

Construction of new C–C bonds via decarboxylative coupling is a potentially powerful synthetic tool since it avoids highly basic reaction conditions and preformed organometallic reagents that produce stoichiometric metal waste.1 In addition, the byproduct (CO2) is non-toxic and requires no special separation procedures. Thus several research groups have demonstrated that decarboxylative couplings are practical alternatives to standard cross-coupling reactions.1-6 For example, Gooβen and co-workers have reported a Pd(II)-catalyzed coupling of benzoic acids with haloaromatics to generate biaryl products.3 Furthermore, Myers has shown a Pd(II)-catalyzed decarboxylative variant of the Heck reaction.4 Our research has focused on the development of decarboxylative allylation and benzylation reactions.5 In this arena we have reported the decarboxylative allylation (DcA) of heteroaromatic coumarin substrates under mild conditions (eq. 1).6
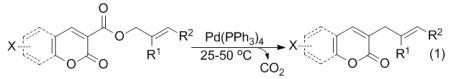 |
(1) |
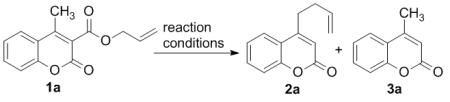
Coumarins are not only ‘privileged’ scaffolds of biological and pharmaceutical interest,7,8 but they are also widely used in dyes because of their photophysical properties.9 In our continuing investigations of the DcA of coumarins, we turned our attention to the investigation of decarboxylative couplings of 4-substituted coumarins. Thus, 4-methyl-3-allylcoumarate 1a was synthesized and subjected to our previous conditions for Pd(0)-catalyzed decarboxylative coupling at 50 °C.6 Disappointingly, the starting material remained intact even after prolonged heating. However, upon heating the substrate at 110 °C in toluene for 6 h, we observed decarboxylation and C–C bond formation (2a) as well as protiodecarboxylation (eq. 2). Closer analysis of the products by 1H NMR spectroscopy revealed that allylation occurs at the methyl terminus providing 4-homoallylcoumarin in lieu of the expected allylation of the 3-postion of the coumarin.6 In every other case of decarboxylative coupling that we have investigated, allylation occurs regiospecifically at the site that bears the carboxylate.10 Thus, the observation of remote decarboxylative allylation warranted further investigation. Herein, we report that many other substituted coumarins exhibit similar regiochemistry in their allylation and we present a mechanism that explains this unusual regiochemical outcome.
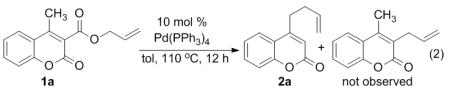 |
(2) |
Encouraged by the unexpected regiochemistry of allylation, we optimized the reaction conditions with the goal of suppressing the undesired protonation product (3a). After rigorous catalyst and solvent screening (Table 1) it was found that 3 mol% Pd2(dba)3 in combination with 6 mol% Xantphos provided excellent yields of 4-butenylcoumarin (2a) when allowed to react with substrate 1a in toluene at 70 °C.
Table 1.
Optimization of Reaction Conditions.[a]
| Entry | Catalyst | Solvent | Yield (%)[b] | 2a:3a [b] |
|---|---|---|---|---|
| 1 | 5 mol% Pd(PPh3)4 | tol-d8 | 99 | 77:23 |
| 2 | 5 mol% Pd(PPh3)4 | CD3CN | 65 | 63:37 |
| 3 | 5 mol% Pd(PPh3)4 | DMF-d7 | 95 | 80:20 |
| 4 | 5 mol% Pd(PPh3)4 | THF-d8 | 87 | 83:17 |
| 5 | 3 mol% Pd2(dba)3, 6 mol% dppe |
tol-d8 | 75 | 70:30 |
| 6 | 3 mol% Pd2(dba)3, 6 mol% dppb |
tol-d8 | 67 | 77:23 |
| 7 | 3 mol% Pd2(dba)3, 6 mol% rac-BINAP |
tol-d8 | 77 | 70:30 |
| 8 |
3 mol% Pd2(dba)3,
6 mol% Xantphos |
tol-d8 | 99 | 94:6 |
| 9 | 3 mol% Pd2(dba)3, 6 mol% Xantphos |
DMF-d7 | 80 | 80:20 |
| 10 | 3 mol% Pd2(dba)3, 6 mol% dppf |
tol-d8 | 75 | 72:28 |
All reactions were carried out at 70 °C for 12 h, 0.1 mmol scale, 0.2 (M).
