

Abstract
C9OF72-hexanucleotide repeat expansions and ubiquilin-2 (UBQLN2) mutations are recently identified genetic markers in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD). We investigate the relationship between C9ORF72 expansions and the clinical phenotype and neuropathology of ALS and FTLD. Genetic analysis and immunohistochemistry (1HC) were performed on autopsy-confirmed ALS (N = 75), FTLD-TDP (N = 30), AD (N = 14), and controls (N = 11). IHC for neurodegenerative disease pathology consisted of C9ORF72, UBQLN, p62, and TDP-43. A C9ORF72 expansion was identified in 19.4 % of ALS and 31 % of FTLD-TDP cases. ALS cases with C9ORF72 expansions frequently showed a bulbar onset of disease (57 %) and more rapid disease progression to death compared to non-expansion cases. Staining with C9ORF72 antibodies did not yield specific pathology. UBQLN pathology showed a highly distinct pattern in ALS and FTLD-TDP cases with the C9ORF72 expansion, with UBQLN-positive cytoplasmic inclusions in the cerebellar granular layer and extensive UBQLN-positive aggregates and dystrophic neurites in the hippocampal molecular layer and CA regions. These UBQLN pathologies were sufficiently unique to allow correct prediction of cases that were later confirmed to have C9ORF72 expansions by genetic analysis. UBQLN pathology partially co-localized with p62, and to a minor extent with TDP-43 positive dystrophic neurites and spinal cord skein-like inclusions. Our data indicate a pathophysiological link between C9ORF72 expansions and UBQLN proteins in ALS and FTLD-TDP that is associated with a highly characteristic pattern of UBQLN pathology. Our study indicates that this pathology is associated with alterations in clinical phenotype, and suggests that the presence of C9ORF72 repeat expansions may indicate a worse prognosis in ALS.
Keywords: Amyotrophic lateral sclerosis, Frontotemporal lobar degeneration, C9ORF72, UBQLN2, UBQLN1
Introduction
Amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD) are fatal neurodegenerative diseases without effective treatments. ALS is the most frequent adult onset motor neuron disease characterized by rapidly progressive paresis leading to death with a mean survival of ~ 3 years following onset of symptoms [21]. FTLD is the second most frequent cause of dementia and in people <65 years old, its prevalence is similar to Alzheimer's disease (AD) [44]. TDP-43 is the major component in ubiquitin-positive neuronal inclusions in sporadic ALS (sALS) and the largest subset of FTLD now known as FTLD-TDP [14, 39, 40]. Indeed, ALS and FTLD-TDP are thought to represent a continuum of different manifestations of the same underlying disease, i.e. TDP-43 proteinopathy. ALS is mostly sporadic but ~ 10 % of cases have a first- or second-degree relative with the disease suggestive of familial inheritance of ALS (fALS). Over ten genes have been found to harbor mutations that cause fALS, with mutations in SOD1, encoding the Cu/Zn superoxide dismutase, TARDBP, fused in sarcoma (FUS) and the optineurin (OPTN) gene accounting for ~ 30 % of these cases [9, 19, 28, 43, 47, 51, 57]. Mutations in TARDBP and FUS are occasionally observed in FTLD cases as well [27, 56], Moreover, mutations in the ubiq-uilin-2 (UBQLN2) gene encoding the ubiquitin-like protein UBQLN2 were shown to cause very rare cases of domi-nantly inherited, chromosome-X-linked ALS and ALS with dementia [8]. UBQLN is a member of the ubiquitin-like protein family (ubiquilins) that participates in degradation of proteins through the proteasome and autophagy pathways [24, 31, 37, 46]. Humans contain four ubiquilin genes, each encoding a separate protein. The expression pattern of these genes in humans is not known, although studies suggest that UBQLN1 has a more widespread expression than UBQLN2, and that UBQLN3 is only expressed in the testis [6, 34, 63].
Importantly, two recent studies identified a non-coding GGGGCC hexanucleotide repeat expansion in the C9ORF72 gene as the most common genetic abnormality in familial and sporadic ALS/FTLD [7, 42]. The repeat expansion was found to be associated with a selective reduced expression of one of the C9ORF72 transcripts [16]. It was suggested that aberrant promoter function, aberrant splicing of C9ORF72 primary transcripts or sequestration of RNA-binding proteins could be possible pathogenic consequences of C9ORF72 mutations [7, 16].
Despite these important findings on identifying disease genes associated with ALS/FTLD, few studies have systematically analyzed the pathology of ALS/FTLD with C9ORF72 repeat expansions [1, 3, 16, 36, 49, 50, 52, 54] or with UBQLN2 mutations [8]. Previous studies have shown that ALS/FTLD cases with C9ORF72 expansion cases do not contain protein aggregates that comprise C9ORF72 protein [7, 42], although TDP-43 inclusions are present and p62 was hypothesized to be the major disease pathology since p62-immunoreactive neuronal cytoplasmic inclusions in the cerebral cortex, basal ganglia, hippocampus, and cerebellum were detected in cases with C9ORF72 repeat expansions [1, 54]. Here, we describe neuropathological findings in a large and clinically well-defined cohort of ALS and FTLD-TDP autopsy cases and controls, and evaluate the relevance of C9ORF72 and UBQLN2 gene mutations to the neuropathological and clinical phenotypes.
Methods
Autopsy cohort
Individuals who underwent autopsy in the Center for Neurodegenerative Disease Research at the University of Pennsylvania from 2001 to 2010 were included. Our cohort was composed of 75 patients with a clinical diagnosis of ALS in accordance with the modified El Escorial Criteria [4] and a confirmed neuropathological diagnosis of ALS (Table 1). Detailed clinical characteristics [age at onset, age at death, site of onset, disease duration, ALS global disease severity as measured by a functional rating score (ALSFRS-R) [5], the Medical Research Council sumscore (MRCS) [25], gender, performance on cognitive tests] were ascertained by retrospective chart review of clinical visits within the University of Pennsylvania Health System; the vast majority of patients were seen by two neurologists (L.E., L.M.). Unless otherwise specified, results of clinical testing used in this study were from the visit most proximate to death, occurring within 3 months of death. Of the ALS cases included here, ten had a clinical history of dementia (ALS-D) and met criteria for FTLD [17, 38, 41]. Thirteen of the ALS cases had a family history of ALS (fALS). We furthermore included 30 cases with a neuropathological diagnosis of FTLD-TDP [33], 14 cases with a neuropathological diagnosis of AD [18], and 11 normal controls (CTRL) age-matched to the ALS group.
Table 1.
Demographic and clinical data of ALS, FTLD-TDP and control autopsy cases
| ALS all | ALS C9+ | ALS C9− | FTLD-TDP all | FTLD-TDP C9+ | FTLD-TDP C9− | AD | CTRL | |
|---|---|---|---|---|---|---|---|---|
| N (female/male) | 75 (30/45) | 14 (5/9) | 61 (25/36) | 30 (10/8) | 9 | 21 | 14 (9/5) | 11 (5/6) |
| Median (interquartile range) | ||||||||
| Age (years) | 61 (55–69) | 58 (55–62) | 65 (56–74) | 70 (63–76) | 65 (54–75) | 70 (63–77) | 83 (78–85) | 68 (60–78) |
| Duration (years) | 2 (1.2–4) | 2 (1–2) | 2 (1.3–4) | 6 (4.3–10) | 7 (5–9) | 7 (4–11) | 12 (9–13) | |
| ALSFRS-R | 19 (15–25) | 23 (17–32) | 11 (6–17) | |||||
| MRCS | 38 (34–45) | 40 (34–45) | 41 (35–48) | |||||
| F-words | 10.5 (8–13) | 11 (8–15) | 10 (7–13) | 3 (2–10) | ||||
| Oral trail | 51 (50–52) | 51 (48–52) | 51 (50–52) | 25 (23–27) | ||||
| FBI | 45 (32–53) | 42 (36–50) | 36 (30–55) | 34 (12–56) | ||||
| MMSE | 30 (28–30) | 30 (29–30) | 30 (27–30) | 20 (11–27) | 8 (2–15) | |||
| Brain weight (kg) | 1.35 (1.12–1.43) | 1.33 (1.24–1.4) | 1.32 (1.22–1.43) | 1.06 (1.0–1.28) | 1.13 (1.0–1.3) | 1.1 (1.0–1.2) | 1.06 (0.9–1.14) | 1.33 (1.23–1.3) |
Data for the neuropsychological tests was not available for FTLD-TDP C9+
Age age at death, ALSFRS-R ALS functional rating scale, C9+ with C9ORF72 hexanucleotide repeat expansion, C9− without C9ORF72 hexanucleotide repeat expansion, duration duration of disease, F-words F-words test, FBI frontal behavioral inventory, Oral trail Oral trail-making test, MMSE Mini-Mental State Examination. MRCS Medical Research Council Sumscore
Basic neuropathological characterization
Pathology was examined in the following regions of the central nervous system (CNS): amygdala, hippocampus (dentate gyrus, molecular layer, and CA regions/subiculum), middle frontal gyrus, superior or middle temporal gyms (SMT), motor cortex, cerebellum, cervical spinal cord (CSC), and lumbar spinal cord (LSC, tissue for this region was available for ALS cases only).
