

1. New insights into the pathophysiology of immune-mediated marrow failure: Neal S. Young
The last several years have seen exciting developments in the study of marrow failure. In the clinic, outcomes for older patients undergoing allogeneic sibling transplant and younger patients receiving hematopoietic stem cells from matched unrelated marrow donors have markedly improved [1-4]. In patients who are treated with immunosuppression, overall hematologic improvement has been increased with tandem courses of antithymocyte globulin (ATG) and survival in primary refractory patients has increased, due to both more aggressive immunosuppression and to supportive care, especially new antifungal agents [5-7]. In the laboratory, sophisticated flow cytometry and molecular biology methods allow immune abnormalities to be detected and monitored over time in individual patients. Clonal abnormalities can be assessed early by sensitive techniques like fluorescent in situ hybridization (FISH) and the application of genomics, as in comparative genomic hybridization (CGH). Finally, discoveries in the marrow failure clinic have been generalized to the origins of a broad group of human cancers and to specific organ failure disease. The linking of constitutional marrow failure to acquired aplastic anemia through genetic defects in telomere repair has provided the likely explanation for the troubling evolution from an inflammatory pathophysiology, T cell-mediated marrow destruction, to malignant hematologic diseases like myelodysplasia (MDS) and acute myeloid leukemia. The regenerative/repair deficit conferred by telomerase deficiency affects not only bone marrow but other tissues, leading to disease in the lung and liver.
Whatever their long-term outcome in the clinic, that the great majority of patients with aplastic anemia will respond to immunosuppressive therapy (usually delivered as at least one and sometimes two repeated course of ATG) implies an underlying immune pathophysiology. Low pre-treatment blood counts (reticulocytes and lymphocytes) decrease the probability of hematologic improvement after immunosuppression and of survival, suggesting that stem cell number (rather than an alternative pathophysiology) limits response to therapy [7]. The persistence of oligoclones of cytotoxic T cells after successful ATG administration, their re-emergence at relapse, and uncertainty as to the mechanism(s) of action of ATGs can be interpreted as evidence of the inadequacy of current immune therapeutic strategies.
Clonal evolution occurs in a subset of patients, about 15% long-term, and is almost always manifest as the emergence of aneuploidy in marrow cytogenetics, accompanied by recurrent pancytopenia and often MDS or leukemia [8,9]. Selection of clones is broadly understood from in vitro and in vivo studies. For the most common abnormality of monosomy 7, in aplastic anemia, as also in congenital neutropenia, elevated G-CSF, either endogenous in the setting of severe neutropenia or exogenous due to chronic administration of the cytokine, favors cells bearing a G-CSF receptor that signals for proliferation and not differentiation [10]. For trisomy 8, an immune response to aneuploid cells leads to selection and persistence of trisomy 8 cells that fail to complete apoptosis.
Defective telomere homeostasis is a feature of aplastic anemia in some cases and helps explain the occurrence of marrow failure, relapse, and clonal evolution. Mutations in genes of the telomere repair complex and of the shelterin proteins occur in children with dyskeratosis congenita; mutations in TERT (the telomerase gene), TERC (the RNA template gene), and also in TINF2 (a shelterin gene) are also present in adults without obvious family histories and in the absence of typical physical findings associated with constitutional marrow failure [11,12]. TERT and TERC mutations reduce telomerase activity in vitro and accelerate telomere attrition in vivo. TERT mutations subsequently have been associated with idiopathic pulmonary fibrosis.
Most recently, we have studied five families in which the probands presented with marrow failure, and TERT and TERC mutations in these pedigrees have tracked with severe liver disease; telomerase repair mutations also have been detected in a large series of patients with a variety of liver diseases. Abnormal telomere biology underlies regenerative diseases of bone marrow, lung, and liver.
In acquired aplastic anemia, independent of known genetic alterations, the presence of short telomeres of leukocytes at time of presentation profoundly affects the clinical course: patients with short telomeres do respond to immunosuppressive interventions, but their relapse rate is almost double that of cases with normal telomere length, and virtually all clonal evolution occurs in patients in the lowest quartile of telomere length. Excess telomere-free ends of chromosomes and genomic instability by spectrakaryotyping (SKY) can be demonstrated in tissue culture of bone marrow from aplastic anemia patients with short telomeres and TERT- and TERC-mutant family members. Hypomorphic TERT mutations can also be detected in patients with de novo acute myeloid leukemia. These data in patients and with human cells confirm predictions made based on yeast experiments and in “knock-outs” of telomerase genes in the mouse. Short telomeres and single nucleotide polymorphisms in TERT have been linked by multiple laboratories to cancer development in other circumstances, suggesting that telomere biology provides the missing link between chronic inflammation and malignant transformation. In marrow failure, sex hormone therapy may be helpful in improving hematopoiesis due to direct modulation of telomerase activity.