Yields and product distributions were determined by 1H NMR.
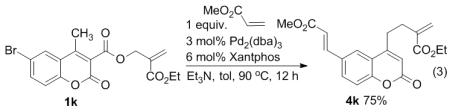
Under the optimized reaction conditions, we explored the substrate scope for this decarboxylative allylation reaction (Table 2). A wide variety of 4-methyl-3-allylcoumarates with substitution on arene were synthesized and examined. Gratifyingly, it was found that a variety of aryl substitutions allow formation of excellent yields of coupling products, irrespective of the electronics. Even halogen substituents, such as Br, are compatible with our coupling conditions (2d, 2g, 2k, Table 2). Thus, one-pot decarboxylative allylation/cross-coupling reactions are feasible (eq. 3). In addition to the couplings of unsubstituted allyl esters, a variety of substituted and functionalized allyl esters undergo coupling to provide products in excellent yields. However, one limitation of the reaction is that allyl esters that possess β-hydrogens preferentially form elimination products (eq. 4).
 |
(3) |
 |
(4) |
Table 2.
Migratory Decarboxylative Allylation
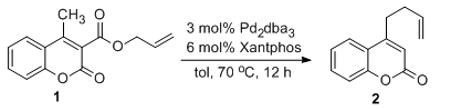
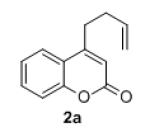
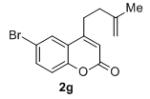
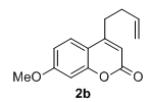
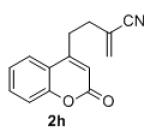
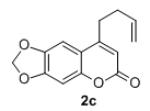
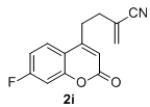
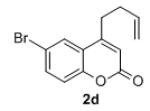
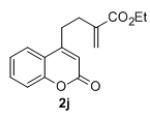
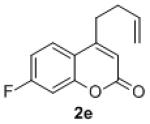
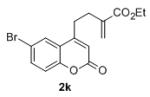
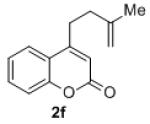
Isolated yields for reactions were performed on at 0.2 M on a 0.5 mmol scale.
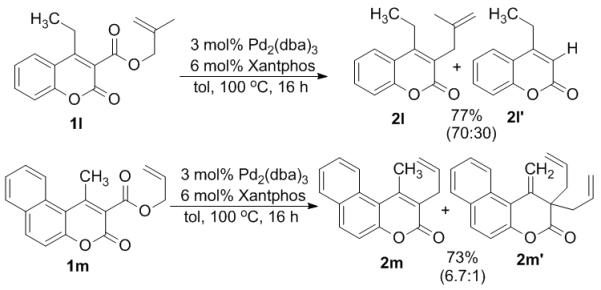
Since the successful substrates for the remote allylation were 4-methyl substituted, we became curious whether larger 4-alkyl groups would participate in migratory allylation as well. Toward this end, the 4-ethyl substituted substrate 1l was prepared and allowed to react under our standard reaction conditions. Interestingly, the substrate underwent typical site-specific allylation as previously reported.6 Thus, it appears that sterics disfavor the mechanism by which the 4-alkyl group is allylated. Similarly, substrate 1m did not undergo remote allylation of the methyl group, rather it resulted in α-allylation to give (2m) along with the diallylation to give 2m’.