Sections were fixed and cut into 6–7 μm sections, stained with hematoxylin and eosin (HE) and Thioflaviu S, and immunohistochemistry (IHC) was performed with antibodies to tau, Aβ, α-synuclein, ubiquitin, and TDP-43 as previously described [13, 14, 39, 40], The extent of TDP-43, tau, Aβ pathology, and neuronal loss (as monitored by HE) was rated for each region on a four-point ordinal scale (0, none; 1, mild; 2, moderate; 3, severe/numerous) as previously described [15, 55].
Characterization of UBQLN and C9ORF72 pathology
IHC for UBQLN was performed using two antibodies to UBQLN2: a commercially available mouse monoclonal antibody (MAb) (clone 5F5, H00029978-M03, Abnova, Walnut, CA, USA) used at 1:20,000 dilution, and a polyclonal rabbit antibody (AP12092PU-N, Acris Antibodies, San Diego, CA, USA), used at 1:100 dilution [8], The two antibodies were generated against C- and N-terminal portions of UBQLN2, respectively, but are likely to crossreact with all four UBQLN proteins because the regions used to generate the antibodies are highly conserved in all UBQLN proteins. In addition to these antibodies we used a polyclonal rabbit anti-UBQLN1-specific antibody raised by our lab to a synthetic peptide corresponding to amino acids 501–510 in human UBQLN1, a sequence that is not present in UBQLN-2, UBQLN-3 or UBQLN-4 proteins, used at 1:10,000 dilution. IHC for C9ORF72 pathology was performed using a commercially available rabbit polyclonal anti-C9ORF72 ab (HPA023873, Sigma-Aldrich, St. Louis, MO, USA), 1:1,000, similar to the procedure as described above [7], For p62 IHC, a commercially available mouse MAb (Abnova, Walnut, CA, USA) was used at 1:500 and with microwave antigen retrieval [1, 23].
Double-labeling immunofluorescence (IF) analyses were performed as previously described [40] using Alexa Fluor 488- and 594-conjugated secondary ab (Molecular Probes, Eugene, OR, USA), treated for autofluorescence with Sudan Black solution [48], and coverslipped with Vecta-shield-DAPI mounting medium (Vector Laboratories). Fluorescence images were obtained using a Leica TCS SPE-II scanning laser confocal microscope.
Neuropsychological testing
Ante-mortem cognitive testing was performed at 3- to 6-month intervals during routine clinic visits. Data for three cognitive tests were available for a subset of the autopsy cohort (n = 55/75 ALS cases, 17/30 FTLD-TDP cases) within 12 months of death. These included a test of letter-guided verbal fluency (F-words test) [29], a test of frontal executive function (Oral Trail-Making Test) [30] assessing information processing speed and the capacity to maintain a complex mental set, and the Mini-Mental State Examination (MMSE) [12]. For the letter fluency test, patients without significant dysarthria were asked to generate as many unique words (proper nouns and numbers excluded) beginning with the letter “F” in 1 min. Patients with dysarthria, but showing preserved limb and hand function, were asked to write the words; 90°s was allotted to these patients. The total number of unique words produced was recorded. For the Oral Trail-Making Test, patients without significant dysarthria were asked to sequentially say letters of the alphabet, beginning with A, alternating with numbers, beginning with 1, in an ascending order (i.e. A–1, B–2) and ending at Z–26. Each error was subtracted from the maximum score of 52. Patients with significant dysarthria, but with relatively preserved hand and limb ability, were asked to perform the same task in writing. We also included the Frontal Behavioral Inventory (FBI), a 24-item caregiver-based behavioral questionnaire designed for the diagnosis and quantification of FTD symptoms [20]. Data for neuropsychological tests were unavailable in the subgroup of FTLD with C9ORF72 expansion, therefore no analysis of FTLD-TDP subgroups with regard to neuropsychological data is provided here.
Genetics methods
Genomic DNA was extracted from peripheral blood or brain tissue following the manufacturer's protocols [Flex-igene (Qiagen, Valencia, CA, USA) or QuickGene DNA whole blood kit L (Autogen, Holliston, MA, USA) for blood, and QIAsymphony DNA Mini Kit (Qiagen) for brain]. Genotyping for a C9ORF72 repeat expansion was performed as described by Renton et al. [42] with some modifications. Briefly, repeat-primed PCR was performed using 100 ng of DNA in a final volume of 28 μl containing (final concentrations): Roche (Indianapolis, IN, USA) FastStart PCR Master Mix (1×), DMSO (7 %, Sigma-Aldrich, St. Louis, MO, USA), betaine (0.93 M, Sigma-Aldrich, St. Louis, MO, USA), deazaGTP (0.18 mM, Roche, Indianapolis, IN, USA), MgCl2 (0.9 mM, Sigma-Aldrich, St. Louis, MO, USA), and 10× primer mix (1×). The 10× primer mix was prepared containing 14 μM 6-FAM-labeled forward primer (6-FAM-5′ AGTCGCTAG AGGCGAAAGC), 7 μM reverse repeat primer (5′TACGC ATCCCAGTTTGAGACGGGGGCCGGGGCCGGGGCC GGGG), and 14 μM anchor tail reverse primer (5′TACG CATCCCAGTTTGAGACG). Touchdown PCR cycling conditions consisted of 4 min at 95 °C followed by cycles of 95 °C for 30 s, annealing between 70 and 56 °C for 1 min, and extension at 72 °C for 3 min, ending with a final extension step of 10 min at 72 °C. The annealing temperature is decreased by 2 °C in each step starting at 70 °C for two cycles, 68 °C for three cycles, 66 °C for four cycles, 64 °C for five cycles, 62 °C for six cycles, 60 °C for seven cycles, 58 °C for eight cycles, and 56 °C for five cycles. PCR product (2 μl) was mixed with 0.5 μl of ROX 500 Size Standard (Life Technologies, Carlsbad, CA, USA) and 7.5 μl Hi-Di formamide (Life Technologies, Carlsbad, CA, USA) and evaluated on an ABI 3130 capillary electrophoresis instrument with POP7 polymer and a 36 cm capillary with a 23 s injection time. Interpretation of a positive expansion case was based on the presence of a stutter pattern while that of a case lacking the expansion produced one or more peaks with an abrupt ending peak. In some cases the absence of an expansion was confirmed using a standard two primer PCR reaction across the repeat region, The presence of two unique peaks was interpreted as negative for a repeat expansion while the identification of only a single peak was not informative. This two primer genotyping was performed using primers from by Dejesus-Hernandez et al. [7] with minor protocol modifications. Briefly, the PCR was performed using 50 ng of DNA in a final volume of 20 μl containing (final concentrations): Amplitaq Gold buffer (1×), DMSO (5 %), betaine (1 M), dNTP mixture with 7-deazaGTP instead of dGTP (0.25 mM each), MgCl2(0.9 mM), forward and reverse primers (1 μM each), and Amplitaq Gold polymerase 0.5 U/reaction (Life Technologies, Carlsbad, CA, USA). PCR cycling conditions consisted of 10 min at 94 °C followed by 36 cycles of 94 μC for 35 s, annealing between 62 μC for 2 min, and extension at 72 °C for 1 min, ending with a final extension step of 10 min at 72 °C. The ABI 3130 electrophoresis conditions for this assay are the same as for the repeat-primed PCR reaction. The coding region of UBQLN2 was sequenced as described by Deng et al. [8] in ALS and FTLD-TDP cases with DNA available. Sequence data were analyzed using Mutation Surveyor software (SoftGenetics, State College, PA, USA).