2. Outcomes after immunosuppressive therapy for aplastic anemia: Judith CW Marsh
2.1 Introduction
The current standard immunosuppressive therapy (IST) for aplastic anemia (AA) is the combination of antithymocyte globulin (ATG) and cyclosporine (CSA). Overall survival after IST for aplastic anemia has improved significantly during the period from early 1970s to 2000, and is currently around 75% at 5 years. Although this equates with survival after hematopoietic stem cell transplantation (HSCT), there are important differences between IST and HSCT for AA, in terms of speed of hematological recovery, later complications and quality of life issues. Survival following IST has, however, failed to improve further during the last decade (see figure 1) [13].
Figure 1.
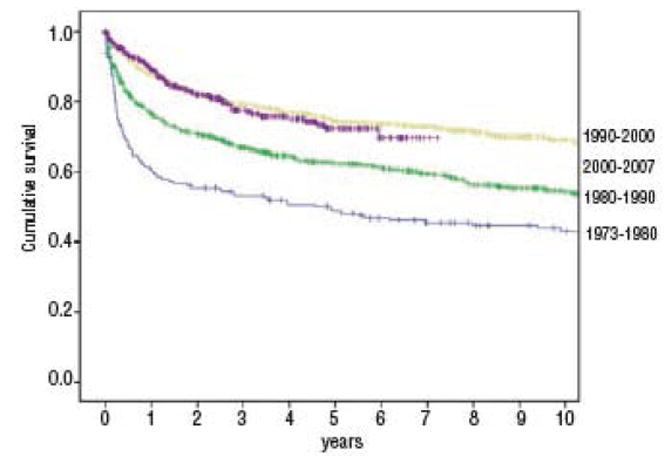
Survival up to 10 years for patients (n=2400) with severe aplastic anemia treated with ATG reported to the European Group for Blood and Marrow Transplantation (EBMT) database having received ATG ± CSA as a first line treatment. Patients were treated between 1973-2007. Five-year survival probabilities are: 49±7% for patients treated between 1973-1980 (n=178), 62 ± 3% for those treated between 1980-1990 (n=850), 74 ± 3% for patients treated between 1990-2000 (n=928) and 72 ± 6% for those treated between 2000-2007 (n=444).
2.2 Indications for IST
ATG with CSA is indicated as first line therapy for non-severe AA patients who are transfusion dependent, severe AA (SAA) patients > 40 years of age and SAA patients < 40 years who lack an HLA identical sibling donor. Patients with SAA who are < 40 years old and who have an HLA compatible sibling, should be offered HSCT as first line treatment. In Europe, unrelated donor (UD) HSCT is now considered after failure to respond to one course of ATG in patients who lack an HLA compatible sibling (see figure 2), as a result of recent improvements in outcome after UD HSCT [14]. Older age is not per se a reason for withholding specific treatment, but depends on severity of the disease, and mainly on the severity of neutropenia, the presence of co-morbidity and the willingness of the patient and his family to be treated, as patients > 60 years old have an increased risk of infection, bleeding and cardiac events after ATG [15].
Figure 2.
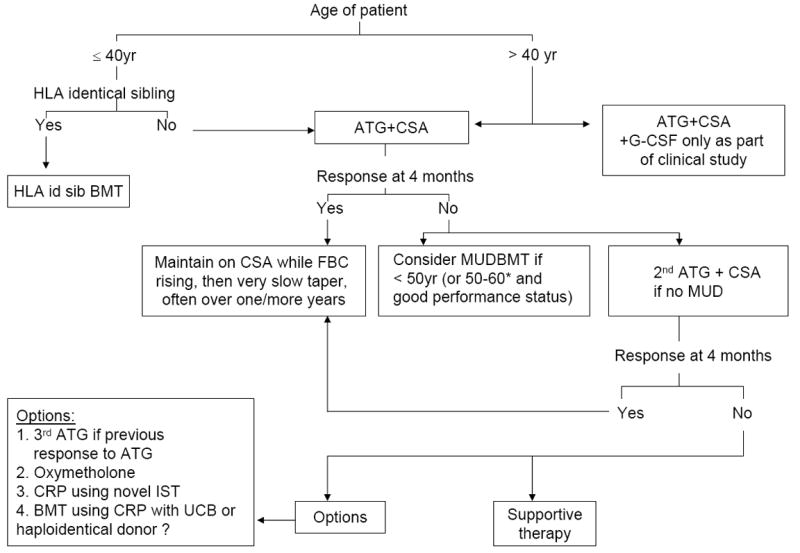
Treatment of acquired severe aplastic anaemia
CRP, clinical research protocol; IST, immunosuppressive therapy; UCB, umbilical cord blood; MUD, matched unrelated donor; CSA, ciclosporin; HLA id sib, HLA identical sibling. * For patients older than 60 years, there is currently insufficient data on the role of HSCT in severe AA although data for MDS suggests that this may be a future option (see text).
