

Abstract
Direct detection of pseudouridine (ψ), an isomer of uridine, in RNA is challenging. The most popular method requires chemical derivatization using N-cyclohexyl-N'-β-(4-methylmorpholinumethyl) carbodiimide p-tosylate (CMCT) followed by radiolabeled primer extension mediated by reverse transcriptase. More recently, mass spectrometry (MS)-based approaches for sequence placement of pseudouridine in RNA have been developed. Nearly all of these approaches, however, only yield qualitative information regarding the presence or absence of pseudouridine in a given RNA population. Here, we have extended a previously developed liquid chromatography tandem mass spectrometry (LC-MS/MS) method to enable both the qualitative and quantitative analysis of pseudouridine. Quantitative selected reaction monitoring (SRM) assays were developed using synthetic oligonucleotides, with or without pseudouridine, and the results yielded a linear relationship between the ion abundance of the pseudouridine-specific fragment ion and the amount of pseudouridine-containing oligonucleotide present in the original sample. Using this quantitative SRM assay, the extent of pseudouridine hypomodification in the conserved T-loop of tRNA isolated from two different Escherichia coli strains was established.
Keywords: Pseudouridine quantification, tRNA, Mass spectrometry, Selected reaction monitoring, SRM, Detection of RNA modifications, Oligonucleotides
Introduction
Pseudouridine (ψ) is the most prevalent post-transcriptional modification in ribonucleic acids (RNA) [1, 2]. This isomer of uridine is commonly found in non-coding RNAs, including transfer RNA (tRNA), ribosomal RNA (rRNA), small nuclear RNA (snRNA), and small nucleolar RNA (snoRNA). Moreover, pseudouridine is found in all phylogenetic domains of life (Archaea, Eubacteria, and Eukarya) [3]. Although a precise molecular role for this modification is unclear, pseudouridine has been shown to confer rigidity and stabilize RNA structure, enhance local base stacking, and is involved in imino proton-H2O coordination [4, 5]. Pseudouridine modulates codon-anticodon interactions between messenger RNA and tRNA [6] and facilitates ribosome assembly [7].
Pseudouridine is formed by site-specific isomerization at the polyribonucleotide level following recognition of the target uridine in the context of the RNA sequence or structure of the site of interest by a group of enzymes called pseudouridine synthases [8, 9]. Studies on prokaryotes and eukaryotes have shown that deletion of pseudouridine synthase genes has a negative impact on cell growth and protein synthesis rates [10, 11] indicating the critical function of this modification in the translational apparatus. However, identifying the exact location of pseudouridine in the context of its parent RNA is key to an improved understanding of the function of this posttranscriptional modification.
Pseudouridine is a mass-silent modification, therefore it does not exhibit a mass shift compared with its unmodified counterpart upon post-transcriptional formation in RNA. The most widely used method for detection and sequence placement of pseudouridine is based on ψ-specific irreversible derivatization using the reagent N-cyclohexyl-N'-β-(4-methylmorpholinium) ethylcarbodiimide p-tosylate (CMCT) followed by reverse transcriptase mediated extension of radiolabeled primer and subsequent separation on a denaturing polyacrylamide gel [12]. Although effective, this method has some drawbacks and limitations, which have spurred development of mass spectrometry-based approaches for the detection and sequence placement of pseudouridine in RNA [13]. The two major mass spectrometry-based approaches involve enzymatic digestion of RNA to either nucleosides or oligonucleotides, yielding samples suitable for analysis.
The presence of pseudouridine can be detected by total enzymatic hydrolysis of RNA into nucleosides and subsequent analysis by liquid chromatography/electrospray ionization mass spectrometry (LC-ESI-MS) [14, 15]. The sensitivity and specificity of pseudouridine detection in a RNA nucleoside mixture was further enhanced upon derivatization with methyl vinyl sulfone [15]. A limitation of this approach is the lack of information relating to the sequence location of pseudouridine in the RNA.
To determine the specific sequence placement of this modification, RNase mapping was employed in combination with chemical derivatization [16–18]. RNase mapping uses base-specific endoribonucleases that generate a mixture of oligoribonucleotides, which are amenable to accurate mass measurements by ESI or MALDI (matrix-assisted laser desorption/ionization) mass spectrometry. As pseudouridine is a mass silent modification, the RNA (before or after enzymatic treatment) is chemically derivatized with either CMCT [16, 17] or acrylonitrile (cyanoethylation) [18] to covalently link a mass tag to pseudouridine that enables differentiation of pseudouridine from uridine by mass spectrometry. Identification of pseudouridine oligonucleotides is accomplished by mass spectrometric comparison of untreated and CMCT or acrylonitrile treated samples. Mass differences of 252 Da (CMCT-treated) or 53 Da (acrylonitrile) between the two samples are used to identify RNase digestion products that contain pseudouridine [16–18].
A more recent development is a tandem mass spectrometry (MS/MS) approach compatible with LC-MS based RNase mapping of nucleic acids [19]. This approach was developed to take advantage of the unique but stable C-C glycosidic bond, which contrasts with the typically labile C–N glycosidic bond in uridine or other nucleosides. The stable C–C glycosidic bond yields unique fragmentation pathways during collision-induced dissociation (CID) leading to the production of pseudouridine-specific fragment ions. These diagnostic ions include the doubly dehydrated nucleoside anion m/z 207 and its MS/MS product ion at m/z 164 as well as the fragmentation product at m/z 165 arising from MS/MS of m/z 225. Selective detection of these fragmentation pathways enabled the identification of pseudouridine in a given oligonucleotide by selected reaction monitoring (SRM). Specifically, pseudouridines can be identified by scanning the SRM transition of m/z 207 to 164. Alternatively, the SRM transition of m/z 225 to 165 can be used to selectively identify the presence of a 5' terminal pseudouridine in the RNase digestion product [19]. McCloskey and co-workers used these SRM assays and chemical derivatization to identify pseudouridines in 23 S rRNAs of Haloarcula marismortui and Deinococcus radiodurans [20], and the 16 S rRNA of Thermus thermophilus [21].