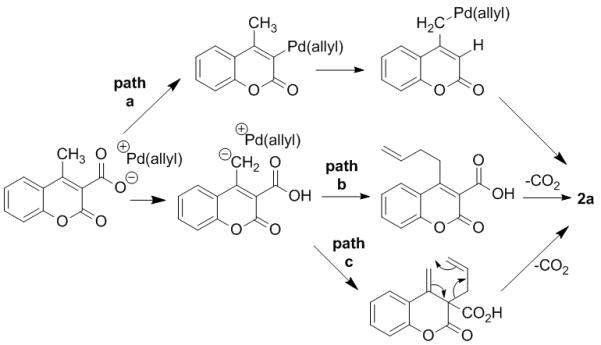
At the outset, several mechanisms seemed reasonable for the formation of the remotely allylated coumarins such as 2a. First, decarboxylative metalation could produce an aryl palladium species that is capable of undergoing a 1,3-migration (path a).11 Alternatively, the intermediate carboxylate may deprotonate the methyl group to generate a stabilized malonic acid dienolate. Such a proposal is reasonable given that the pKa values of carboxylates (~12 in DMSO) and malonates (~14 in DMSO) are comparable.12 Allylation and decarboxylation would then produce 2a. Lastly, the observation of the diallylated product (2m’, Scheme 1) suggested that the allylation of the methyl group might be proceeding via an α-allylation/Cope rearrangement mechanism (path c, Scheme 2).13
Scheme 1.
Sterics inhibit migration.
Scheme 2.
α-Allylation vs γ-allylation.
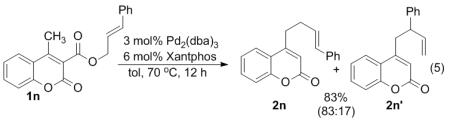
The mechanism illustrated by path c is easily probed, since such a mechanism predicts that a substituted allyl ester will react to form the branched allylated product rather than the linear product.5e,14 To test this, the coumaryl cinnamyl ester 1n was treated with Pd catalyst (eq. 5). The resulting product forms in high yield with a 83:17 l:b ratio. The regioselective formation of the linear allylated product (2n) suggests that the methyl group is directly allylated and that α-allylation/Cope rearrangement is not the dominant mechanism for product formation. However, most decarboxylative cinnamylations provide product with l:b ratios of >95:5,2a,2g,5f,5h,6,15 so the relatively low selectivity in this case may indicate a minor contribution of the allylation/Cope-rearrangement mechanism.
 |
(5) |
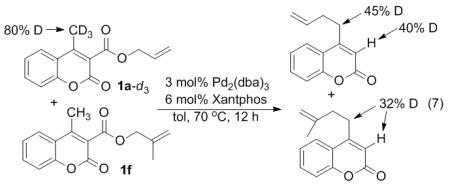
With the above information in hand, further mechanistic studies were necessary to refine our mechanistic hypothesis. To begin, a deuterium labelling study was performed to determine the origin of proton that comes at the α-carbon after decarboxylation. Toward this end, 1a-d3 was prepared and allowed to undergo decarboxylative coupling. The α-carbon of the resulting product was 75% deuterated at the α-position. Thus, deuterium is clearly transferred from the methyl group to the α-carbon.
 |
(6) |
Next, a crossover experiment showed extensive crossover between the deuterated reactant 1a-d3 and a protio coumarin (eq 7). In addition to the observation of crossover, some mechanistic insight was obtained from the preparation of the requisite deuterated coumarin (1a-d3). Specifically, it was observed that treatment of the allyl ester 1a with K2CO3 and D2O did not lead to any appreciable deuterium incorporation (Scheme 3). However, treatment of the carboxylic acid under the same conditions led to extensive deuterium incorporation. Thus, the carboxylate group is necessary to facilitate deprotonation of the 4-methyl coumarin.
 |
(7) |
Scheme 3.
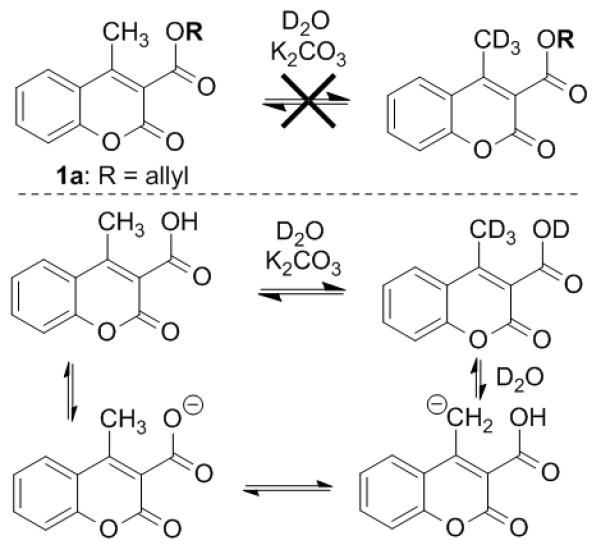
Carboxylate-assisted deuteration.