2.3 Response rates and predictive factors after ATG and CSA
Response to ATG occurs in around 50% by 3 months and 70-75% by 6 months, and supports an autoimmune basis for the disease in many patients. Lack of response may be due to (i) inadequate immunosuppression (ii) irreversible stem cell deficiency or (iii) non-immune mediated AA. Patients with non-severe AA are more likely to respond than patients with severe or very severe AA. However, the classical Camitta criteria do not reliably predict for response to ATG, as they were established in the pre-ATG era to assess who may benefit from HSCT. Recent work has shown that only younger age, absolute reticulocyte count (ARC) and absolute lymphocyte count (ALC), correlate with response to ATG [7]. The lack of association with the absolute neutrophil response reflected a high number of early deaths in patients with very severe neutropenia. Several studies have examined whether the presence of a PNH clone is associated with response to ATG, with conflicting results. This may reflect differences in sensitivity of the test used to detect a PNH clone. Using very sensitive tests to detect GPI-deficient clones of < 0.003 cells, a strong correlation exists with response among Japanese patients [16]. Shortened telomere length (which occurs in around a third of AA patients) does not preclude initial response to IST, but predicts relapse after ATG and is a risk factor for later cytogenetic abnormalities and evolution to MDS and AML [17].
2.4 Can response to ATG and CSA be improved further?
Two prospective randomised studies have examined the use of daily G-CSF for 3 months after ATG and CSA [18]. The addition of G-CSF conferred no benefit in terms of survival, durable response or reduction in infections. Follow up was too short to evaluate the long term risk of clonal evolution to myelodysplastic syndrome (MDS), AML or PNH. A retrospective study from EBMT showed a trend for more MDS/AML when G-CSF is used with ATG and CSA, and G-CSF was a significant risk factor for MDS/AML in older patients (> 40 years) [9]. In vitro studies have shown that G-CSF causes expansion of small pre-existing monosomy 7 cells and up-regulation of the isoform IV G-CSF receptor, resulting in defective differentiation signalling and expansion of undifferentiated cells [10].
The rationale for using additional immunosuppressive agents with ATG and CSA is that the combination of ATG and CSA alone may provide insufficient immunosuppression for some patients. A retrospective study showed no improvement in response or reduction in relapse using mycophenolate mofetil (MMF) in combination with ATG and CSA compared without MMF [19]. The addition of sirolimus in a prospective randomised study resulted in no significant difference in response to ATG, CSA (51% at 6 months in the arm with sirolimus and 62% without sirolimus) [20].
A second course of ATG may be given if there is relapse after the first course or no response to the first course. When rabbit ATG is given for the second course following an initial course of horse ATG, the response rate was 30% for non-responders and 65% for relapsing patients [21]. A recent study from Japan has examined prospectively the outcome of 52 children who failed one course of IST. The response to a second course of ATG was only 11%.
2.5 Alternative immunosuppressive agents
High dose cyclophosphamide (200mg/kg) given in the absence of stem cell support, results in durable responses were seen in just over 50% of patients who have failed ATG, but the predictable and markedly prolonged cytopenias exposes patients to a high risk of fatal fungal infections, and prolonged in-patient days in hospital. Furthermore, it does not eliminate the risk of clonal events. The anti-CD52 monoclonal antibody, alemtuzumab is currently being evaluated in the treatment of AA. A small prospective study using a single course of alemtuzumab (100mg over 5 days) and CSA in AA, showed response in 9/18 patients. Relapses were common but successfully treated with a further course [22]. Daclizumab (anti-IL-2R) shows response in almost 40% of patients with non-severe AA [6].
2.6 Late complications after IST
Relapse occurs in up to 30-35% of patients when CSA is withdrawn at 6 months. A more prolonged course of CSA with a later slow tapering of the drug reduces the relapse risk to around 13-16%, and about a third of patients are CSA dependent and required a small dose long term. Long term follow up of the prospective German national study of 84 patients treated with either the combination of ATG and CSA or ATG alone, between 1986-1989, reported an actuarial probability of developing hemolytic PNH at 11 years was 10%, MDS or AML 8% and a solid tumor 11% [8]. Risk factors for developing MDS/AML include (i) repeated courses of ATG (ii) older age (iii) high doses and prolonged duration of G-CSF with ATG and CSA and (iv) short telomeres.