Statistical analysis
Data analysis was performed using SPSS (Version 17.0 SPSS Inc., Chicago, IL, USA). The “average” (and “range”) of data on patient characteristics was estimated by calculating the median (and 25th–75th percentiles). Differences between two groups were compared using Wilcoxon Mann–Whitney test. To compare raw data of multiple groups, Kruskal–Wallis analysis of variance on ranks was applied, followed in case of significance by Dunn's Method. Trend analysis was conducted using the Mantel–Haenszel Chi-square test. All correlations were studied using Spearman's rank order correlation coefficient. Bonferroni-correction for multiple testing was applied when contrasts were not driven by a specific hypothesis. For all other tests, P values <0.05 were considered significant. All statistical tests were two-sided.
Results
Genetic analysis of C9ORF72 and UBQLN2
To identify FTLD and ALS cases with either a C9ORF72 hexanucleotide repeat expansion or USQLN2 gene mutation in our cohort, we analyzed all sporadic and familial autopsy cases with FTLD-TDP (n = 29) and ALS (n = 72) for which a DNA sample was available (for 1 FTLD-TDP case and 3 ALS cases, no DNA sample was available).
A C9ORF72 hexanucleotide repeat expansion was identified in 19,4 % (14/72) of ALS cases, and in 31.0 % (9/29) of FTLD-TDP cases. For the subset of autopsy cases in which information about family history was known, a C9ORF72 expansion was identified in 42.9 % (6/14) of ALS cases with a family history of ALS and/or FTLD in first- or second- degree relatives. The C9ORF72 expansion rate was 23.1 % (3/13) in ALS cases with a family history of neurodegenerative diseases other than ALS or FTLD suggesting that the phenotype for C9ORF72 expansions may be broad. C9ORF72 expansions were also identified in 11.6 % (5/43) apparent sporadic ALS cases; however, this number may be high due to lack of complete family history. Similarly, a C9ORF72 expansion was identified in 33.3 % (6/18) of FTLD-TDP cases with a family history of any neurodegenerative disease, although the number of total cases with information is too small to draw conclusions or make comparisons. No expansions were identified in a small number of apparent sporadic FTLD-TDP cases (n = 6). The ethnic composition of the cohort was approximately 26 % white Latino, 67 % Caucasian, and 2 % Black, and 5 % other or unknown. Of those with known ethnicity or race (n = 99), C9ORF72 expansions were identified in 8.1 % white Latinos (8/99) and 15.1 % Caucasian non-Latinos (15/99). The cohort included five cases with progranulin (GRN) mutations and two with SOD1 mutations, none of which were found to have a C9ORF72 expansion.
Since UBQLN2 mutations have been associated with ALS, the UBQLN2 gene was sequenced in all ALS and FTLD-TDP cases (n = 101) for which DNA was available, to rule out mutations as a cause of disease. No pathogenic UBQLN2 mutations were identified in the cases sequenced; however, a variant of uncertain significance (c.998G>T, p.Ala330Ser) was identified in a FTLD-TDP case. An affected family member was tested and did not carry the variant, therefore it is not likely to be the cause of FTLD in the family.
Clinical phenotype in ALS and FTLD-TDP with/without C9ORF72 expansion
Having identified several (n = 14) ALS and (n = 9) FTLD-TDP autopsy cases with a C9ORF72 repeat expansion within our cohort, we determined if the presence of this repeat expansion was linked to the clinical phenotype. We observed the presence of the C9ORF72 expansion to correlate with several clinical variables, especially in the subgroup of cases with ALS. Data on disease duration were available for 66 ALS autopsy cases (it was missing for 1 C9ORF72 expansion case and for 8 non-expansion cases). Disease duration to death was significantly shorter in ALS with C9ORF72 expansion as compared to non-expansion cases (P = 0.016; Fig. 1). In contrast, no significant difference regarding disease duration, age of onset or age at death was observed in FTLD-TDP with/without C9ORF72 expansion (P > 0.05).
Fig. 1.
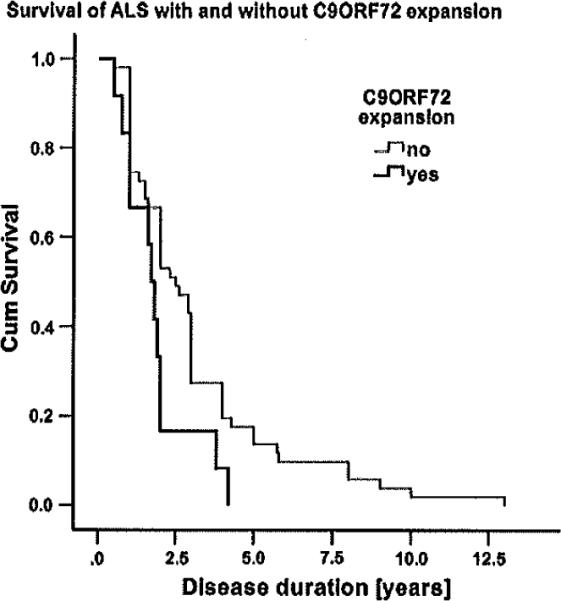
Survival in ALS with and without C9ORF72 expansions. Kaplan-Meier plot shows survival of ALS autopsy cases with C9ORF72 repeat expansion (black curve, n = 14) and without C9ORF72 repeat expansion (grey curve, n = 61). Survival time was significantly shorter in cases with C9ORF72 repeat expansion (P = 0.016)
A bulbar onset of disease was significantly more frequent in the C9ORF72 expansion ALS cases as compared to non-expansion cases (P = 0.04, data on site of onset was missing for 1 non-expansion case). Of 14 ALS cases with C9ORF72 expansion, 8 (57.1 %) showed a bulbar onset of disease, as compared to 21 % in the subgroup of non-expansion cases. Four of the eight ALS C9ORF72 expansion cases with a bulbar onset showed bulbar LMN clinical signs, two presented with a bulbar UMN clinical picture and for two patients the data were incomplete. No significant difference with regard to gender or clinical rating scales (ALSFRS-R, MRCS) was observed between ALS or FTLD-TDP cases with/without C9ORF72 expansion. Furthermore, no difference with regard to neuropsychological tests of executive function (F-words, Oral Trail-making test) or memory function (MMSE) could be detected between ALS with/without C9ORF72 expansion (P > 0.05 each).
Description of C9ORF72 pathology
As the presence of C9ORF72 repeat expansions in ALS was associated with distinct alterations in clinical phenotype, we examined if a neuropathological disease signature exists that could distinguish ALS and FTLD-TDP cases with and without C9ORF72 expansion. We first analyzed C9ORF72 immunoreactivity in IHC, using a commercially available rabbit polyclonal anti-C9ORF72 ab which detects both the long (481 amino acid protein, isoform a) and the shorter isoform (222 amino acid protein, isoform b) of proteins encoded by C9ORF72 [7] (Supplementary figure 1). Similar to Dejesus et al. [7], we observed a coarse punctate staining of synaptic terminals mainly in the CA4 region of the hippocampus in ALS, FTLD-TDP, AD, and CTRL without any difference in extent or regional distribution between the groups (Supplementary figure 2).