While the observations of extensive crossover and carboxylate assisted deprotonation seemed to implicate a mechanism that follows path b, path a could not be ruled out based on these experiments alone. The mechanistic ambiguity was further clarified by a simple but crucial experiment. When a typical reaction was arrested after 2 h, the γ-allylated, α-coumaric acid 5f was isolated in good yield. When the coumaric acid 5f was resubjected to the reaction conditions, the decarboxylated γ-allylation product 2f was obtained (Scheme 4).
Scheme 4.
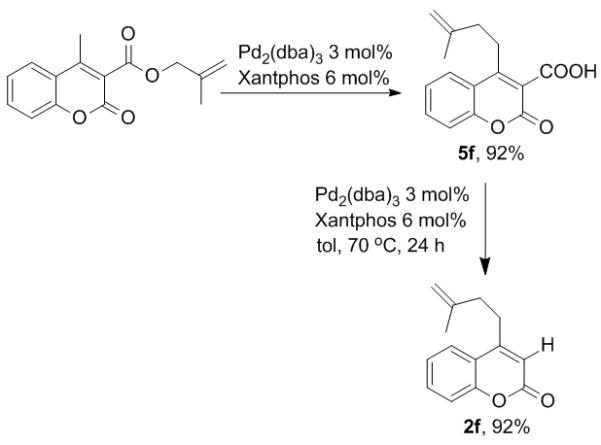
Isolation of an intermediate.
It is noteworthy that a Pd(II) source, Pd(OAc)2, failed to catalyze decarboxylation of 5f under the reaction conditions.2b,2d,14 However, Pd(0) sources such as Pd(PPh3)4 and Pd2(dba)3/Xantphos were effective catalysts for decarboxylation of 5f. Thus, it appears that decarboxylation is catalyzed by Pd(0).
Combination of all of the mechanistic studies suggests the following mechanism for this unusual decarboxylative γ-allylation. First palladium undergoes oxidative addition to form a π-allylpalladium complex and the coumarin carboxylate counterion. Then 1,5-proton transfer occurs to generate a stabilized carbanion at the methyl terminus. Next, nucleophilic substitution of the π-allylpalladium complex forms the C–C bond. The C–C bond could form via nucleophilic attack on the allyl ligand from the stabilized carbanion (Scheme 5) or via reductive elimination from a bisallyl-like Pd complex.16 We favour the former mechanism because enolate nucleophiles that are related to our coumarin nucleophiles are known to react via backside attack on Pd(π-allyl) cations.17,18 Lastly, protonation of palladium results in a palladium carboxylate that can undergo decarboxylation followed by C–H bond forming reductive elimination (Scheme 5).19
Scheme 5.
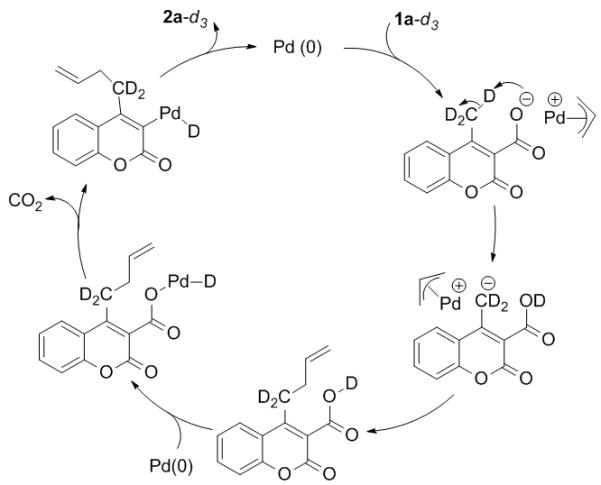
Mechanistic Pathway of γ-Allylation.
In conclusion, we have observed remote decarboxylative allylation for the first time and developed a simple method for the γ-allylation of coumarins based on this finding. Mechanistic studies suggest that the remote allylation is made possible by a carboxylate-assisted deprotonation to generate the nucleophile prior to decarboxylation. After allylation, decarboxylation of the carboxylic acid is catalyzed by Pd(0), contrary to more commonly observed Pd(II)-catalyzed decarboxylations.2,5,13
Experimental Section
General Procedure for the Palladium-Catalyzed Decarboxylative γ-allylation of 3-allylcoumarates
In an oven dried Schlenk flask, 1a (0.50 mmol) was dissolved in toluene (2.5 mL) under argon followed by the addition of Pd2(dba)3 (0.015 mmol, 3 mol%) and Xantphos (0.03 mmol, 6 mol%). The resulting reaction mixture was heated at 70 °C for 12 h. The solution was then concentrated on a rotary evaporator and the residue was purified directly via flash chromatography on silica gel (EtOAc: hexane, 20:80).