3. Bone Marrow Transplantation (BMT): Andrea Bacigalupo
3.1 HLA identical siblings
3.1.1 Current outcomes
An allogeneic bone marrow transplant (BMT) from an HLA identical sibling remains the treatment of choice for patients with acquired aplastic anemia (SAA). The current survival for children (<16 years) receiving a bone marrow transplant (BMT) from an HLA identical sibling, following a preparative regimen with cyclophosphamide (CY) 200 mg/kg, is 91%, significantly better than survival for patients over 16 years of age (74%) [2]. As to the stem cell source, a recent EBMT/IBMTR report suggests that the use of peripheral blood (PB) reduces survival when compared to bone marrow, in patients <20 years, from 85% to 73%, and in patients >20 years from 64% to 52%: the major cause of excess mortality in the PB arm was chronic graft versus host disease (GvHD) [23]. This study suggests that PB transplants should not be used in patients with acquired AA, possibly because the increased incidence of chronic GvHD does not translate in improved survival in aplastic patients, in the way it does benefit patients with leukemia. A suitable marrow cell dose is recommended, since results of transplantation are highly dependent on the number nucleated cells infused. CY alone remains the best conditioning regimen for young patients, and ATG would seem to reduce the risk of graft failure, although a recent randomized trial has shown some advantage, but no significant difference in survival [24]. The combination of cyclosporine (CSA) and methotrexate (MTX) offers a survival advantage over CSA alone (84% vs 75%), when used for graft versus host disease prophylaxis. Therefore standard CY 50 mg/kg/day x4, with 3 days of ATG conditioning, followed by unmanipulated marrow as a stem cell source, and CSA+MTX as GvHD prophylaxis is still standard of care for patients with acquired AA undergoing an HLA identical sibling transplant.
3.1.2 Irradiation
In the recent EBMT analysis of SAA patients undergoing and HLA identical sibling BMT, the use of irradiation was a significant negative predictor for outcome [2]. Although some centers continue to use total lymphoid irradiation (TLI) for HLA matched sibling transplants, with excellent results an no second tumors, the consensus in the EBMT Working Party is that, in the setting of an HLA identical sibling transplants, the use of radiation, peripheral blood, or other conditioning regimens should all be tested in prospective trials, because of an unproven benefit for the patient.
3.1.3 HLA identical BMT for patients over 30 years
Despite the excellent results with standard BMT in AA (see Figure 3), there is a strong age effect: the actuarial 10 year survival for patients grafted from HLA identical siblings in the last decade is respectively 83%, 73%, 68% and 51% for ages 1-20 (n=681), 21-30 (n=339), 31-40 (n=146) and =>40 (n=111) (EBMT data unpublished). Unfortunately the use of PB as a stem cell source did not solve the problem, and actually worsened outcome in older patients, though not significantly [23]. There have been 3 reports in the literature suggesting that the combination of fludarabine and CY (FLU-CY) produces a high rate of engraftment and very encouraging survival [25]. We have therefore explored the use of FLU-CY and ATG, in patients over the age of 30: the initial results are encouraging. with transplant mortality lower than 30%; we have also compared the FLU-CY regimen given to 30 patients (median age 45) with a matched pair of SAA patients receiving CY 200 (median age 39): actuarial survival at 5 years was 76% for FLU-CY vs 56% for CY200 (EBMT data unpublished).
Figure 3.