While effective, the approaches described above can only yield qualitative information about the presence or absence of pseudouridine in a given RNA. Most often, the modification at a given position may not be present at stoichiometric amounts as the degree or level of modification may be modulated for regulatory purposes like growth or stress response [22, 23]. Several methods have, therefore, been developed to quantify pseudouridine in RNA. In one, RNase H mediated cleavage of RNA, 32P substitution of phosphate backbone, thin layer chromatography (TLC) of hydrolyzed nucleotides followed by phosphorimaging allows for the quantification of this post-transcriptional modification [24]. A ligation-based method was developed that allows recognition of the modification site and quantification on denaturing PAGE [25]. Although these methods are highly specific, in that modification at a single sequence location can be quantified, the overall assay is not readily applied to multiple modifications. Beyond the LC-MS analysis of nucleoside digests mentioned above, which can be performed quantitatively [26, 27], a novel method enabling the relative quantification of pseudouridine from the base hydrolysis of oligoribonucleotides has recently appeared [28]. Here we have developed a quantitative assay for the routine sequence-specific analysis of pseudouridine. This SRM-based assay utilizes the pseudouridine-specific transitions identified by McCloskey and coworkers [19] to enable one to determine the degree of pseudouridine modification in RNA via RNase mass mapping and LC-MS/MS, where the oligonucleotides are less than 20 nt. Further, we have applied this quantitative assay to document the variability of pseudouridine hypomodification in the conserved T- loop of E. coli tRNAs.
Experimental
Materials
Oligoribonucleotides were obtained from Dharmacon RNAi Technologies (Lafayette, CO, USA) and deprotected as per the manufacturer's instructions. Escherichia coli strain K12 was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA), and E. coli Rosetta 2(DE3) strain was purchased from Novagen (Gibbstown, NJ, USA). 1,1,1,3,3,3-Hexafluoroisopropanol (HFIP), ammonium acetate, Tri-Reagent, lysozyme chloride from chicken egg white, chloroform, 2-propanol, absolute ethanol, 1,2-diaminocyclohexyltetraacetic acid monohydrate (CDTA), and E. coli were used as received from Sigma-Aldrich (St. Louis, MO). HPLC-MS grade methanol, water and acetonitrile were obtained from Honeywell Burdick & Jackson, Inc. (Muskegon, MI, USA). Triethylamine (TEA), sodium chloride, sodium acetate, sodium hydroxide, and sodium dodecyl sufate (SDS) were purchased from Fisher Scientific (Fairlawn, NJ, USA). Molecular biology grade Tris base (tris(hydroxymethyl)aminomethane), and tris-hydrochloride were purchased from Promega (Madison, WI, USA). Bacto yeast extract and bacto tryptone were used as received from BD Biosciences (Sparks, MD, USA). Sodium citrate was purchased from Mallinckrodt Baker, Inc. (Paris, KY, USA). UltraPure agarose, bacterial alkaline phosphatase (BAP), and Dyna beads M-280 were purchased from Invitrogen Corporation (Carlsbad, CA, USA). RNase T1 was purchased from Roche Molecular Biochemicals (Indianapolis, IN, USA). Sep-Pak C18 cartridges were obtained from Waters (Milford, MA, USA). The 5'-biotin labeled oligodeoxynucleotide 5'biotin-GGCAGAGGTCT CAGGTTCGAATCCTGTCGGGCGCG-3′ was obtained from Integrated DNA Technologies, Inc. (Coralville, IA, USA).
Isolation of E. coli tRNAs
E. coli K12 cells were cultured with regular Luria-Bertani (LB) medium, while the medium was supplemented with chloramphenicol (25 μg/mL) for culturing Rosetta 2(DE3) cells as per manufacturer's instructions. The cells were harvested at the end of log phase (0.8–0.9 OD600) and either used straightaway or frozen at −80 °C until further use. The tRNAs were isolated using Tri-Reagent as described [29].
Purification of tRNA Arg (CCG)
Rosetta 2(DE3) cells (Novagen) are designed to alleviate codon bias by complementing the insufficient tRNA pools in the parental cell line K12 through overexpression of rare tRNAs by a multicopy plasmid. One of the overexpressed tRNAs is . This tRNA was affinity purified by using a biotinylated oligodeoxynucleotide whose sequence is complementary to the 3′-end of . The biotinylated oligodeoxynucleotide was coupled to streptavidin-coated magnetic beads (M-280, Invitrogen) following the manufacturer's instructions. Hybridization of the Tri-Reagent isolated tRNAs with the magnetic beads was performed at 70 °C for 10 min followed by washing with stirring at 37 °C for 2 h. The affinity purified was eluted using 15 mM sodium chloride, 1.7 mM sodium citrate, and a 0.1% SDS solution.
Enzymatic Digestion of tRNAs
Guanosine specific ribonuclease RNase T1 was precipitated and purified as described [30]. For digestion, 500 units of RNase T1 and 0.0005 units BAP were added to 10 μg of tRNA and 5 μL of 220 mM ammonium acetate buffer. The reaction mixture was incubated in a 37 °C water bath for 2 h.