UBQLN pathology in subgroups of ALS and FTLD-TDP with/without C9ORF72 repeat expansion
As C9ORF72 staining itself did not reveal any compelling differences between ALS and FTLD-TDP cases with or without C9ORF72 expansion, we examined other types of pathology including UBQLN, TDP-43, and p62 through IHC in the entire cohort of autopsy cases.
Intriguingly, we observed UBQLN pathology in ALS and FTLD-TDP cases with a C9ORF72 expansion to be highly distinct and to allow the differentiation of expansion cases from non-expansion cases with the pathologist blinded to the genetics results and based on the analysis of UBQLN pathology only. In the hippocampus of FTLD-TDP and ALS with C9ORF72 expansion, dystrophic neurites that showed focal swellings and dot-like stipples (Fig. 2) and irregular, aggregate-like formations were extensive in the hippocampal molecular layer and in the CA1–CA4 region. In contrast, non-expansion cases hardly showed any hippocampal molecular layer pathology; instead, UBQLN pathology in non-expansion cases was restricted to the CA regions and the entorhinal cortex. Furthermore, dentate gyrus cytoplasmic neuronat inclusions (Fig. 2) were much more frequent in ALS and FTLD-TDP with C9ORF72 expansion as compared to non-expansion cases (Fig. 3). These inclusions were observed in both ALS-D (n = 10) as well as in ALS without dementia (n = 65), though they were not observed in any AD or CTRL case.
Fig. 2.

UBQLN pathology in ALS and FTLD-TDP. a–c Hippocampal dentate gyrus and molecular layer of an ALS case with C9ORF72 expansion, a Dentate gyrus shows cytoplasmic inclusions that are depicted in b in higher resolution, dystrophic neurites with focal swellings as well as irregular, aggregate-like formations, which are depicted in c in higher resolution, d Cerebeliar molecular layer of an ALS case with a C9ORF72 expansion showing cytoplasmic inclusions, which are shown in e in high resolution, f CSC anterior horn α-motoneuron of an ALS case witli a C9ORF72 expansion showing skein-like to more solid cytopiasmic inclusions. Scale bars a–c, e, f 20 μm; d 200 μm
Fig. 3.

UBQLN pathology in subgroups of ALS and FTLD-TDP with/without C9ORF72 repeat expansion. The figure illustrates the differentiation of ALS and FTLD-TDP with C9ORF72 expansion (ALS/FTLD-C9+) from non-expansion cases (ALS/FTLD-C9−) by UBQLN pathology. a Hippocampal dentate gyrus and molecular layer of a representative ALS case without C9ORF72 repeat expansion. White dentate gyrus neuronal inclusions are detectable, the molecular layer is free of any pathology (d provides higher resolution image), b, c Extensive UBQLN pathology presenting with dystrophic neurites and aggregate-like formations in the molecular layer of two cases with C9ORF72 repeat expansion (column from b downwards shows an ALS case, column from c downwards shows an FTLD-TDP case). c, f Higher resolution images of aggregate-like formations in the hippocampal molecular layer, g, j Cerebellar granular layer of a FTLD-TDP case without C9ORF72 repeat expansion. The cerebellar granular layer does not show any pathology, h, k Cerebellar granular layer of an ALS case and an FTLD-TDP case (i, l) with C9ORF72 repeat expansion shows cytoplasmic neuronal inclusions which were observed in all ALS/FTLD with C9ORF72 repeat expansion. Scale bars a–c, g–i 200 μm; d, j 50 μm; e, f, k, 1 20 μm
In the cerebellum, all ALS and FTLD-TDP C9ORF72 expansion cases showed round to crescent-shaped cytoplasmic neuronal inclusions in the granular layer (Fig. 3). In contrast, such inclusions were not observed in any of the ALS or FTLD-TDP non-expansion cases or in any control case. No further inclusions in the Purkinje cells or other cerebellar layers were observed, neither was there any relevant cerebellar neuronal loss.
In neocortical areas, FTLD-TDP and ALS with C9ORF72 expansions showed dystrophic neurites and (to a lesser extent) aggregate-iike formations throughout the neocortex, while in non-expansion cases pathology was largely limited to superficial neocortical layers (e.g. cortical layers 2 and 3). In addition, neocortical pathology (as shown in Fig, 2) was much more extensive in the C9ORF72 expansion cases. Neocortical dystrophic neurites were also found in AD cases located close to amyloid plaques. Spinal cord anterior horn skein-like to solid cytoplasmic inclusions were observed in both ALS with and without C9ORF72 expansion and did not contribute to the differentiation of expansion versus non-expansion cases. They were not observed in FTLD-TDP or normal controls.
As UBQLN pathology showed a characteristic signature in ALS and FTLD-TDP with C9ORF72 repeat expansion, we quantified UBQLN pathology in expansion and non-expansion cases using a semi-quantitative rating scale. UBQLN pathology was significantly more extensive in ALS and FTLD-TDP cases with C9ORF72 expansion as compared to non-expansion cases in all the regions analyzed here P < 0.05 for each area) with the exception of the spinal cord (Fig. 4). Cases with a GRN or SOD1 mutation did not show differences in UBQLN pathology to cases with ALS or FTLD without C9ORF72 expansion.
Fig. 4.

Extent of UBQLN pathology in ALS and controls. a Bar plot shows mean extent of UBQLN pathology in different regions of the central nervous system in ALS, FTLD-TDP and CTRL, b Bar plot shows mean extent of UBQLN pathology in different regions of the CNS in ALS cases with/without C9ORF72 expansions, c Bar plot shows mean extent of UBQLN pathology in different regions of the CNS in FTLD-TDP case with/without C9ORF72 expansions. Whiskers in bar plot indicate 95 % confidence interval of mean. Amyg amygdala, CSC cervical spinal cord, CA htppocampal CA regions/subiculum, Cer cerebellum, Dent hippocampal dentate gyrus, LSC lumbar spinal cord, MF middle frontal gyrus, Mol hippocampal molecular layer, Mol motor cortex, SMT superior or midtemporal gyrus
We also analyzed a possible difference of other types of pathology in subgroups of ALS and FTLD-TDP with and without C9ORF72 repeat expansion. The extent of ubiquitin pathology was significantly more extensive in ALS and FTLD-TDP cases with a C9ORF72 expansion as compared to non-expansion cases in the middle frontal gyrus and SMT (P < 0.05 each). No significant difference of tau, Aβ, or α-synuclein pathology was observed between ALS and FTLD-TDP with and without a C9ORF72 expansion.
Characterization of UBQLN pathology by UBQLN subtypes, TDP-43 and p62
To further characterize the composition of the distinct pathology in ALS and FTLD-TDP with C9ORF72 repeat expansion, we performed IHC and double-labeling IF for UBQLN subtypes, TDP-43 and p62.
The UBQLN2 antibodies used here were likely to cross-react with other UBQLN proteins because the regions used to generate the antibodies are highly conserved in all four UBQLN subtypes. We observed the UBQLN2 antibodies to detect both UBQLN2 and UBQLN1 (Supplementary figure 3), whereas the polyclonal rabbit anti-UBQLN1-specific antibody did not cross-react with UBQLN2 (antibodies specific for the other UBQLN proteins are not available). Immunostaining with the UBQLN1-specific antibody exhibited considerable overlap with immunostaining using the antibody that does not discriminate between the different UBQLN isotypes, especially in irregular, aggregate-like formations (e.g. in the hippocampus; Fig. 5) and focal swellings of dystrophic neurites. However, the UBQLN1 antibody did not appear to stain the neuronal cytoplasmic inclusions in certain regions of the brain that were easily seen with the Pan-UBQLN antibody (e.g. in the hippocampal dentate gyrus; Fig. 5). It is possible that these inclusions do not contain UBQLN1 or that the UBQLN1 epitope is masked.
Fig. 5.