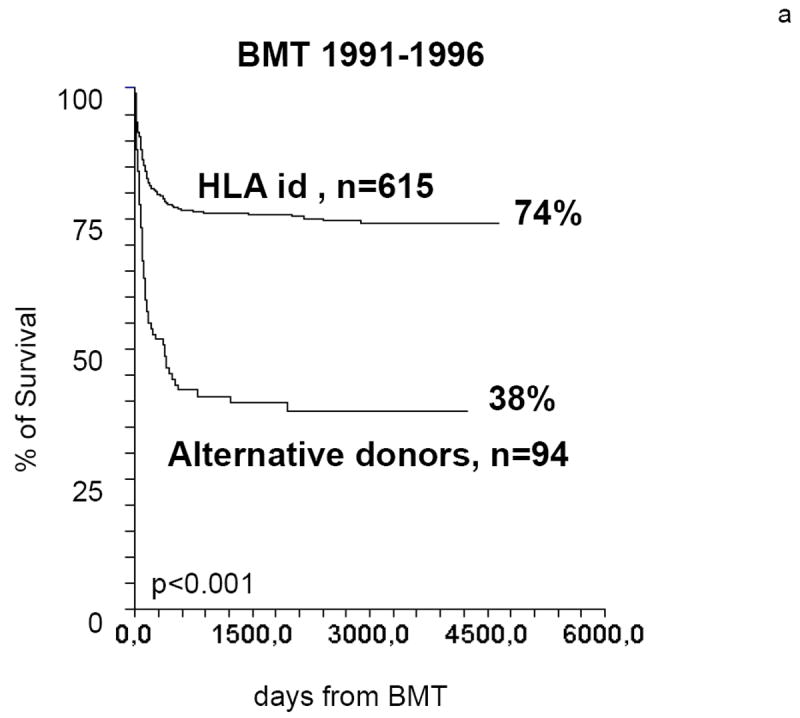
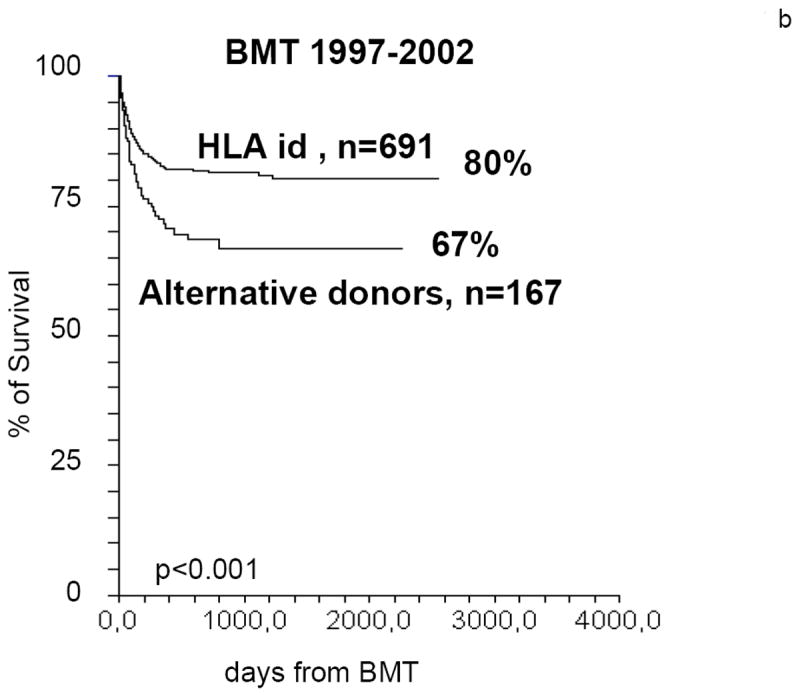
Actuarial survival of SAA patients undergoing a BMT from HLA identical siblings or from alternative donors. In the period 1991-96 the difference in actuarial survival is greater than 35%. In the period 1997-2002 the difference is reduced to less than 15% (data from the EBMT WPSAA)
3.1.4 Engraftment, rejection, mixed chimerism
One of the historic issues in SAA transplants has always been engraftment/rejection, the reason being that (a) this is an autoimmune disorder, and (b) CY 200 alone is not immuno-ablative. Rejection is reported in the range of 10-25%, according to the conditioning regimen and the duration of transfusion support before transplant. In some countries, where patients are highly sensitized, busulfan as been introduced in the conditioning regimen. There are several forms of rejection: (A) failure to achieve a neutrophil count of 500/cmm (so called primary graft failure or failure to engraft) (B) acute/early rejection after initial engraftment (classic early rejection), (C) late –often progressive- graft failure, and (D) acute rejection following discontinuation of CSA therapy: the 3 latter are referred to as secondary graft failure. The CIBMTR has recently analyzed 166 patients receiving a second graft from sibling donors, to study risk factors affecting outcome [26] : best survival was seen in patients with good performance status grafted beyond 3 months from 1st transplant (76%) compared to 33% for patients with poor performance status grafted within 3 months from 1st transplant. The predominant cause of failure was non engraftment. The conclusion of this study is that novel conditioning regimens should be investigated for second transplants in SAA patients with primary or secondary graft failure [26].
With improved methods to identify donor/recipient cells, we have learned that many patients have mixed chimerism post-transplant. A recent EBMT analysis stratifies patients into 5 separate groups: (a) complete donor chimerism (b) transient mixed chimeras (c) stable mixed chimeras (d) progressive mixed chimeras and (e) recipient cells and early rejection [27]. The complete donor chimeras have more GvHD, whereas progressive mixed chimeras are at high risk of late graft failure, especially after discontinuation of CSA therapy. Therefore, monitoring chimeric status after transplantation in SAA is important, and may give relevant information on optimal immunosuppressive therapy to be administered: a significant proportion of patients (approximately 30%) will become permanent mixed chimeras, with normal cell counts and usually no GvHD.