Supplementary Material
Footnotes
See supporting information for full experimental procedures,1H NMR, 13C NMR, and GC/MS data.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
This work was supported by the National Institute of General Medical Sciences (NIGMS 1R01GM079644).
References
- [1] a).Rayabarapu DK, Tunge JA. J. Am. Chem. Soc. 2005;127:13510. doi: 10.1021/ja0542688. [DOI] [PubMed] [Google Scholar]; b) Trost BM, Xu J. J. Am. Chem. Soc. 2005;127:17180. doi: 10.1021/ja055968f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Shimizu I, Yamada T, Tsuji J. Tetraheron Lett. 1980;21:3199. [Google Scholar]; b) Tsuda T, Chujo Y, Nishi S.-i., Tawara K, Saegusa T. J. Am. Chem. Soc. 1980;102:6381. [Google Scholar]; c) Tsuji J, Ohashi Y, Minami O. Tetrahedron Lett. 1987;28:2397. [Google Scholar]; d) Stoltz BM, Behenna DC. J. Am. Chem. Soc. 2004;126:15044. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]; e) Craig D, Grellepois F. Org. Lett. 2005;7:463. doi: 10.1021/ol047577w. [DOI] [PubMed] [Google Scholar]; f) Kuwano R, Ishida N, Murakami M. Chem. Commun. 2005:3951. doi: 10.1039/b505105c. [DOI] [PubMed] [Google Scholar]; g) Mellegaard-Waetzig SR, Rayabarapu DK, Tunge JA. Synlett. 2005:2759. [Google Scholar]; h) Nakamura M, Hajra A, Endo K, Nakamura E. Angew. Chem. Int. Ed. 2005;44:7248. doi: 10.1002/anie.200502703. [DOI] [PubMed] [Google Scholar]; i) Suryaprakash GK, Beier P. Angew. Chem. Int. Ed. 2006;45:2172. doi: 10.1002/anie.200503783. [DOI] [PubMed] [Google Scholar]; j) Patil NT, Huo Z, Yamamoto Y. J. Org. Chem. 2006;71:6991. doi: 10.1021/jo061110c. [DOI] [PubMed] [Google Scholar]; k) You S-L, Dai L-X. Angew. Chem. Int. Ed. 2006;45:5246. doi: 10.1002/anie.200601889. [DOI] [PubMed] [Google Scholar]; l) Imao D, Itoi A, Yamazaki A, Shirakura M, Ohtoshi R, Ogata K, Ohmori Y, Ohta T, Ito Y. J. Org. Chem. 2007;72:1652. doi: 10.1021/jo0621569. [DOI] [PubMed] [Google Scholar]; m) Yeagley AA, Chruma JJ. Org. Lett. 2007;9:2879. doi: 10.1021/ol071080f. [DOI] [PubMed] [Google Scholar]; n) Baudoin O. Angew. Chem.. Int. Ed. 2007;46:1373. doi: 10.1002/anie.200604494. [DOI] [PubMed] [Google Scholar]; o) Trost BM, Xu J, Schmidt T. J. Am. Chem. Soc. 2008;130:11852. doi: 10.1021/ja8038954. [DOI] [PMC free article] [PubMed] [Google Scholar]; p) Becht J-M, Drian CL. Org. Lett. 2008;10:3161. doi: 10.1021/ol8011293. [DOI] [PubMed] [Google Scholar]; q) Moon J, Jeong M, Nam H, Ju J, Moon JH, Jung HM, Lee S. Org. Lett. 2008;10:945. doi: 10.1021/ol703130y. [DOI] [PubMed] [Google Scholar]; r) Tardibono LP, Jr., Partzner J, Cesario C, Miller MMJ. Org. Lett. 2009;11:4076. doi: 10.1021/ol901518g. [DOI] [PMC free article] [PubMed] [Google Scholar]; s) Wang Z, Ding Q, He X, Wu J. Org. Biomol. Chem. 2009;7:863. doi: 10.1039/b821870f. [DOI] [PubMed] [Google Scholar]