Relation of UBQLN pathology to UBQLN1, TDP-43 and p62. Double-labeling IF analyzed by confocal microscopy shows a hippocampal dystrophic neurites and aggregate-like formations as detected in ALS/FTLD-TDP with C9ORF72 expansion to show a partial co-localization of immunoreactivity for UBQLN and TDP-43 in the aggregate-like alteration, b Higher resolution image shows UBQLN pathology to be surrounded by TDP-43 immunoreactive material. c Hippocampal dentate cytoplasmic neuronal inclusions show immunoreacfivity with a general UBQLN antibody, but not for UBQLN1, d In contrast, hippocampal molecular layer aggregate-like formations show immunoreactivity for general UBQLN and UBQLN1. e Cerebellar granular layer cytoplasmic neuronal inclusions as detected in ALS/FTLD-TDP case with C9ORF72 expansions partially show immunoreactivity for UBQLN and p62. f Hippocampal molecular layer aggregate-like formations only partially show immunoreactivity for both UBQLN and p62, though UBQLN pathology is more extensive than p62 pathology. Scale bars a, c, f 10 μm; b, e 5 μm; d 20 μm
We next analyzed the relation of UBQLN pathology to TDP-43, for which distinct subtypes regarding the distribution of pathology in the cortices of FTLD-TDP have been described [32]. While three different types of TDP-43 pathology were observed in FTLD cases without the expansion (7 type A, 9 type B, and 5 type C), curiously, FTLD-TDP with C9ORF72 expansion mainly (8/9) showed type B. In ALS and FTLD-TDP cases with C9ORF72 expansion, UBQLN pathology was much more extensive than TDP-43 pathology in the regions examined here. Double-labeling IF analyzed by confocal microscopy showed that hippocampal dentate gyrus inclusions and molecular layer aggregate-like formations immunoreactive for UBQLN did not co-stain with TDP-43 antibodies (Fig. 5). In focal swellings of dystrophic neurites in neocortical areas of ALS and FTLD-TDP, UBQLN and TDP-43 pathology were observed to be located close to one another, though they rarely overlapped. When they did, high-resolution confocal microscopy demonstrated that in several of these lesions, a core of UBQLN-positive structures seemed to be surrounded by TDP-43 immunoreactive material (Fig. 5).
As p62 pathology has previously been reported [1, 52, 54] in ALS and FTLD with C9ORF72 expansion, we analyzed its relation to UBQLN pathology. In both the hippocampus and cerebellum of ALS and FTLD-TDP cases with C9ORF72 expansion, several inclusions exhibited colocalization of UBQLN and p62 pathology in both hippocampal aggregate-like formations as well as in cerebellar granular layer neuronal inclusions (Fig. 5). However, UBQLN pathology was more extensive than p62 pathology throughout the areas analyzed here (only a minor part of the pathology immunoreactive for UBQLN also showed immunoreactivity to p62, as is shown for cerebellar granular layer inclusions and hippocampal molecular layer aggregate-like formations in Fig. 5).
Relation of UBQLN pathology to other pathology and clinical phenotype
As we observed distinct UBQLN pathology in ALS and FTLD-TDP with C9ORF72 expansion, we analyzed its relation to other pathology as well as to clinical parameters in the entire cohort of ALS and FTLD-TDP.
The extent of UBQLN pathology as determined on a semi-quantitative rating scale correlated with neuronal loss in the hippocampus (R = 0.37, P = 0.03) and in the motor cortex (r = 0.59, P = 0.028) of ALS. The extent of ubiquitin pathology increased with the extent of UBQLN pathology in the amygdala (R = 0.6, P = 0.022) and primary motor cortex (R = 0.82, P = 0.014) of ALS. We also analyzed a possible relation of UBQLN pathology to the clinical phenotype in the entire cohort of ALS and FTLD-TDP. In ALS, more extensive SMT UBQLN pathology correlated with a shorter duration of disease (R = −0.41, P = 0.004), though this relation failed to reach significance in the motor cortex (P = 0.05) or in any other region. In contrast, no correlation of UBQLN pathology with age at onset or age at death could be observed (R < 0.4, P > 0.5 each). No direct correlation of UBQLN pathology with tests of executive function (F-words test, Oral Trail-making test) or memory (MMSE) was observed in ALS or FTLD-TDP (R < 0.4, P > 0.05 each). The extent of UBQLN pathology showed no correlation with clinical rating scales (MRCS, ALSFRS-R) in ALS (R < 0.4, P > 0.05 each).
Discussion
This study confirms C9ORF72 repeat expansions to be frequent in ALS and FTLD-TDP. It furthermore shows the presence of C9ORF72 expansions in ALS to be linked to alterations in clinical phenotype, including a shorter duration of disease. Importantly, this study demonstrates that while staining for C9ORF72 does not identify expansion cases, UBQLN pathology provides a characteristic neuropathological disease signature that distinguishes ALS and FTLD-TDP cases with and without C9ORF72 expansion, suggesting a pathophysiological link between C9ORF72 and UBQLN pathology.
As we expected based on previous studies, the frequency of identified C9ORF72 repeat expansions in autopsy confirmed ALS and FTLD-TDP was relatively high, particularly in familial cases. The frequency of 42.9 % in fALS was comparable to that seen by Gijselinck et al. [16] (47 %). For FTLD there is a wider variation of expansion frequency in the literature by cohort (22.5–61.5 % in FTLD-TDP [7] and 16 % in clinical FTLD [16], yet our finding of 31 % in FTLD-TDP in our relatively small cohort is similar.
We observed C9ORF72 repeat expansion not only to be frequent in ALS and FTLD-TDP but also to be associated with alterations in clinical phenotype of ALS. Although previous studies have not linked C9ORF72 expansions to a shorter survival in ALS [7, 16, 42], our study observed that ALS cases with C9ORF72 expansions showed a significantly shorter disease duration as compared to non-expansion cases (Fig. 1), suggesting that the presence of C9ORF72 expansions could be a negative prognostic indicator in ALS. However, the shorter disease duration of C9ORF72 expansion ALS observed here could well have been influenced by the specific composition of our cohort as well as by missing data in nine of the ALS autopsy cases. Generally, a bulbar onset of disease is clinically associated with speech and swallowing difficulties early-on in the disease, and is usually observed in approximately 25 % of all ALS cases [21]. Remarkably, our subgroup of ALS with C9ORF72 expansion showed a bulbar onset in over 50 % of cases, as compared to a significantly lower frequency in the subgroup of non-expansion cases (21 %). This high proportion of bulbar onset cases is in line with a previous study [52] and may contribute to the shorter disease duration of C9ORF72 expansion cases observed here, since a bulbar onset is a well-known risk factor for a shorter survival in ALS [21]. As our finding of a shorter survival in ALS with a C9ORF72 repeat expansion may have been influenced by the high proportion of bulbar onset cases in the expansion group and by some missing data, future studies will be necessary to confirm the relevance of the C9ORF72 repeat expansion as a risk factor for a shorter survival in ALS.
To determine neuropathological correlates of the C9ORF72 repeat expansion, we analyzed C9ORF72 pathology in ALS and FTLD-TDP. Using a commercially available antibody for IHC, we observed no difference in C9ORF72 immunoreactivity between cases with or without C9ORF72 expansion (Supplementary figure 2). A previous study suggested the transcription of C9ORF72 leads to the expression of two different protein isoforms of C9ORF72 [7]. While a significant reduction of transcripts encoding the longer (481 amino acid) isoform was observed, no significant changes in total protein levels could be detected. As a possible explanation, the antibody used here was detecting both alternative isoforms of the protein C9ORF72 (Supplementary figure 1), and may therefore not be sensitive enough to reflect regional differences in protein composition of C9ORF72 affecting only a single isoform.