3.2 Unrelated donor (UD) transplants
The outcome of unrelated donor transplants for patients with AA, has improved in the last decade [28], see Figure 3: better donor/recipient HLA matching has probably played a major role, but also significant changes in the conditioning regimen have occurred [29, 30]. The Japanese study reported 154 SAA patients undergoing an UD transplant, the majority receiving 3Gy TBI [30]: unfavorable factors for survival were older age (>20), conditioning without anti-thymocyte globulin (ATG) and a long (>3 years) interval from diagnosis to transplant. The American Seattle study [29] tested different doses of TBI and found that 2 Gy was the best dose. The EBMT group tested a non-radiation based program: results were overall encouraging with 70% surviving, although rejection was high in young adults over the age of 14. The EBMT is currently testing a conditioning regimen which is very similar to the Japanese regimen: FLU-CY-ATG and low dose TBI (2 Gy) (unpublished). As a consequence of less toxic conditioning regimens and improved donor/recipient matching, survival has almost doubled in the past decade [2,28] from 38% in 1991-1996 to 65% in the period 1997-2002, and in the latter period survival after UD transplants in children is 75% vs 63% for adults >16 years of age. Results of UD transplants have improved to such an extent, that treatment strategy may be affected: in children without a matched sibling donor, an UD search should be started at diagnosis, and transplantation should be seriously considered after one course of IS in the presence of a suitable donor. In young adults between 20 and 30 years old, the same may be true. Adults over the age of 30 years should be entered on a prospective trial, alternative donor transplant is an option for second line treatment in patients failing 1 or 2 courses of immunosuppressive treatment.
3.3 Cord Blood Transplants
A proportion of patients will not have a matched donor in the family, and will not find a suitable unrelated donor in the world wide network (Bone Marrow Donors World wide, BMDWW). The percentage of patients who lack a donor will vary between 5% and 40%, according to the ethnic origin of the patient. Cord blood transplantation (CBT) is an alternative option which has been successfully explored in patients with hematologic malignancies. Due to the high rate of rejection in AA patients and the low cell numbers of cord blood units, transplants of unrelated cord blood have been usually been discouraged in this setting. However, a recent study from the Japanese group [3] reports 31 CBT with an overall survival of 42%, but a more encouraging 80% survival for patients receiving the FLU-CY-TBI 2 Gy combination as a conditioning regimen. Thus, cord blood may not be the first option in AA patients lacking a family donor, but some investigators are exploring this stem cell source, and results may be encouraging with appropriate cell dosing, double units [31], alternative routes of administration, namely intra-osseous, and new conditioning regimens [3].
3.4 Conclusions
Children grafted from HLA identical siblings, should receive a conventional CY200 conditioning regimen, followed by bone marrow as a stem cell source and CSA plus MTX for GvHD prophylaxis. Adults, grafted from HLA identical siblings, may explore the use of FLU-CY with ATG. Patients who lack an HLA identical sibling should be considered for an alternative donor transplant: an 8/8 matched unrelated donor would be the first choice, although it may be possible to use also a 7/8 donor or a one antigen mismatched family donor. The preferred conditioning regimen for alternative donor transplants, is FLU-CY ATG with the addition of low dose (2 Gy) TBI. Young children with an 8/8 matched UD may also be grafted with FLU-CY-ATG without TBI. The use of unrelated cord blood remains investigational.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fuhrer M. Risk-adapted procedures for HSCT from alternative donors in children with severe aplastic anemia. Bone Marrow Transpl. 2008;42(Suppl 2):S97–100. doi: 10.1038/bmt.2008.293. [DOI] [PubMed] [Google Scholar]
- 2.Locasciulli A, et al. Outcome of patients with acquired aplastic anemia given first line bone marrow transplantation or immunosuppressive treatment in the last decade: a report from the European Group for Blood and Marrow Transplantation (EBMT) Haematologica. 2007;92:11–18. doi: 10.3324/haematol.10075. [DOI] [PubMed] [Google Scholar]