- [3] a).Goossen LJ, Deng G, Levy LM. Science. 2006;313:662. doi: 10.1126/science.1128684. [DOI] [PubMed] [Google Scholar]; b) Goossen LJ, Rodriguez N, Melzer B, Linder C, Deng G, Levy LM. J. Am. Chem. Soc. 2007;129:4824. doi: 10.1021/ja068993+. [DOI] [PubMed] [Google Scholar]; c) Goossen LJ, Zimmermann B, Knauber T. Angew. Chem. Int. Ed. 2008;47:7103. doi: 10.1002/anie.200800728. [DOI] [PubMed] [Google Scholar]; d) Goossen LJ, Rodriguez N, Linder C. J. Am. Chem. Soc. 2008;130:15248. doi: 10.1021/ja8050926. [DOI] [PubMed] [Google Scholar]; e) Goossen LJ, Linder C, Rodriguez N, Lange PP. Chem. Eur. J. 2009:9336. doi: 10.1002/chem.200900892. [DOI] [PubMed] [Google Scholar]; f) Satoh T, Miura M. Synthesis. 2010:3395. [Google Scholar]
- [4] a).Myers AG, Tanaka D, Mennion MR. J. Am. Chem. Soc. 2002;124:11250. doi: 10.1021/ja027523m. [DOI] [PubMed] [Google Scholar]; b) Tanaka D, Myers AG. Org. Lett. 2004;6:433. doi: 10.1021/ol0363467. [DOI] [PubMed] [Google Scholar]; c) Tanaka D, Romeril SP, Myers AG. J. Am. Chem. Soc. 2005;127:10323. doi: 10.1021/ja052099l. [DOI] [PubMed] [Google Scholar]
- [5] a).Burger EC, Tunge JA. Org. Lett. 2004;6:4113. doi: 10.1021/ol048149t. [DOI] [PubMed] [Google Scholar]; b) Tunge JA, Burger EC. Eur. J. Org. Chem. 2005:1715. [Google Scholar]; c) Burger EC, Barron BR, Tunge JA. Synlett. 2006:2824. [Google Scholar]; d) Burger EC, Tunge JA. J. Am. Chem. Soc. 2006;128:10002. doi: 10.1021/ja063115x. [DOI] [PubMed] [Google Scholar]; e) Waetzig SR, Tunge JA. J. Am. Chem. Soc. 2007;129:4138. doi: 10.1021/ja070116w. [DOI] [PubMed] [Google Scholar]; f) Waetzig SR, Tunge JA. J. Am. Chem. Soc. 2007;129:14860. doi: 10.1021/ja077070r. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Waetzig SR, Tunge JA. Chem. Commun. 2008:3311. doi: 10.1039/b806949b. [DOI] [PubMed] [Google Scholar]; h) Weaver JD, Tunge JA. Org. Lett. 2008;10:4657. doi: 10.1021/ol801951e. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Wang C, Tunge JA. J. Am. Chem. Soc. 2008;130:8118. doi: 10.1021/ja801742h. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Wang C, Pahadi N, Tunge JA. Tetrahedron. 2009;65:5102. doi: 10.1016/j.tet.2009.04.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jana R, Trivedi R, Tunge JA. Org. Lett. 2009;11:3434. doi: 10.1021/ol901288r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7] a).Estevez-Craun A, Gonzalez AG. Nat. Prod. Rep. 1997:465. doi: 10.1039/np9971400465. [DOI] [PubMed] [Google Scholar]; b) Horton DA, Bourne GT, Smythe ML. Chem. Rev. 2003;103:893. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]; c) Ngameni B, Touaibia M, Patnam R, Belkaid A, Sonna P, Ngadjui BT, Annabi B, Roy R. Phytochemistry. 2006;67:2573. doi: 10.1016/j.phytochem.2006.09.017. [DOI] [PubMed] [Google Scholar]; d) Chun K, Park S-K, Kim HM, Choi Y, Kim M-H, Park C-H, Joe B-Y, Chun TG, Choi H-M, Lee H-Y, Hong SH, Kim MS, Nam K-Y, Han G. Bioorg. Med. Chem. 2008;16:530. doi: 10.1016/j.bmc.2007.09.014. [DOI] [PubMed] [Google Scholar]