Ribonuclease Digestion Products
tRNA sequences were obtained from the Transfer RNA Database [31]. Sequences were theoretically digested using Mongo Oligo Mass Calculator (ver. 2.06; http://library.med. utah.edu/masspec/).
Mass Spectrometry
Direct sample infusion (ESI-MS) and LC-MS/MS experiments were performed on a Thermo Scientific (Waltham, MA, USA) LTQ XL linear ion trap mass spectrometer fitted with an electrospray ionization (ESI) probe at a fixed vertical spray angle (60°) in the Ion Max Source. The electrospray ion source was coupled to a Finnegan Surveyor MS Pump Plus HPLC system and Finnegan Micro AS autosampler for automated injections.
For direct infusion, synthetic oligoribonucleotide samples, combined with CDTA (0.01 nmol) and TEA (0.0015%), were dissolved to achieve a final concentration of 10 pmol/μL in 50:50 water/methanol. Samples were infused via a syringe pump at a flow rate of 3 μL/min and the spectra were acquired in negative ion mode. The settings for pseudouridine detection were: 275 °C ESI source; 4.5 kV spray voltage; sheath, auxiliary and sweep gas to 25, 14, and 10 arbitrary units; ions were tuned for m/z 207 to enable automatic adjustment of capillary, tube lens, and other ion optics voltages. This tune file was used for detection and quantification of all the ψ-specific transitions for all the tested oligonucleotides.
For LC-MS/MS analysis, reverse phase ion-pairing chromatography was performed on an Xbridge MS C18 1.0×150 mm column, 3.5 μm particle size and 135Å pore size (Waters, Milford, MA, USA) at a flow rate of 40 μL/min. Mobile phase A consisted of 16.3 mM TEA/400 mM HFIP at pH 7.0 in water; equal amounts of mobile phase A and methanol were combined to produce mobile phase B. The mobile phase gradient starting at 5 % B increased to 10 % B at 2 min, and 95 % B at 52 min. The mobile phase composition was held at 95 % B for 5 min prior to reequilibrating the column for 15 min under initial mobile phase conditions before the next injection. The entire column effluent was coupled to the electrospray probe via PEEK tubing (1/16 in o.d. 0.005 in i.d., Upchurch Scientific). Negative ion mass spectra were recorded in continuum mode from m/z 450 to 2000 in scan event 1 to acquire the m/z values of intact oligonucleotides. To identify the ion charge states, data were acquired in enhanced scan mode. For detecting pseudouridine diagnostic ions, two additional scan events were used with an elevated source voltage of 90 V for in-source fragmentation. The presence of ψ in the dissociated ions was detected by two consecutive scan events using a reaction monitoring function that recorded the transition from m/z 207 to 164 for internal pseudouridines (scan event 2) and the transition from m/z 225 to 165 for 5′ terminal ψ (scan event 3) at a normalized collision energy of 30%. Vendor supplied Xcalibur software was used for all data acquisition and processing.
Quantitative Analysis
To evaluate the suitability of this method to quantitate unknowns, a set of standard solutions with varied but known concentrations of ψ-containing oligonucleotides were subjected to LC-MS analysis. Matching the retention time of a specific m/z value of an oligonucleotide ion with the ψ-specific transition occurring at the same time indicates the presence of pseudouridine containing oligonucleotide in the eluent. SRM peak area values were used to generate calibration curves in Microsoft Excel from which the pseudouridine concentrations of the sample of interest could be determined.
Results and Discussion
Identification of Pseudouridine-specific Fragment Ions in Oligonucleotides
The SRM approach for detecting ψ-diagnostic fragment ions was originally developed on a triple quadrupole instrument [19]. To confirm that an ion trap mass spectrometer also generates identical SRM transitions for pseudouridine detection, the oligoribonucleotide AψCAG (monoisotopic masscalculated 1552.26) was studied. This oligonucleotide upon in-source dissociation was expected to yield m/z 207, and selection of this precursor and further ion trap fragmentation should yield the diagnostic ion at m/z 164. The AψCAG oligonucleotide exhibited predominantly a deprotonated doubly charged ion at m/z 775.25 (Figure 1a). In-source fragmentation followed by selection and fragmentation of m/z 207 in the ion trap yielded an abundant product ion at m/z 164 (Figure 1b). The negative control, AUCAG, was subjected to the same experimental conditions (Figure 1c)with no product ion at m/z 164 detected (Figure 1d). These data demonstrate that the m/z 207 to 164 SRM transition can be observed with an ion trap mass spectrometer, and this transition is specific to pseudouridine-containing oligoribonucleotides as reported previously [19]. Next, we sought to establish parameters by which this SRM transition could be used to quantify pseudouridine within RNAs.
Figure 1.

(a) ESI-MS of a 10 pmol/μL solution of AψCAG. (b) ESI-MS/MS of a 10 pmol/μL solution of AψCAG. In-source fragmentation (90 V source lens) was applied to dissociate AψCAG. The precursor ion (m/z 207) was then selected for collision-induced dissociation (CID) yielding the mass spectrum seen. The pseudouridine-specific fragment ion at m/z 164 is readily detected. (c) ESI-MS of a 10 pmol/μL solution of AUCAG. (d) ESI-MS/MS of a 10 pmol/μL solution of AUCAG. In-source fragmentation (90 V source lens) was applied to dissociate AUCAG. The precursor ion (m/z 207) was then selected for collision-induced dissociation (CID) yielding the mass spectrum seen. As expected, no pseudouridine-specific fragment ion at m/z 164 can be detected
Quantification Studies
To establish whether a quantitative SRM assay could be developed for pseudouridine, pairs of synthetic oligonucleotides that share an identical sequence but differ for the presence or absence of pseudouridine at a defined site were used. In general, a set of standard solutions containing known amounts of a pseudouridine-containing oligonucleotide was mixed with the same sequence lacking pseudouridine where the total concentration of the two oligonucleotides does not exceed 70 pmol. The samples consisted of 0, 5, 10, 20, 35, 50, 60, 70 pmol of a pseudouridine-containing oligonucleotide and 70, 65, 60, 50, 35, 20, 10, 0 pmol of a pseudouridine-lacking oligonucleotide. These samples were injected onto a C-18 column and analyzed by LC-MS/MS.