Having found that C9ORF72 pathology itself could not distinguish between ALS and FTLD-TDP cases with or without C9ORF72 expansion, we examined other pathology including UBQLN. Though none of the ALS and FTLD-TDP cases in our cohort showed an UBQLN mutation, we observed UBQLN pathology to be widespread in ALS and FTLD-TDP (Fig. 3). This confirms findings of a previous study that UBQLN pathology occurs not only in fALS with UBQLN2 mutations but also in sporadic cases [8]. One of the antibodies used here to identify UBQLN pathology was the same one that was used in the previous study and may cross-react with all four UBQLN proteins. Our results suggest that UBQLN1 might accumulate in a subset of the inclusions in which UBQLN proteins are found, or that its epitope might be masked under certain circumstances. It is also possible that different UBQLN proteins accumulate in the different inclusions. It will be important to determine if this is the case using antibodies specific for each of the isoforms, UBQLN proteins contain an N-terminal ubiquitin-like domain, which mediates interaction with the 19S subunit of the proteasome, and a C-terminal ubiquitin-associated domain, which binds poly-ubiquitinated proteins [26]. This structural organization is characteristic of proteins that deliver ubiquitinated proteins to the proteasome for degradation [11]. However, the proteins also function in autophagy, suggesting they play dual roles in disposing misfolded proteins through the proteasome and lysosomal pathways [45]. There is increasing evidence that different members of this family may play an important role in neurodegenerative diseases, including ALS [8], Huntington's disease [59] and AD [2, 22, 34, 35, 53, 58], Our study demonstrates that while some of the pathological alterations (e.g. dystrophic neurites, aggregate-like formations) seen in hippocampal and neo-cortical areas of ALS and FTLD-TDP contain material immunoreactive for UBQLN1, others (e.g. dentate gyrus cytoplasmic neuronal inclusions) are distinguishable by general UBQLN accumulation and/or pathology.
Importantly, and in contrast to staining with C9ORF72 antibodies, we observed ALS and FTLD-TDP cases with C9ORF72 expansions to show a highly distinct pattern of UBQLN pathology that enabled the correct classification of cases with C9ORF72 repeat expansions by a pathologist blinded to the genetic analysis results. This finding suggests a pafhophysiological link between C9ORF72 mutations and UBQLN pathology in ALS and FTLD-TDP. It is so far unclear how the C9ORF72 hexamicleotide repeat expansion leads to pathology and how this may, in turn, affect other proteins like UBQLN. It is interesting to note that UBQLNs have been shown to bind proteins containing polyglutamine and polyalanine expansions [10, 60, 61], which might be related to the pathology seen with C9ORF72 hexanucleotide repeat expansions. In normal individuals at least three alternatively spliced C9ORF72 transcripts (variants 1–3) are expressed in the brain. Quantitative mRNA expression analysis indicated that the GGGGCC repeat expansion abolished C9ORF72 transcript variant 1 expression, leading to an overall reduction in C9ORF72 transcripts [7]. On a speculative level, changes caused by C9ORF72 transcription could (through a yet unknown pathomechanism) be active further downstream in the pathological pathway of ALS and FTLD-TDP and affect the regional expression of proteins involved in handing over polyubiquitinated proteins to the shuttle factors for degradation. In line with this, previous studies analyzing the pathology of C9ORF72 ALS and FTLD expansion cases observed inclusions immunoreactive of p62 (also called sequestosome 1), another protein that is believed to be involved in protein degradation in similar regions that showed extensive UBQLN pathology here [1, 23, 49, 50, 54, 62]. Another possibility is that the expansions cause the encoded C9ORF72 protein to mis-fold, which might attract and dedicate UBQLN to its disposal thus diverting it UBQLN's vital functions. Proteins involved in shuttling proteins disposal like UBQLN and p62 could be reactively upregulated as a consequence of protein alterations further upstream in the pathological pathway activated by C9ORF72 expansions. Further studies will be needed to elucidate the pathophysiological link between C9ORF72 and proteins involved in protein degradation.
On a speculative level, extensive UBQLN pathology in ALS C9ORF72 expansion cases may also contribute to a shorter survival: supporting this hypothesis, we observed more extensive SMT UBQLN pathology to correlate with shorter disease duration in the entire ALS cohort. However, this relation just failed to reach significance in the primary motor cortex, and it could not be shown for other areas analyzed here, but this may reflect the limitations of a semi-quantitative rating system. Furthermore, we observed the extent of UBQLN pathology to correlate with neuronal loss in the primary motor cortex and hippocampus. Larger, multi-center studies linking genetics and pathology to survival will be necessary to clarify if the presence of C9ORF72 expansions and/or UBQLN pathology is of prognostic relevance in ALS.
Taken together, our data suggest that C9ORF72 gene mutations are pathophysiologically linked to UBQLN pathology in ALS and FTLD-TDP and that there are distinct patterns of UBQLN pathology in ALS and FTLD-TDP that predict the presence of C9ORF72 repeat expansions. Our study furthermore indicates that this pattern of pathology is associated with alterations in clinical phenotype (including a frequent bulbar onset of disease), and suggests that the presence of a C9ORF72 repeat expansion may indicate a worse prognosis in ALS.
Acknowledgments
We thank many patients who contributed samples for this study. We acknowledge Robert Greene, Kevin Raible, Terry Schuck and Charles Dunn for technical assistance, and Elisabeth McCarty-Wood and Dana Falcone for their support in patient recruitment and gathering family history information. This work was supported by the NIH (AG033101, AG017586, AG010124, AG032953, AG039510, NS044266), the Wyncote Foundation, and the Koller Family Foundation. VMYL is the John H. Ware, 3rd, Professor of Alzheimer's Disease Research. JQT is the William Maul Measey-Truman G. Schnabel, Jr., Professor of Geriatric Medicine and Gerontology. JB is supported by a grant of the Deutsche Forschungsgemeinschaft DFG (AOBJ586910). JBT is supported by a grant of the Fundación Alfonso Martin Escudero.
Footnotes
J. Brettschneider and V.M. Van Deerlin contributed equally to this work.
Electronic supplementary material The online version of this article (doi:10.1007/s00401-012-0970-z) contains supplementary material, which is available to authorized users.
Conflict of interest The authors declare they have no conflict of interest.
References
- 1.Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, Rogelj B, Al-Chalabi A, Hortobagyi T, Shaw CE. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011;122:691–702. doi: 10.1007/s00401-011-0911-2. [DOI] [PubMed] [Google Scholar]
- 2.Bertram L, Hiltunen M, Parkinson M, Ingelsson M, Lange C, Ramasamy K, Mullin K, Menon R, Sampson AJ, Hsiao MY, Elliott KJ, Velicelebi G, Moscarillo T, Hyman BT, Wagner SL, Becker KD, Blacker D, Tanzi RE. Family-based association between Alzheimer's disease and variants in UBQLN1. N Engl J Med. 2005;352:884–894. doi: 10.1056/NEJMoa042765. [DOI] [PubMed] [Google Scholar]
- 3.Boxer AL, Mackenzie IR, Boeve BF, Baker M, Seeley WW, Crook R, Feldman H, Hsiung GY, Rutherford N, Laluz V, Whitwell J, Foti D, McDade E, Molano J, Karydas A, Wojtas A, Goldman J, Mirsky J, Sengdy P, Dearmond S, Miller BL, Rademakers R. Clinical, neuroimaging and neuropathological features of a new chromosome 9p-linked FTD-ALS family. J Neurol Neurosurg Psychiatry. 2011;82:196–203. doi: 10.1136/jnnp.2009.204081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 5.Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B, Nakanishi A. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III) J Neurol Sci. 1999;169:13–21. doi: 10.1016/s0022-510x(99)00210-5. [DOI] [PubMed] [Google Scholar]