- [8] a).Govindachari TR, Pai BR, Subramaniam PS, Rao UR, Muthukumaraswamy N. Tetrahedron. 1967;23:4161. [Google Scholar]; b) Crombie L, Games DE, McCormick A. J. Chem. Soc. Sec. C, Organic. 1967:2545. [Google Scholar]; c) Ito C, Itoigawa M, Mishina Y, Filho VC, Enjo F, Tokuda H, Nishino H, Furukawa H. J. Nat. Prod. 2003;66:368. doi: 10.1021/np0203640. [DOI] [PubMed] [Google Scholar]; d) Yang H, Jiang B, Reynertson KA, Basile MJ, Kennelly EJ. J. Agric. Food Chem. 2006;54:4114. doi: 10.1021/jf0532462. [DOI] [PubMed] [Google Scholar]
- [9] a).Bhide BH, Parikh SP, Patel SJ. Chem. Indust. 1974:306. [Google Scholar]; b) Trenor SR, Shultz AR, Love BJ, Long TE. Chem. Rev. 2004;104:3059. doi: 10.1021/cr030037c. [DOI] [PubMed] [Google Scholar]; c) Wang B-Y, Liu X-Y, Hu Y-L, Su Z-X. Polym. Int. 2009;58:703. [Google Scholar]
- [10].Decarboxylative allylation can occur at multiple mesomeric sites in resonance stabilized carbanions. Shintani R, Tsuji T, Park S, Hayashi T. Chem. Commun. 2010:1697. doi: 10.1039/b924416f.
- [11] a).Li B, Shi ZJ. Chin. Sci. Bull. 2010;55:2807. [Google Scholar]; b) Mota AJ, Dedieu A. J. Org. Chem. 2007;72:9669. doi: 10.1021/jo701701s. [DOI] [PubMed] [Google Scholar]; c) Shi F, Larock RC. Topp. Curr. Chem. 2010;292:123. doi: 10.1007/128_2008_46. [DOI] [PubMed] [Google Scholar]; d) Zhao J, Campo M, Larock RC. Angew. Chem. Int. Ed. 2005;44:1873. doi: 10.1002/anie.200462327. [DOI] [PubMed] [Google Scholar]; e) Kesharwani T, Larock RC. Tetrahedron. 2008;64:6090. [Google Scholar]
- [12].Bordwell FG. Acc. Chem. Res. 1988;21:456. [Google Scholar]
- [13] a).Nakamura H, Iwama H, Ito M, Yamamoto Y. J. Am. Chem. Soc. 1999;121:10850. [Google Scholar]; b) Waetzig SR, Rayabarapu DK, Weaver JD. J. A. Tunge, Angew. Chem. Int. Ed. 2006;45:4977. doi: 10.1002/anie.200600721. [DOI] [PubMed] [Google Scholar]
- [14].Recio A, III, Tunge JA. Org. Lett. 2009;12:5630. doi: 10.1021/ol902065p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Grenning AJ, Tunge JA. Org. Lett. 2010;12:740. doi: 10.1021/ol902828p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16] a).For bisallyl palladium chemistry see: Szabo K. Chem. Eur. J. 2004;10:5268. doi: 10.1002/chem.200400261. Gollaszewskl A, Schwartz J. Organometallics. 1985;4:415. Nakamura H, Iwama H, Yamamoto Y. J. Am. Chem. Soc. 1996;118:6641. Nakamura H, Shim J-G, Yamamoto Y. J. Am. Chem. Soc. 1997;119:8113. Zhang P, Brozek LA, Morken JP. J. Am. Chem. Soc. 2010;132:10686. doi: 10.1021/ja105161f.
- [17] a).Trost BM, Verhoeven TR. J. Org. Chem. 1976;41:3215. [Google Scholar]; b) Trost BM, Xu J, Schmidt T. J. Am. Chem. Soc. 2009;131:18343. doi: 10.1021/ja9053948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Additionally, allowing the reaction of 1a to take place in the presence of 1 equiv. of benzaldehyde led only to product 2a. A palladium bisallyl-like complex would be expected to be trapped by the aldehyde (footnote 16).
- [19].Trost BM. Chem. Eur. J. 1998;4:2405. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.