An example of the process used to generate quantitative SRM assays is illustrated using the oligonucleotide G(ψ/U) G. Injection of an equimolar mixture of the two oligonucleotides yielded two partially resolved chromatographic peaks, with singly charged ions (m/z 933.08) being more abundant than doubly charged ions (m/z 466.16) (Supplemental Figure S1). Monitoring the pseudouridine specific SRM transition of m/z 207 to 164 identified the earlier eluting peak as the pseudouridine-containing oligonucleotide. An SRM calibration curve for GψG was generated using the peak area for the m/z 207 to 164 transition. Three injections of each mixture were analyzed. As seen in Figure 2a, a linear response (R2=0.998) was obtained over the concentration range of interest (0 to 60 pmol).
Figure 2.

Calibration curves for the selected reaction monitoring (SRM) transition m/z 207 to 164 for (a)GψG, (b)m5UψCG, (c) AψCAG, (d) UAACψAUGACG, and (e) UAACψAψGACG. Calibration curves were generated by LC-MS/MS analysis of defined amounts of each pseudouridine-containing oligonucleotide in combination with its uridine-containing analog, where the total oligonucleotide amount was 70 pmol. SRM transition peak areas, determined in triplicate, were plotted against the amount of pseudouridine-containing oligonucleotide and the resulting data fit to a linear least squares function. Although the sensitivities are sample dependent, all oligonucleotides yielded linear calibration curves with high precision
Similar results were obtained from other oligonucleotides. The TψCG:TUCG (R2=0.975) and AψCAG:AUCAG (R2= 0.991) mixtures yielded linear responses (Figure 2b, c) along with chromatographic resolution of the two components (Supplemental Figures S2 and S3). Differences in assay sensitivity were found among these three pairs of oligonucleotides with GψG being the most sensitive and TψCG the least sensitive, although the differences among these shorter oligonucleotides were less than an order of magnitude.
Next we sought to determine whether the SRM quantification assay could be extended to larger oligonucleotides as well as oligonucleotides containing more than one pseudouridine. An 11-mer sequence containing two internal uridines, UAACUAUGACG, was used along with the single and double pseudouridine analogs UAACψAUGACG and UAACψAψGACG. For these larger oligonucleotides, the pseudouridine containing analogs were not separated from the 11-mer containing only uridines (Supplemental Figure S4). During LC-MS, the doubly charged ions (m/z 1735.6) were consistently of higher abundance than the triply charged anions (m/z 1156.8).
While both pseudouridine analogs yielded linear calibration curves (Figure 2 d, e), quantification was only possible up to 60 pmol. At 70 pmol, the response began to saturate yielding a non-linear response (data not shown). Not unexpectedly, the sensitivity for UAACψAUGACG was reduced compared with the shorter oligonucleotides discussed above. However, the sensitivity essentially doubled between UAACψAUGACG and UAACψAψGACG, consistent with this SRM assay yielding a linear response to the amount of pseudouridine within a particular oligonucleotide. While the SRM assay response does appear to reflect the amount of pseudouridine within a particular oligonucleotide, it is noted that UAACψAψGACG was less consistent, particularly at higher concentrations, than UAACψAUGACG or any of the smaller oligonucleotides. Moreover, because this method relies on peak area measurements from the mass spectral data, the precision will be less than methods where internal standards are used.
Having confirmed that internal pseudouridines can be quantified via an SRM assay, we next examined whether the m/z 225 to 165 SRM transition, which is unique to terminal pseudouridines, could also be used in a quantitative fashion. The first oligonucleotide pair investigated was ψGG and UGG. As developed by McCloskey and co-workers, terminal pseudouridines yield the unique m/z 225 to 165 SRM transition, while all pseudouridine-containing oligonucleotides generate the m/z 207 to 164 SRM transition. As shown in Supplemental Figure S5, ψGG can be identified by either SRM transition. Comparing these transitions for use in quantitation of pseudouridine in ψGG, no significant differences between them were found (Figure 3a). Neither SRM transition yielded a sensitive response; in particular, quantification was not possible below 20 pmol of the pseudouridine containing oligonucleotide. Moreover, comparing the results from GψG with ψGG (where the two oligonucleotides are isomers), the terminal pseudouridine results in a much less reproducible fragmentation, reducing the quality of the calibration curve (GψG R2=0.998 versus ψGG R2=0.947). These results suggest quantification of small oligonucleotides with terminal pseudouridines may be less accurate and less sensitive than comparable oligonucleotides with an internal pseudouridine.
Figure 3.