- 3.Yoshimi A, et al. Unrelated cord blood transplantation for severe aplastic anemia. Biol Blood Marrow Transplant. 2008;14:1057–1063. doi: 10.1016/j.bbmt.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 4.Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108:2509–2519. doi: 10.1182/blood-2006-03-010777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valdez JM, Scheinberg P, Young NS, Walsh TJ. Infections in patients with aplastic anemia. Semin Hematol. 2009;46:269. doi: 10.1053/j.seminhematol.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 6.Maciejewski JP, et al. Recombinant humanized anti-IL2 receptor antibody (daclizumab) produces responses in patients with moderate aplastic anemia. Blood. 2003;102:3584–3586. doi: 10.1182/blood-2003-04-1032. [DOI] [PubMed] [Google Scholar]
- 7.Scheinberg P, Wu C, Nunez O, Young NS. Predicting response to immunosuppressive therapy and survival in severe aplastic anaemia. Br J Haematol. 2009;144:206–216. doi: 10.1111/j.1365-2141.2008.07450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frickhofen N, Heimpel H, Kaltwasser JP, Schrezenmeier H for the German Aplastic Anaemia Study Group. Antithymocyte globulin with or without cyclosporin A: 11-year follow-up of a randomised trial comparing treatments of aplastic anemia. Blood. 2003;101:1236–1242. doi: 10.1182/blood-2002-04-1134. [DOI] [PubMed] [Google Scholar]
- 9.Socie G, et al. Granulocyte-stimulating factor and severe aplastic anemia: a survey by the European Group for Blood and Marrow Transplantation (EBMT) Blood. 2007;109:2794–2796. doi: 10.1182/blood-2006-07-034272. [DOI] [PubMed] [Google Scholar]
- 10.Sloand EM, Yong ASM, Ramkissoon S, et al. Granulocyte colony-stimulating factor preferentially stimulates proliferation of monosomy 7 cells bearing the isoform IV receptor. PNAS. 2006;103:14483–14488. doi: 10.1073/pnas.0605245103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calado RT, Young NS. Telomere maintenance and human bone marrow failure. Blood. 2008;111:4446–4455. doi: 10.1182/blood-2007-08-019729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirwan M, Dokal I. Dyskeratois congenita, stem cells, and telomeres. Biochim Biophys Acta. 2009;1792:371–379. doi: 10.1016/j.bbadis.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Passweg JR, Tichelli A. Immunosuppressive treatment for aplastic anemia: are we hitting the ceiling? Haematologica. 2009;94:310–312. doi: 10.3324/haematol.2008.002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marsh JCW, Ball SE, Cavenagh J, Darbyshire P, Dokal I, Gordon-Smith EC, Keidan AJ, Laurie A, Martin A, Mercieca J, Killick SB, Stewart R, Yin JAL British Committee for Standards in Haematology (BCSH) Guidelines for the diagnosis and management of aplastic anaemia. Brit J Haematol. doi: 10.1111/j.1365-2141.2009.07842.x. Published Online: Aug 10 2009 11:38PM. [DOI] [PubMed] [Google Scholar]
- 15.Kao SY, Xu W, Brandwein JM, Lipton JH, Messner HA, Minden MD, Schimmer AD, Schuh AC, Yee K, Gupta V. Outcomes of older patients (≥ 60 years) with acquired aplastic anaemia treated with immunosuppressive therapy. Brit J Haematol. 2008;143:738–743. doi: 10.1111/j.1365-2141.2008.07389.x. [DOI] [PubMed] [Google Scholar]
- 16.Sugimori C, Chuhjo T, Feng X, et al. Minor populations of CD55-CD59- blood cells predicts response to immunosuppressive therapy and prognosis in patients with aplastic anemia. Blood. 2005;107:1308–1314. doi: 10.1182/blood-2005-06-2485. [DOI] [PubMed] [Google Scholar]
- 17.Cooper JN, Calado R, Wu C, et al. Telomere Length of peripheral blood leukocytes predicts relapse and clonal evolution after immunosuppressive therapy in severe aplastic anemia. Blood. 2008;112:442. abstract. [Google Scholar]
- 18.Gurion R, Gafter-Gvili A, Paul M, Vidal L, Ben-Bassat I, Yeshurun M, Schilberg O, Raanani P. Hematopoietic growth factors in aplastic anemia patients treated with immunosuppressive therapy-systematic review and meta-analysis. Haematologica. 2009;94:712–719. doi: 10.3324/haematol.2008.002170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scheinberg P, Nunez O, Wu C, Young N. Treatment of severe aplastic anaemia with combined immunosuppression: antithymocyte globulin, ciclosporin and mycophenolate mofetil. Brit J Haematol. 2006b;133:606–611. doi: 10.1111/j.1365-2141.2006.06085.x. [DOI] [PubMed] [Google Scholar]