- 6.Conklin D, Holderman S, Whitmore TE, Maurer M, Feldhaus AL. Molecular cloning, chromosome mapping and characterization of UBQLN3 a testis-specific gene that contains an ubiquitin-like domain. Gene. 2000;249:91–98. doi: 10.1016/s0378-1119(00)00122-0. [DOI] [PubMed] [Google Scholar]
- 7.Dejesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Gesehwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, Shi Y, Zhai H, Jiang H, Hirano M, Rampersaud E, Jansen GH, Donkervoort S, Bigio EH, Brooks BR, Ajroud K, Sufit RL, Haines JL, Mugnaini E, Pericak-Vance MA, Siddique T. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477:211–215. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, Getzoff ED, Hu P, Herzfeldt B, Roos RP, et al. Amyotrophic lateral sclerosis and structural defects in Cu, Zn superoxide dismutase. Science. 1993;261:1047–1051. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- 10.Doi H, Mitsui K, Kurosawa M, Machida Y, Kuroiwa Y, Nukina N. Identification of ubiquitin-interacting proteins in purified polyglutamine aggregates. FEBS Lett. 2004;571:171–176. doi: 10.1016/j.febslet.2004.06.077. [DOI] [PubMed] [Google Scholar]
- 11.Elsasser S, Finley D. Delivery of ubiquitinated substrates to protein-unfolding machines. Nat Cell Biol. 2005;7:742–749. doi: 10.1038/ncb0805-742. [DOI] [PubMed] [Google Scholar]
- 12.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 13.Geser F, Lee VM, Trojanowski JQ. Amyotrophic lateral sclerosis and frontotemporal lobar degeneration: a spectrum of TDP-43 proteinopathies. Neuropathology. 2010;30:103–112. doi: 10.1111/j.1440-1789.2009.01091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geser F, Martinez-Lage M, Robinson J, Uryu K, Neumann M, Brandmeir NJ, Xie SX, Kwong LK, Elman L, McCluskey L, Clark CM, Malunda J, Miller BL, Zimmerman EA, Qian J, Van Deerlin V, Grossman M, Lee VM, Trojanowski JQ. Clinical and pathological continuum of multisystem TDP-43 proteinopathies. Arch Neurol. 2009;66:180–189. doi: 10.1001/archneurol.2008.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geser F, Robinson JL, Malunda JA, Xie SX, Clark CM, Kwong LK, Moberg PJ, Moore EM, Van Deerlin VM, Lee VM, Arnold SE, Trojanowski JQ. Pathological 43-kDa transactivation response DNA-binding protein in older adults with and without severe mental illness. Arch Neurol. 2010;67:1238–1250. doi: 10.1001/archneurol.2010.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, Janssens J, Bettens K, Van Cau-wenberghe C, Pereson S, Engelborghs S, Sieben A, De Jonghe P, Vandenberghe R, Santens P, De Bleecker J, Maes G, Baumer V, Dillen L, Joris G, Cuijt I, Corsmit E, Elinck E, Van Dongen J, Vermeulen S, Van den Broeck M, Vaerenberg C, Mattheijssens M, Peeters K, Robberecht W, Cras P, Martin JJ, De Deyn PP, Cruts M, Van Broeckhoven C. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2012;11:54–65. doi: 10.1016/S1474-4422(11)70261-7. [DOI] [PubMed] [Google Scholar]
- 17.Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, Ogar JM, Rohrer JD, Black S, Boeve BF, Manes F, Dronkers NF, Vandenberghe R, Rascovsky K, Patterson K, Miller BL, Knopman DS, Hodges JR, Mesulam MM, Grossman M. Classification of primary progressive aphasia and its variants. Neurology. 2011;76:1006–1014. doi: 10.1212/WNL.0b013e31821103e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hyman BT, Trojanowski JQ. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:1095–1097. doi: 10.1097/00005072-199710000-00002. [DOI] [PubMed] [Google Scholar]
- 19.Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- 20.Kertesz A, Davidson W, Fox H. Frontal behavioral inventory: diagnostic criteria for frontal lobe dementia. Can J Neurol Sci. 1997;24:29–36. doi: 10.1017/s0317167100021053. [DOI] [PubMed] [Google Scholar]
- 21.Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardi-man O, Burrell JR, Zoing MC. Amyotrophic lateral sclerosis. Lancet. 2011;377:942–955. doi: 10.1016/S0140-6736(10)61156-7. [DOI] [PubMed] [Google Scholar]
- 22.Kim SH, Shi Y, Hanson KA, Williams LM, Sakasai R, Bowler MJ, Tibbetts RS. Potentiation of amyotrophic lateral sclerosis (ALS)-associated TDP-43 aggregation by the protea-some-targeting factor, ubiquilin 1. J Biol Chem. 2009;284:8083–8092. doi: 10.1074/jbc.M808064200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.King A, Maekawa S, Bodi I, Troakes C, Al-Sarraj S. Ubiquitinated, p62 immunopositive cerebellar cortical neuronal inclusions are evident across the spectrum of TDP-43 proteinopathies but are only rarely additionally immunopositive for phosphorylation-dependent TDP-43. Neuropathoiogy. 2011;31:239–249. doi: 10.1111/j.1440-1789.2010.01171.x. [DOI] [PubMed] [Google Scholar]
- 24.Kleijnen MF, Shih AH, Zhou P, Kumar S, Soccio RE, Kedersha NL, Gill G, Howley PM. The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol Cell. 2000;6:409–419. doi: 10.1016/s1097-2765(00)00040-x. [DOI] [PubMed] [Google Scholar]
- 25.Kleyweg RP, van der Meche FG, Schmitz PI. Interobserver agreement in the assessment of muscle strength and functional abilities in Guillain-Barre syndrome. Muscle Nerve. 1991;14:1103–1109. doi: 10.1002/mus.880141111. [DOI] [PubMed] [Google Scholar]
- 26.Ko HS, Uehara T, Tsuruma K, Nomura Y. Ubiquilin interacts with ubiquitylated proteins and proteasome through its ubiquitin-associated and ubiquitin-like domains. FEBS Lett. 2004;566:110–114. doi: 10.1016/j.febslet.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 27.Kovacs GG, Murrell JR, Horvath S, Haraszti L, Majtenyi K, Molnar MJ, Budka H, Ghetti B, Spina S. TARDBP variation associated with frontotemporal dementia, supranuclear gaze palsy, and chorea. Mov Disord. 2009;24:1843–1847. doi: 10.1002/mds.22697. [DOI] [PubMed] [Google Scholar]
- 28.Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Van-derburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH., Jr Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 29.Lezak M. Neuropsychological assessment. Oxford University Press; New York: 1983. [Google Scholar]
- 30.Libon DJ, Massimo L, Moore P, Coslett HB, Chatterjee A, Aguirre GK, Rice A, Vesely L, Grossman M. Screening for frontotemporal dementias and Alzheimer's disease with the Philadelphia Brief Assessment of Cognition: a preliminary analysis. Dement Geriatr Cogn Disord. 2007;24:441–447. doi: 10.1159/000110577. [DOI] [PubMed] [Google Scholar]
- 31.Lim PJ, Danner R, Liang J, Doong H, Harman C, Srinivasan D, Rothenberg C, Wang H, Ye Y, Fang S, Monteiro MJ. Ubiquilin and p97/VCP bind erasin, forming a complex involved in ERAD. J Cell Biol. 2009;187:201–217. doi: 10.1083/jcb.200903024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, Perry RH, Trojanowski JQ, Mann DM, Lee VM. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011;122:111–113. doi: 10.1007/s00401-011-0845-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mackenzie IR, Neumann M, Bigio EH, Caims NJ, Alafuzoff I, Kril J, Kovacs GG, Ghetti B, Halliday G, Holm IE, Ince PG, Kamphorst W, Revesz T, Rozemuller AJ, Kumar-Singh S, Akiyama H, Baborie A, Spina S, Dickson DW, Trojanowski JQ, Mann DM. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119:1–4. doi: 10.1007/s00401-009-0612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mah AL, Perry G, Smith MA, Monteiro MJ. Identification of ubiquilin, a novel presenilin interactor that increases presenilin protein accumulation. J Cell Biol. 2000;151:847–862. doi: 10.1083/jcb.151.4.