(a) Calibration curves for the selected reaction monitoring (SRM) transition m/z 207 to 164 (filled triangle) and the SRM transition m/z 225 to 165 (filled square) for ψGG. Both SRM transitions yielded similar results as evaluated by their sensitivity and reproducibility (linearity). (b) Calibration curve for the selected reaction monitoring (SRM) transition m/z 225 to 165 for ψAACUAUAACG. While the sensitivity of the SRM assay is similar to that found for the smaller ψGG oligonucleotide, the 11-mer did yield improved linearity. Calibration curves were generated by LC-MS/MS analysis of defined amounts of each pseudouridine-containing oligonucleotide in combination with its uridine-containing analog, where the total oligonucleotide amount was 70 pmol. SRM transition peak areas, determined in triplicate, were plotted against the amount of pseudouridine-containing oligonucleotide and the resulting data fit to a linear least squares function
As before, a longer oligonucleotide was also analyzed. The same 11-mer as above, UAACUAUAACG, was used with ψAACUAUAACG to enable comparisons with the internal pseudouridine SRM behavior. A mixture of the two 11-mers was not resolved on the C-18 column (Supplemental Figure S6), consistent with the chromatographic behavior of the internal pseudouridine analogs. However, while the sensitivity of the m/z 225 to 165 SRM transition for ψAACUAUAACG was less than the sensitivity of the m/z 207 to 164 SRM transition for UAACψAUGACG, monitoring the terminal pseudouridine SRM yielded a linear response (R2=0.991) with reasonable precision (Figure 3b). Thus, it appears that quantification of terminal pseudouridines by the specific SRM transition would be more appropriate for longer, rather than shorter, oligonucleotides.
Application of the SRM Assay for Quantification of Pseudouridine
Having established that the pseudouridine specific SRM transitions m/z 207 to 164 and m/z 225 to 165 could be used to quantify pseudouridine in oligonucleotides, we next sought to apply this method for the quantification of pseudouridine in various biological samples. The samples investigated were Escherichia coli transfer RNAs.
Pseudouridine is one of the highly conserved and most common modifications in the 7-base T-loop of tRNAs. The T-loop has four strongly conserved bases, which include ψ55, and two semi-conserved bases [22, 32]. Pseudouridine can be found less frequently at many other sites in tRNA in a domain specific manner [33]. Nucleosides Gm18 in the conserved D loop and ψ55 interact in the tRNA tertiary structure by hydrogen bonding, thereby stabilizing the L shape of tRNA [34]. m5UψCG (where m5U=rT) is part of the conserved pentanucleotide Gm5UψCG present in the T-loop of all E. coli tRNAs [22] except those tRNAs encoding isoleucine, leucine, and initiator tRNAs. Digestion of E. coli tRNAs containing the conserved Gm5UψCG T-loop sequence with RNase T1 results in the tetranucleotide m5UψCGp. Initial studies on the nucleotide composition of this tetranucleotide indicated that both pseudouridine and uridine appeared to be present in the electrophoretically purified RNase T1 sample. These data suggested that uridine is not stoichiometrically modified to pseudouridine at this sequence location [22]. Thus, we decided to examine the extent of pseudouridine modification in E. coli tRNAs that yield the tetranucleotide m5UψCGp using the SRM quantitative assay.
The SRM calibration curve for m5UψCG generated previously (Figure 2b) was used for quantification of pseudouridine in the T-loop of E. coli tRNAs. A commercially available E. coli was analyzed. The sequence of , including expected posttranscriptional modifications, is shown in Figure 4a. Digestion with RNase T1 and alkaline phosphatase will yield m5UψCG, which can then be quantitatively analyzed. Quantitative analysis was done by two separate SRM assay measurements, with the amount of starting material doubled between assays.
Figure 4.

RNA sequence, including posttranscriptional modifications, for (a) E. coli and (b) E. coli
Results from LC-MS/MS analysis of E. coli are presented in Figure 5. Figure 5a is the total ion chromatogram (TIC) from LC-MS analysis of the RNase T1 digest. Figure 5b is the SRM assay for the m/z 207 to 164 transition. Three peaks were detected from the RNase T1 digest. The first peak arises from m/z 606.2 (Figure 5c), which is the doubly charged anion of m5UψCG. The second peak corresponds to an RNase T1 oligonucleotide from E. coli tRNA His (m2AψψCCAG) that has previously been found as a co-purified contaminant tRNA in commercial E. coli samples [17]. The final peak arises from the RNase T1 digestion product ACUQUAms2i6AAψCUG (Q=queuosine), which is found in the anticodon stem and loop region of E. coli . Quantitative analysis of two separate samples (1× and 2×) yielded consistent results for the amount of pseudouridine in the T-loop of the commercial E. coli sample (Table 1). Based on these analyses, pseudouridine is present 67% of the time in the T-loop, consistent with the prior report that uridine is not stoichiometrically modified to pseudouridine at this sequence location [22].
Figure 5.
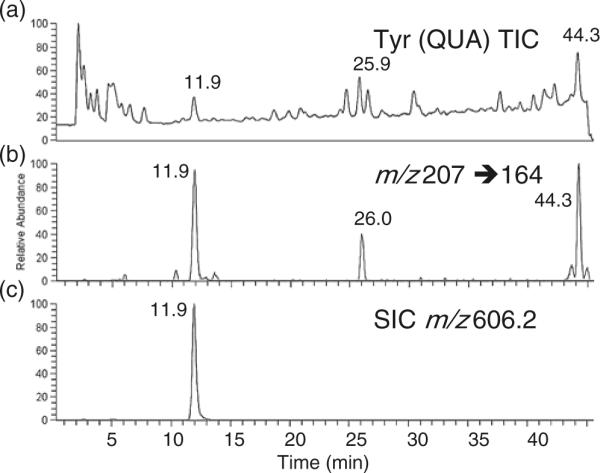
Quantitative analysis of pseudouridine in the T-loop tetramer m5UψCG of E. coli . The tRNA was digested using RNase T1 and BAP and analyzed by LC-MS/MS. Quantification of pseudouridine was performed using the SRM transition m/z 207 to 164 and the m5UψCG calibration curve in Figure 2b. Results are listed in Table 1. (a) Total ion chromatogram (TIC) of LC-MS analysis of RNase T1 digest of . (b) Pseudouridine-specific SRM transition. Three RNase T1/BAP digestion products contain pseudouridine (see text). (c) Selected ion chromatogram (SIC) of m/z 606.2, which arises from the doubly charged anion of m5UψCG
Table 1.
