lindqvist AKB, et al. Gene expression profiling of arthritis using a QTL chip reveals a complex gene regulation of the Cia5 region in mice. Genes and Immunity. 2005;6(7):575–583. doi: 10.1038/sj.gene.6364242. [DOI] [PubMed] [Google Scholar]
- 98.Lindvall T, Karlsson J, Holmdahl R, et al. Dissection of a locus on mouse chromosome 5 reveals arthritis promoting and inhibitory genes. Arthritis Research and Therapy. 2009;11(1, article R10) doi: 10.1186/ar2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ibrahim SM, Koczan D, Thiesen HJ. Gene-expression profile of collagen-induced arthritis. Journal of Autoimmunity. 2002;18(2):159–167. doi: 10.1006/jaut.2001.0580. [DOI] [PubMed] [Google Scholar]
- 100.Gulko PS, Kawahito Y, Remmers EF, et al. Identification of a new non-major histocompatibility complex genetic locus on chromosome 2 that controls disease severity in collagen-induced arthritis in rats. Arthritis and Rheumatism. 1998;41(12):2122–2131. doi: 10.1002/1529-0131(199812)41:12<2122::AID-ART7>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 101.Kawahito Y, Remmers EF, Wilder RL, et al. A genetic linkage map of rat chromosome 20 derived from five F2 crosses. Immunogenetics. 1998;48(5):335–338. doi: 10.1007/s002510050440. [DOI] [PubMed] [Google Scholar]
- 102.Shepard JS, Remmers EF, Chen S, et al. A genetic linkage map of rat Chromosome 15 derived from five F2 crosses. Mammalian Genome. 1999;10(2):186–188. doi: 10.1007/s003359900966. [DOI] [PubMed] [Google Scholar]
- 103.Joe B, Remmers EF, Dobbins DE, et al. Genetic dissection of collagen-induced arthritis in chromosome 10 quantitative trait locus speed congenic rats: evidence for more than one regulatory locus and sex influences. Immunogenetics. 2000;51(11):930–944. doi: 10.1007/s002510000231. [DOI] [PubMed] [Google Scholar]
- 104.Wester L, Olofsson P, Ibrahim SM, Holmdahl R. Chronicity of pristane-induced arthritis in rats is controlled by genes on chromosome 14. Journal of Autoimmunity. 2003;21(4):305–313. doi: 10.1016/s0896-8411(03)00136-7. [DOI] [PubMed] [Google Scholar]
- 105.Wester L, Koczan D, Holmberg J, et al. Differential gene expression in pristane-induced arthritis susceptible DA versus resistant E3 rats. Arthritis Research & Therapy. 2003;5(6):R361–R372. doi: 10.1186/ar993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Adarichev VA, Nesterovitch AB, Bárdos T, et al. Sex effect on clinical and immunologic quantitative trait loci in a murine model of rheumatoid arthritis. Arthritis and Rheumatism. 2003;48(6):1708–1720. doi: 10.1002/art.11016. [DOI] [PubMed] [Google Scholar]
- 107.Otto JM, Chandrasekeran R, Vermes C, et al. A genome scan using a novel genetic cross identifies new susceptibility loci and traits in a mouse model of rheumatoid arthritis. Journal of Immunology. 2000;165(9):5278–5286. doi: 10.4049/jimmunol.165.9.5278. [DOI] [PubMed] [Google Scholar]
- 108.Johannesson M, Karlsson J, Wernhoff P, et al. Identification of epistasis through a partial advanced intercross reveals three arthritis loci within the Cia5 QTL in mice. Genes and Immunity. 2005;6(3):175–185. doi: 10.1038/sj.gene.6364155. [DOI] [PubMed] [Google Scholar]
- 109.Firneisz G, Zehavi I, Vermes C, Hanyecz A, Frieman JA, Glant TT. Identification and quantification of disease-related gene clusters. Bioinformatics. 2003;19(14):1781–1786. doi: 10.1093/bioinformatics/btg252. [DOI] [PubMed] [Google Scholar]
- 110.Yang HT, Jirholt J, Svensson L, et al. Identification of genes controlling collagen-induced arthritis in mice: striking homology with susceptibility loci previously identified in the rat. Journal of Immunology. 1999;163(5):2916–2921. [PubMed] [Google Scholar]
- 111.Van Houten N, Blake SF. Direct measurement of anergy of antigen-specific T cells following oral tolerance induction. Journal of Immunology. 1996;157(4):1337–1341. [PubMed] [Google Scholar]
- 112.Studelska DR, Mandik-Nayak L, Zhou X, et al. High affinity glycosaminoglycan and autoantigen interaction explains joint specificity in a mouse model of rheumatoid arthritis. Journal of Biological Chemistry. 2009;284(4):2354–2362. doi: 10.1074/jbc.M806458200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hughes LB, Reynolds RJ, Brown EE, et al. Most common single-nucleotide polymorphisms associated with rheumatoid arthritis in persons of European ancestry confer risk of rheumatoid arthritis in African Americans. Arthritis and Rheumatism. 2010;62(12):3547–3553. doi: 10.1002/art.27732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cui J, Saevarsdottir S, Thomson B, et al. Rheumatoid arthritis risk allele PTPRC is also associated with response to anti-tumor necrosis factor α therapy. Arthritis and Rheumatism. 2010;62(7):1849–1861. doi: 10.1002/art.27457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Thompson SD, Sudman M, Ramos PS, et al. The susceptibility loci juvenile idiopathic arthritis shares with other autoimmune diseases extend to PTPN2, COG6, and ANGPT1. Arthritis and Rheumatism. 2010;62(11):3265–3276. doi: 10.1002/art.27688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Morgan AW, Robinson JI, Conaghan PG, et al. Evaluation of the rheumatoid arthritis susceptibility loci HLA-DRB1, PTPN22, OLIG3/TNFAIP3, STAT4 and TRAF1/C5 in an inception cohort. Arthritis Research and Therapy. 2010;12(2, article R57) doi: 10.1186/ar2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Freudenberg J, Lee HS, Han BG, et al. Genome-Wide Association Study of Rheumatoid Arthritis in Koreans: population-specific loci as well as overlap with European Susceptibility Loci. Arthritis and Rheumatism. 2011;63(4):884–893. doi: 10.1002/art.30235. [DOI] [PubMed] [Google Scholar]