- 20.Scheinberg P, Wu CO, Nunez O, et al. Treatment of severe aplastic anemia with a combination of horse antithymocyte globulin and cyclosporine, with or without sirolimus: a prospective randomized study. Haematologica. 2009;94:348–354. doi: 10.3324/haematol.13829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scheinberg P, Nunez O, Young N. Re-treatment with rabbit antithymocyte globulin and ciclosporin for patients with relapsed or refractory severe aplastic anaemia. Brit J Haematol. 2006a;133:622–627. doi: 10.1111/j.1365-2141.2006.06098.x. [DOI] [PubMed] [Google Scholar]
- 22.Risitano AM, Seneca E, Marando L, Serio B, Selleri C, Scalia G, Vecchio LD, Iori A, Kulagin A, Maury S, Halter J, Gupta V, Bacigalupo A, Socié G, Tichelli A, Marsh J, Schrezenmeier H, Passweg JR, Rotoli B. Subcutaneous Alemtuzumab is a safe and effective treatment for global or single-lineage immune-mediated marrow failures: a survey from the EBMT-WPSAA. Blood (ASH Annual Meeting Abstracts) 2008;112:1042. [Google Scholar]
- 23.Schrezenmeier H, Passweg JR, Marsh JC, et al. Worse outcome and more chronic GVHD with peripheral blood progenitor cells than bone marrow in HLA-matched sibling donor transplants for young patients with severe acquired aplastic anemia: A report from the European Group for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research. Blood. 2007;110(4):1397–400. doi: 10.1182/blood-2007-03-081596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Champlin RE, Perez WS, Passweg JR, et al. Bone marrow transplantation for severe aplastic anemia: a randomized controlled study of conditioning regimens. Blood. 2007;109:4582–5. doi: 10.1182/blood-2006-10-052308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.George B, Methews V, Viswabandya A, Kavitha ML, Srivastava A, Chandy M. Fludarabine and cyclophosphamide based reduced intensity conditioning (RIC) regimens reduce rejection and improve outcome in Indian patients undergoing allogeneic stem cell transplantation for severe aplastic anemia. Bone Marrow Transplant. 2007;40:13–8. doi: 10.1038/sj.bmt.1705669. [DOI] [PubMed] [Google Scholar]
- 26.Horan JT, Carreras J, Tarima S, Camitta BM, Gale RP, Hale GA, Hinterberger W, Marsh J, Passweg JR, Walters MC, Eapen M. Risk factors affecting outcome of second HLA-matched sibling donor transplantations for graft failure in severe acquired aplastic anemia. Biol Blood Marrow Transplant. 2009;15:626–31. doi: 10.1016/j.bbmt.2009.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawler M, McCann SR, Marsh JC, Ljungman P, Hows J, Vandenberghe E, O’Riordan J, Locasciulli A, Socié G, Kelly A, Schrezenmeier H, Marin P, Tichelli A, Passweg JR, Dickenson A, Ryan J, Bacigalupo A. Severe Aplastic Anaemia Working Party of the European Blood and Marrow Transplant Group. Serial chimerism analyses indicate that mixed haemopoietic chimerism influences the probability of graft rejection and disease recurrence following allogeneic stem cell transplantation (SCT) for severe aplastic anaemia (SAA): indication for routine assessment of chimerism post SCT for SAA. Brit J Haematol. 2009;144:933–45. doi: 10.1111/j.1365-2141.2008.07533.x. Epub 2009 Jan 9. [DOI] [PubMed] [Google Scholar]
- 28.Maury S, Balere-Appert ML, Chir Z, et al. French Society of Bone Marrow Transplantation and Cellular Therapy (SFGM-TC). Unrelated stem cell transplantation for severe acquired aplastic anemia: improved outcome in the era of high-resolution HLA matching between donor and recipient. Haematologica. 2007;92:589–96. doi: 10.3324/haematol.10899. [DOI] [PubMed] [Google Scholar]
- 29.Kojima S, Matsuyama T, Kato S, et al. Outcome of 154 patients with severe aplastic anemia who received transplants from unrelated donors: the Japan Marrow Donor Program. Blood. 2002;100:799–803. doi: 10.1182/blood.v100.3.799. [DOI] [PubMed] [Google Scholar]
- 30.Deeg HJ, Amylon ID, Harris RE, et al. Marrow transplants from unrelated donors for patients with aplastic anemia: minimum effective dose of total body irradiation. Biol Blood Marrow Transplant. 2001;7:208–15. doi: 10.1053/bbmt.2001.v7.pm11349807. [DOI] [PubMed] [Google Scholar]
- 31.Ruggeri A, de Latour RP, Rocha V, Larghero J, Robin M, Rodrigues CA, Traineau R, Ribaud P, Ferry C, Devergie A, Gluckman E, Socié G. Double cord blood transplantation in patients with high risk bone marrow failure syndromes. Brit J Haematol. 2008;143:404–8. doi: 10.1111/j.1365-2141.2008.07364.x. Epub 2008 Aug 10. [DOI] [PubMed] [Google Scholar]