Quantitative Analysis of Pseudouridine in the T-loop tetramer m5UψCG of Two E. coli tRNAs. The tRNAs were Digested using RNase T1 and BAP and Analyzed by LC-MS/MS. Quantification of Pseudouridine was Performed using the SRM Transition m/z 207 to 164 and the m5UψCG Calibration Curve in Figure 2b. Mass Spectral Data are Found in Figures 5 and 6. Both tRNAs were Hypomodified, with the Overexpressed Significantly Less Modified at ψ55 than the Normally Expressed
| Sample | Assay amount (pmol) | Experimentally observed (pmol) | Modifications/pmol |
|---|---|---|---|
| m5UψCG/m5UUCG | 50/50 pmol | 50±2.78 | 1.00 |
| Tyr (QUA) | 29 pmol | 20±1.56 | 0.68 |
| 14.2 pmol | 9±1.3 | 0.66 | |
| Arg (CCG) | 32 pmol | 9 | 0.28 |
| 16 pmol | 4 | 0.25 |
Hypomodified E. coli tRNAs can be generated when a tRNA gene is overexpressed by transforming cells with a plasmid containing one or more copies of the tRNA gene [23, 35]. Previous studies on overexpression of E. coli tRNAs Leu [35] and Glu [23] both reported reduced levels of pseudouridinylation, although precise quantitative measurements and sequence-specific assignments of hypomodification were not discussed. Thus, quantitative analysis of pseudouridine levels in the T-loop of an overexpressed tRNA should reveal an even more significant level of hypomodification than that found for the commercial E. coli tRNA. To test this possibility, E. coli was purified from a Rosetta2 strain, which contains the Arg (CCG) gene on a multicopy plasmid. The sequence of , including expected post-transcriptional modifications, is shown in Figure 4b. Results from LC-MS/MS analysis of E. coli are shown in Figure 6. Only a single peak for the m/z 207 to 164 SRM assay transition was detected in the RNase T1 digest of this tRNA (Figure 6b), consistent with the presence of only a single pseudouridine in the T-loop [31], which was confirmed by detection of the doubly charged anion for the tetranucleotide at m/z 606.2 (Figure 6c). Quantitative analysis of two separate samples (1× and 2×) yielded consistent results for the amount of pseudouridine in the T-loop of the overexpressed E. coli sample (Table 1). In this case, only 25% of tRNA contained a pseudouridine posttranscriptional modification in place of uridine. These data support prior reports describing undermodification of tRNAs in overexpression systems, and, importantly, this SRM quantitative assay now allows for the sequence-specific characterization of pseudouridine levels in RNAs.
Figure 6.
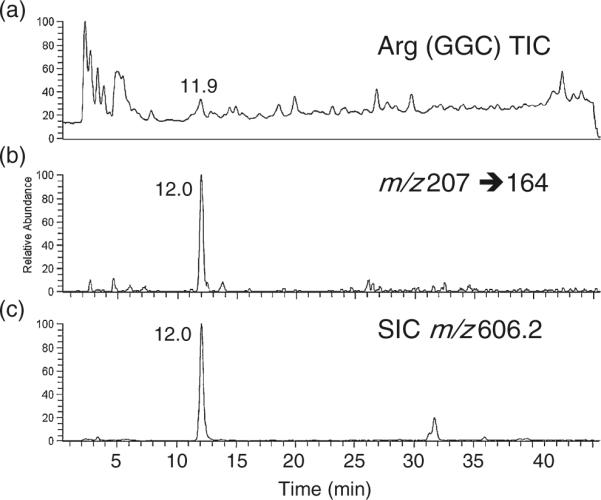
Quantitative analysis of pseudouridine in the T-loop tetramer m5UψCG of E. coli . The tRNA was digested using RNase T1 and BAP and analyzed by LC-MS/MS. Quantification of pseudouridine was performed using the SRM transition m/z 207 to 164 and the m5UψCG calibration curve in Figure 2b. Results are listed in Table 1. (a) Total ion chromatogram (TIC) of LC-MS analysis of RNase T1 digest of . (b) Pseudouridine-specific SRM transition. Only one RNase T1/BAP digestion product contains pseudouridine (see text). (c) Selected ion chromatogram (SIC) of m/z 606.2, which arises from the doubly charged anion of m5UψCG
Conclusions
A quantitative mass spectrometry-based assay to characterize pseudouridine, the most prevalent post-transcriptional modification in RNA, has been developed. The amount of pseudouridine in a given isomeric oligonucleo-tide mixture can be determined even in a background of its uridine-containing analog via a pseudouridine-specific SRM assay. Both pseudouridine-specific SRM transitions (m/z 207 to 164 and m/z 225 to 165) were developed into quantitative assays. The 5'-terminus specific transition (m/z 225 to 165) is less sensitive than the m/z 207 to 164 transition, although the sensitivity for the 5'-terminus pseudouridine does not appear to be affected by the length of the oligonucleotide. The sensitivity of the m/z 207 to 164 transition was, in general, found to be inversely dependent on the length of the oligonucleotide. The linear range for quantification was found to be 5 pmol to 60 pmol, although this range was more limited for oligonucleotides that contained multiple pseudouridines and for the 5'-terminus transition. Although these data demonstrate the site-specific quantification of pseudouridine by mass spectrometry, the method is dependent on the ability to generate a calibration curve for each pseudouridine-containing oligonucleotide of interest. While particular oligonucleotide sequences, especially RNase digestion products containing multiple post-transcriptional modifications, may not be available commercially, for those sequences where calibration standards can be obtained, this assay is easily amenable to automation for the quantitative analysis of pseudouridine in a specific sequence context from a large number of samples.