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Massey LK, Mah AL, Monteiro MJ. Ubiquilin regulates presenilin endoproteolysis and modulates gamma-secretase components, Pen-2 and nicastrin. Biochem J. 2005;391:513–525. doi: 10.1042/BJ20050491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murray ME, Dejesus-Hernandez M, Rutherford NJ, Baker M, Duara R, Graff-Radford NR, Wszolek ZK, Ferman TJ, Josephs KA, Boylan KB, Rademakers R, Dickson DW. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol. 2011;122:673–690. doi: 10.1007/s00401-011-0907-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.N'Diaye EN, Kajihaia KK, Hsieh I, Morisaki H, Debnath J, Brown EJ. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep. 2009;10:173–179. doi: 10.1038/embor.2008.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 39.Neumann M, Kwong LK, Lee BB, Kremmer E, Flatley A, Xu Y, Forman MS, Troost D, Kretzsehmar HA, Trojanowski JQ, Lee VM. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 2009;117:137–149. doi: 10.1007/s00401-008-0477-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 41.Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, van Swieten JC, Seelaar H, Dopper EG, Onyike CU, Hillis AE, Josephs KA, Boeve BF, Kertesz A, Seeley WW, Rankin KP, Johnson JK, Gomo-Tempini ML, Rosen H, Prioleau-Latham CE, Lee A, Kipps CM, Lillo P, Piguet O, Rohrer JD, Rossor MN, Warren JD, Fox NC, Galasko D, Salmon DP, Black SE, Mesulam M, Weintraub S, Dickerson BC, Diehl-Schmid J, Pasquier F, Deramecourt V, Lebert F, Pijnenburg Y, Chow TW, Manes F, Grafman J, Cappa SF, Freedman M, Grossman M, Miller BL. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134:2456–2477. doi: 10.1093/brain/awr179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Holtta-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chio A, Restagno G, Borghero G, Sabatelli M, Heckemian D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21 -linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familia amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 44.Rosso SM, Donker Kaat L, Baks T, Joosse M, de Koning I, Pijnenburg Y, de Jong D, Dooijes D, Kamphorst W, Ravid R, Niermeijer MF, Verheij F, Kremer HP, Scheltens P, van Duijn CM, Heutink P, van Swieten JC. Frontotemporal dementia in The Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain. 2003;126:2016–2022. doi: 10.1093/brain/awg204. [DOI] [PubMed] [Google Scholar]
- 45.Rothenberg C, Monteiro MJ. Ubiquilin at a crossroads in protein degradation pathways. Autophagy. 2010;6:979–980. doi: 10.4161/auto.6.7.13118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rothenberg C, Srinivasan D, Mah L, Kaushik S, Peterhoff CM, Ugolino J, Fang S, Cuervo AM, Nixon RA, Monteiro MJ. Ubiquilin functions in autophagy and is degraded by chaperone-mediated autophagy. Hum Mol Genet. 2010;19:3219–3232. doi: 10.1093/hmg/ddq231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rutherford NJ, Zhang YJ, Baker M, Gass JM, Finch NA, Xu YF, Stewart H, Kelley BJ, Kuntz K, Crook RJ, Sreedharan J, Vance C, Sorenson E, Lippa C, Bigio EH, Geschwind DH, Knopman DS, Mitsumoto H, Petersen RC, Cashman NR, Hutton M, Shaw CE, Boylan KB, Boeve B, Graff-Radford NR, Wszolek ZK, Caselli RJ, Dickson DW, Mackenzie IR, Petrucelli L, Rademakers R. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008;4:el000193. doi: 10.1371/journal.pgen.1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schnell SA, Staines WA, Wessendorf MW. Reduction of lipofuscin-like autofluorescence in ftuorescently labeled tissue. J Histochem Cytochem. 1999;47:719–730. doi: 10.1177/002215549904700601. [DOI] [PubMed] [Google Scholar]
- 49.Simon-Sanchez J, Dopper EG, Cohn-Hokke PE, Hukema RK, Nicolaou N, Seelaar H, de Graaf JR, de Koning I, van Schoor NM, Deeg DJ, Smits M, Raaphorst J, van den Berg LH, Schel-haas HJ, De Die-Smulders CE, Majoor-Krakauer D, Rozemuller AJ, Wiltemsen R, Pijnenburg YA, Heutink P, van Swieten JC. The clinical and pathological phenotype of C9orf72 hexanucleotide repeat expansions. Brain. 2012 doi: 10.1093/brain/awr353. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 50.Snowden JS, Rollinson S, Thompson JC, Harris JM, Stopford CL, Richardson AM, Jones M, Gerhard A, Davidson YS, Robinson A, Gibbons L, Hu Q, Duplessis D, Neary D, Mann DM, Pickering-Brown SM. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain. 2012 doi: 10.1093/brain/awr355. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Dumnll JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stewart H, Rutherford NJ, Briemberg H, Krieger C, Cashman N, Fabros M, Baker M, Fok A, Dejesus-Hernandez M, Bisen A, Rademakers R, Mackenzie IR. Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on cliromosome 9p. Acta Neuropathol. 2012;123:409–417. doi: 10.1007/s00401-011-0937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stieren ES, El Ayadi A, Xiao Y, Siller E, Landsverk ML, Oberhauser AF, Barral JM, Boehning D. Ubiquilin-1 is a molecular chaperone for the amyloid precursor protein. J Biol Chem. 2011;286:35689–35698. doi: 10.1074/jbc.M111.243147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Troakes C, Maekawa S, Wijesekera L, Rogelj B, Siklos L, Bell C, Smith B, Newhouse S, Vance C, Johnson L, Hortobagyi T, Shatunov A, Al-Chalabi A, Leigh N, Shaw CE, King A, Al-Sarraj S. An MND/ALS phenotype associated with C9orf72 repeat expansion: abundant p62-positive, TDP-43-negative inclusions in cerebral cortex, hippocampus and cerebellum but without associated cognitive decline. Neuropathology. 2011 doi: 10.1111/j.1440-1789.2011.01286.x. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 55.Uryu K, Nakashima-Yasuda H, Forman MS, Kwong LK, Clark CM, Grossman M, Miller BL, Kretzschmar HA, Lee VM, Tro-janowski JQ, Neumann M. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J Neuro-palhol Exp Neurol. 2008;67:555–564. doi: 10.1097/NEN.0b013e31817713b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Van Langenhove T, van der Zee J, Sleegcrs K, Engelborghs S, Vandenberghe R, Gijselinck I, Van den Broeck M, Mattheijssens M, Peeters K, De Deyn PP, Cruts M, Van Broeckhoven C. Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology. 2010;74:366–371. doi: 10.1212/WNL.0b013e3181ccc732. [DOI] [PubMed] [Google Scholar]
- 57.Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chatabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Viswanathan J, Haapasalo A, Bottcher C, Miettinen R, Kurkinen KM, Lu A, Thomas A, Maynard CJ, Romano D, Hyman BT, Berezovska O, Bertram L, Soininen H, Dantuma NP, Tanzi RE, Hiltunen M. Alzheimer's disease-associated ubiquilin-1 regulates presenilin-1 accumulation and aggresome formation. Traffic. 2011;12:330–348. doi: 10.1111/j.1600-0854.2010.01149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang H, Lim PJ, Yin C, Rieckher M, Vogel BE, Monteiro MJ. Suppression of polyglutamine-induced toxicity in cell and animal models of Huntington's disease by ubiquilin. Hum Mol Genet. 2006;15:1025–1041. doi: 10.1093/hmg/ddl017. [DOI] [PubMed] [Google Scholar]
- 60.Wang H, Monteiro MJ. Ubiquilin interacts and enhances the degradation of expanded-polyglutamine proteins. Biochem Biophys Res Commun. 2007;360:423–427. doi: 10.1016/j.bbrc.2007.06.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang H, Monteiro MJ. Ubiquilin overexpression reduces GFP-polyalaniue-induced protein aggregates and toxicity. Exp Cell Res. 2007;313:2810–2820. doi: 10.1016/j.yexcr.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wooten MW, Hu X, Babu JR, Seibenhener ML, Geetha T, Paine MG, Wooten MC. Signaling, polyubiquitination, trafficking, and inclusions: sequestosome l/p62's role in neurodegenerative disease. J Biomed Biotechnol. 2006;2006:62079. doi: 10.1155/JBB/2006/62079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu AL, Wang J, Zheleznyak A, Brown EJ. Ubiquitin-related proteins regulate interaction of vimentin intermediate filaments with the plasma membrane. Mol Cell. 1999;4:619–625. doi: 10.1016/s1097-2765(00)80212-9. [DOI] [PubMed] [Google Scholar]