- 118.Plenge RM, Cotsapas C, Davies L, et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nature Genetics. 2007;39(12):1477–1482. doi: 10.1038/ng.2007.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annual Review of Immunology. 1996;14:397–440. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- 120.Menard L, Saadoun D, Isnardi I, et al. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. The Journal of Clinical Investigation. 2011;121(9):3635–3644. doi: 10.1172/JCI45790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jirholt J, Cook A, Emahazion T, et al. Genetic linkage analysis of collagen-induced arthritis in the mouse. European Journal of Immunology. 1998;28(10):3321–3328. doi: 10.1002/(SICI)1521-4141(199810)28:10<3321::AID-IMMU3321>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 122.Sundvall M, Jirholt J, Yang HT, et al. Identification of murine loci associated with susceptibility to chronic experimental autoimmune encephalomyelitis. Nature Genetics. 1995;10(3):313–317. doi: 10.1038/ng0795-313. [DOI] [PubMed] [Google Scholar]
- 123.Karlsson J, Johannesson M, Lindvall T, et al. Genetic interactions in Eae2 control collagen-induced arthritis and the CD4+/CD8+ T cell ratio. Journal of Immunology. 2005;174(1):533–541. doi: 10.4049/jimmunol.174.1.533. [DOI] [PubMed] [Google Scholar]
- 124.Meng HC, Griffiths MM, Remmers EF, et al. Identification of two novel female-specific non-major histocompatibility complex loci regulating collagen-induced arthritis severity and chronicity, and evidence of epistasis. Arthritis and Rheumatism. 2004;50(8):2695–2705. doi: 10.1002/art.20366. [DOI] [PubMed] [Google Scholar]
- 125.Brenner M, Meng HC, Yarlett NC, et al. The non-major histocompatibility complex quantitative trait locus Cia10 contains a major arthritis gene and regulates disease severity, pannus formation, and joint damage. Arthritis and Rheumatism. 2005;52(1):322–332. doi: 10.1002/art.20782. [DOI] [PubMed] [Google Scholar]
- 126.Xu W, Lan H, Hu P, et al. Evidence of linkage to chromosome 1 for early age of onset of rheumatoid arthritis and HLA marker DRB1 genotype in NARAC data. BMC Proceedings. 2007;1(supplement 1):p. S78. doi: 10.1186/1753-6561-1-s1-s78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liu C, Batliwalla F, Li W, et al. Genome-wide association scan identifies candidate polymorphisms associated with differential response to anti-TNF treatment in rheumatoid arthritis. Molecular Medicine. 2008;14(9-10):575–581. doi: 10.2119/2008-00056.Liu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gharaee-Kermani M, Denholm EM, Phan SH. Costimulation of fibroblast collagen and transforming growth factor β1 gene expression by monocyte chemoattractant protein-1 via specific receptors. Journal of Biological Chemistry. 1996;271(30):17779–17784. doi: 10.1074/jbc.271.30.17779. [DOI] [PubMed] [Google Scholar]
- 129.van der Helm-van Mil AHM, Huizinga TWJ. Advances in the genetics of rheumatoid arthritis point to subclassification into distinct disease subsets. Arthritis Research and Therapy. 2008;10(2, article 205) doi: 10.1186/ar2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bowes J, Barton A. Recent advances in the genetics of RA susceptibility. Rheumatology. 2008;47(4):399–402. doi: 10.1093/rheumatology/ken005. [DOI] [PubMed] [Google Scholar]
- 131.Choi SJ, Rho YH, Ji JD, Song GG, Lee YH. Genome scan meta-analysis of rheumatoid arthritis. Rheumatology. 2006;45(2):166–170. doi: 10.1093/rheumatology/kei128. [DOI] [PubMed] [Google Scholar]
- 132.Morgan AW, Barrett JH, Griffiths B, et al. Analysis of Fcgamma receptor haplotypes in rheumatoid arthritis: FCGR3A remains a major susceptibility gene at this locus, with an additional contribution from FCGR3B. Arthritis Research & Therapy. 2006;8(1):p. R5. doi: 10.1186/ar1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kochi Y, Yamada R, Suzuki A, et al. A functional variant in FCRL3, encoding Fc receptor-like 3, is associated with rheumatoid arthritis and several autoimmunities. Nature Genetics. 2005;37(5):478–485. doi: 10.1038/ng1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Licatalosi DD, Mele A, Fak JJ, et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456(7221):464–469. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460(7254):479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Malterer G, Dölken L, Haas J. The miRNA-targetome of KSHV and EBV in human B-cells. RNA Biology. 2011;8(1):30–34. doi: 10.4161/rna.8.1.13745. [DOI] [PubMed] [Google Scholar]
- 137.Wen J, Parker BJ, Jacobsen A, Krogh A. MicroRNA transfection and AGO-bound CLIP-seq data sets reveal distinct determinants of miRNA action. RNA. 2011;17(5):820–834. doi: 10.1261/rna.2387911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhang C, Darnell RB. Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nature Biotechnology. 2011;29(7):607–614. doi: 10.1038/nbt.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]