Using this assay, the levels of pseudouridine at ψ55 in the conserved m5UψCG tetramer present in the T-loop of E. coli tRNAs were determined for two isoaccepting tRNAs. Not surprisingly, ψ55 levels were reduced from an overexpressed tRNA compared with that from a normally expressed tRNA. However, while earlier results suggested hypomodification of ψ55 in E. coli tRNAs, this is the first time a quantitative measure of ψ55 has been reported. This assay will enable additional studies into pseudouridine amounts at a sequence-specific level, which can be useful to further elucidate the functional role(s) of this post-transcriptional modification. Further, these results demonstrate that quantitative SRM assays for other RNA modifications are feasible, and additional efforts are on-going to extend quantitative SRM assays into methylation levels of RNAs as well as to use this existing assay to examine pseudouridine modification from larger RNAs, such as rRNAs.
Supplementary Material
Acknowledgments
The authors acknowledge financial support of this work by the National Institutes of Health (GM 58843) and the University of Cincinnati. The authors thank Thomas Riffelmacher for assistance with tRNA purification.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s13361-011-0137-5) contains supplementary material, which is available to authorized users.
References
- 1.Limbach PA, Crain PF, McCloskey JA. Summary: The modified nucleosides of RNA. Nucleic Acids Res. 1994;22:2183–2196. doi: 10.1093/nar/22.12.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rozenski J, Crain PF, McCloskey JA. The RNA Modification Database. Nucleic Acids Res. 1999;1999(27):196–197. doi: 10.1093/nar/27.1.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Charette M, Gray MW. Pseudouridine in RNA: What, where, how, and why. IUBMB Life. 2000;49:341–351. doi: 10.1080/152165400410182. [DOI] [PubMed] [Google Scholar]
- 4.Arnez JG, Steitz TA. Crystal structure of unmodified tRNA(Gln) complexed with glutaminyl-tRNA synthetase and ATP suggests a possible role for pseudo-uridines in stabilization of RNA structure. Biochemistry. 1994;33:7560–7567. doi: 10.1021/bi00190a008. [DOI] [PubMed] [Google Scholar]
- 5.Davis DR. Stabilization of RNA stacking by pseudouridine. Nucleic Acids Res. 1995;23:5020–5026. doi: 10.1093/nar/23.24.5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis DR, Veltri CA, Nielsen L. An RNA model for investigation of pseudouridine stabilization of the codon–anticodon interaction in tRNA(Lys), tRNA(His), and tRNA(Tyr) J Biomol Struct Dyn. 1998;15:1121–1132. doi: 10.1080/07391102.1998.10509006. [DOI] [PubMed] [Google Scholar]
- 7.Cunningham PR, Richard RB, Weitzmann CJ, Nurse K, Ofengand J. The absence of modified nucleotides affects both in vitro assembly and in vivo function of the 30 S ribosomal subunit of Escherichia coli. Biochimie. 1991;73:789–796. doi: 10.1016/0300-9084(91)90058-9. [DOI] [PubMed] [Google Scholar]
- 8.Gu X, Liu Y, Santi DV. The mechanism of pseudouridine synthase I as deduced from its interaction with 5-fluorouracil-tRNA. Proc. Natl. Acad. Sci. U.S.A. 1999;96:14270–14275. doi: 10.1073/pnas.96.25.14270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spedaliere CJ, Ginter JM, Johnston MV, Mueller EG. The pseudouridine synthases: Revisiting a mechanism that seemed settled. J. Am. Chem. Soc. 2004;126:12758–12759. doi: 10.1021/ja046375s. [DOI] [PubMed] [Google Scholar]
- 10.Raychaudhuri S, Conrad J, Ofengand J. A pseudouridine synthase required for the formation of two universally conserved pseudouridines in ribosomal RNA is essential for normal growth of Escherichia coli. RNA. 1998;4:1407–1417. doi: 10.1017/s1355838298981146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lecointe F, Simos G, Sauer A, Hurt EC, Motorin Y, Grosjean H. Characterization of yeast protein Deg1 as pseudouridine synthase (Pus3) catalyzing the formation of C38 and C39 in tRNA anticodon loop. J. Biol. Chem. 1999;273:1316–1323. doi: 10.1074/jbc.273.3.1316. [DOI] [PubMed] [Google Scholar]
- 12.Ofengand J, Del Campo M, Kaya Y. Mapping pseudouridines in RNA molecules. Methods. 2001;25:365–373. doi: 10.1006/meth.2001.1249. [DOI] [PubMed] [Google Scholar]
- 13.Durairaj A, Limbach PA. Mass spectrometry of the fifth nucleoside: A review of the identification of pseudouridine in nucleic acids. Anal. Chim. Acta. 2008;623:117–125. doi: 10.1016/j.aca.2008.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pomerantz SC, McCloskey JA. Analysis of RNA hydrolysates by liquid chromatography-mass spectrometry. Methods Enzymol. 1990;193:796–824. doi: 10.1016/0076-6879(90)93452-q. [DOI] [PubMed] [Google Scholar]
- 15.Emmerechts G, Herdewijn P, Rozenski J. Pseudouridine detection improvement by derivatization with methyl vinyl sulfone and capillary HPLC-mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2005;825:233–238. doi: 10.1016/j.jchromb.2005.06.041. [DOI] [PubMed] [Google Scholar]
- 16.Patteson KG, Rodicio LP, Limbach PA. Identification of the mass-silent post-transcriptionally modified nucleoside pseudouridine in RNA by matrix-assisted laser desorption/ionization mass spectrometry. Nucleic Acids Res. 2001;29:E49. doi: 10.1093/nar/29.10.e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Durairaj A, Limbach PA. Improving CMC-derivatization of pseudouridine in RNA for mass spectrometric detection. Anal. Chim. Acta. 2008;612:173–181. doi: 10.1016/j.aca.2008.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mengel-Jorgensen J, Kirpekar F. Detection of pseudouridine and other modifications in tRNA by cyanoethylation and MALDI mass spectrometry. Nucleic Acids Res. 2002;30:e135. doi: 10.1093/nar/gnf135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pomerantz SC, McCloskey JA. Detection of the common RNA nucleoside pseudouridine in mixtures of oligonucleotides by mass spectrometry. Anal. Chem. 2005;77:4687–4697. doi: 10.1021/ac058023p. [DOI] [PubMed] [Google Scholar]
- 20.Del Campo M, Recinos C, Yanez G, Pomerantz SC, Guymon R, Crain PF, McCloskey JA, Ofengand J. Number, position, and significance of the pseudouridines in the large subunit ribosomal RNA of Haloarcula marismortui and Deinococcus radiodurans. RNA. 2005;11:210–219. doi: 10.1261/rna.7209905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guymon R, Pomerantz SC, Crain PF, McCloskey JA. Influence of phylogeny on posttranscriptional modification of rRNA in thermophilic prokaryotes: The complete modification map of 16 S rRNA of Thermus thermophilus. Biochemistry. 2006;45:4888–4899. doi: 10.1021/bi052579p. [DOI] [PubMed] [Google Scholar]
- 22.Zamir A, Holley RW, Marquisee M. Evidence for the occurrence of a common pentanucleotide sequence in the structures of transfer ribonucleic acids. J. Biol. Chem. 1965;240:1267–1273. [PubMed] [Google Scholar]
- 23.Madore E, Florentz C, Giegé R, Sekine S, Yokoyama S, Lapointe J. Effect of modified nucleotides on Escherichia coli tRNAGlu structure and on its aminoacylation by glutamyl-tRNA synthetase. Predominant and distinct roles of the mnm5 and s2 modifications of U34. Eur J Biochem. 1999;266:1128–1135. doi: 10.1046/j.1432-1327.1999.00965.x. [DOI] [PubMed] [Google Scholar]
- 24.Zhao X, Yu YT. Detection and quantitation of RNA base modifications. RNA. 2004;10:996–1002. doi: 10.1261/rna.7110804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dai Q, Fong R, Saikia M, Stephenson D, Yu YT, Pan T, Piccirilli JA. Identification of recognition residues for ligation-based detection and quantitation of pseudouridine and N6-methyladenosine. Nucleic Acids Res. 2007;35:6322–6329. doi: 10.1093/nar/gkm657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan CTY, Dyavaiah M, DeMott MS, Taghizadeh K, Dedon PC, Begley TJ. A Quantitative Systems Approach Reveals Dynamic Control of tRNA Modifications during Cellular Stress. PLoS Genet. 2010;6:e1001247. doi: 10.1371/journal.pgen.1001247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brückl T, Globisch D, Wagner M, Müller M, Carell T. Parallel isotope-based quantification of modified tRNA nucleosides. Angew. Chem. Int. Ed. 2009;48:7932–7934. doi: 10.1002/anie.200902740. [DOI] [PubMed] [Google Scholar]
- 28.Taucher M, Ganisl B, Breuker K. Identification, localization, and relative quantitation of pseudouridine in RNA by tandem mass spectrometry of hydrolysis products. Int. J. Mass Spectrom. 2011 doi: 10.1016/j.ijms.2010.05.024. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee C, Kramer G, Graham DE, Appling DR. Yeast mitochondrial initiator tRNA is methylated at guanosine 37 by the Trm5-encoded tRNA (guanine-N1-)-methyltransferase. J. Biol. Chem. 2007;282:27744–27753. doi: 10.1074/jbc.M704572200. [DOI] [PubMed] [Google Scholar]
- 30.Castleberry CM, Limbach PA. Relative quantitation of transfer RNAs using liquid chromatography mass spectrometry and signature digestion products. Nucleic Acids Res. 2010;38:e162. doi: 10.1093/nar/gkq578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Juhling F, Morl M, Hartmann RK, Sprinzl M, Stadler PF, Putz J. tRNAdb 2009: Compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009;2009(37):D159–D162. doi: 10.1093/nar/gkn772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sprinzl M, Hartmann T, Meissner F, Moll J, Vorderwulbeckbecke T. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 1987;15:r53–r188. doi: 10.1093/nar/15.suppl.r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis DR. Biophysical and conformational properties of modified nucleosides in RNA (nuclear magnetic resonance studies) In: Grosjean H, Benne R, editors. Modification and editing of RNA. ASM Press; Washington, DC: 1998. pp. 103–112. [Google Scholar]
- 34.Quigley GJ, Wang AHJ, Seeman NC. Hydrogen bonding in yeast phenylalanine transfer RNA. Proc. Natl. Acad. Sci. U.S.A. 1975;72:4866–4870. doi: 10.1073/pnas.72.12.4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wahab S, Rowley K, Holmes W. Effects of tRNALeu I overproduction in Escherichia coli. Mol Microbiol. 1993;7:253–263. doi: 10.1111/j.1365-2958.1993.tb01116.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.