

Abstract
This short review describes new developments in Pd-catalyzed aminoarylation reactions between aryl halides and alkenes bearing pendant nitrogen nucleophiles. These transformations provide a novel and powerful method for accessing numerous 3-, 5-, 6-, and 7-membered nitrogen heterocycles.
Keywords: alkenes, amines, arylation, catalysis, heterocycles, palladium, stereoselective synthesis
1 Introduction
The stereocontrolled construction of saturated nitrogen heterocycles is of great importance due to the presence of these units in both pharmaceuticals and natural products. Compounds that bear 2-(arylmethyl) substituents constitute an important subclass of nitrogen heterocycles, as these motifs are prevalent in numerous biologically active compounds including those illustrated in Figure 1.
Figure 1.

Biologically active compounds with 2-(arylmethyl)heterocyclic subunits highlighted in blue
Over the past several years palladium-catalyzed alkene aminoarylation reactions have emerged as powerful tools for the synthesis of 2-(arylmethyl)pyrrolidines and related nitrogen heterocycles.1,2,3 These transformations effect the cross-coupling of simple aminoalkene substrates with aryl or alkenyl halides to generate the heterocyclic ring with formation of a C–N bond, a C–C bond, and one or more stereocenters, with good to excellent stereocontrol. Moreover, these methods are quite useful for generating analogs of a particular scaffold, as a wide variety of aryl electrophiles are readily available. This short review will highlight recent developments in this field from 2008–2011.
2 Synthesis of Pyrrolidines via Pd-Catalyzed Alkene Aminoarylation Reactions
2.1 Pd(0)-Catalyzed Alkene Aminoarylation Reactions
In 2004 our group first reported a new method for the stereoselective synthesis of pyrrolidines via Pd-catalyzed cross-coupling reactions between aryl halides and γ-aminoalkene derivatives.4 The reactions are effective with a number of different aryl or alkenyl halide coupling partners, and substrates bearing N-aryl, N-acetyl, N-Boc, and N-Cbz groups can be employed (Scheme 1).1,a,b,5 This method provides access to cis-2,5- and trans-2,3-disubstituted pyrrolidines with good to excellent diastereoselectivity, and enantiomerically enriched substrates are converted to the heterocyclic products without loss of optical purity. Moreover, starting materials bearing internal alkenes are stereoselectively transformed into products that result from suprafacial addition to the alkene.5 The scope and limitations of this strategy have been outlined in previous reviews.1a,b
Scheme 1.

Synthesis of pyrrolidines via Pd-catalyzed alkene aminoarylation reactions
The Pd-catalyzed aminoarylation reactions have been shown to proceed via the catalytic cycle illustrated in Scheme 2.1a,b,4 The transformations are initiated by oxidative addition of the aryl bromide to Pd(0) to afford 1, which is converted to the key intermediate palladium(aryl)amido complex 2 via reaction with the amine substrate and base. Complex 2 undergoes intramolecular syn-migratory insertion of the alkene into the Pd–N bond (syn-aminopalladation) to yield 3.6 The pyrrolidine product is then generated by C–C bond-forming reductive elimination from 3.
Scheme 2.

Catalytic cycle
The stereochemical outcome of these reactions is substrate controlled, and is determined during the alkene syn-aminopalladation event.1a,b As shown in Scheme 3, substrates 4 bearing a substituent at C1 are selectively transformed to cis-2,5-disubstituted pyrrolidines 6 by way of transition state 5, where axial orientation of the R-group minimizes A(1,3)-strain with the nitrogen protecting group. In contrast, analogous reactions of substrates bearing allylic substituents (7) provide trans-2,3-disubstitued products 9 via transition state 8 in which the R-group is equatorial. The generation of 2,4-disubstituted pyrrolidines from substrates that contain a homoallylic group typically proceeds with modest (ca. 2–3:1) diastereoselectivity.1a,b,7
Scheme 3.

Stereochemical model
2.1.1 Synthesis of trans-2,5-Disubstituted Pyrrolidines
Although the transformations described above provide efficient access to cis-2,5-disubstituted pyrrolidines, a stereocontrolled route to the analogous trans-2,5-disubstituted isomers is highly desirable. These latter compounds have found many applications as ligands, chiral auxiliaries, and catalysts.8 In addition, this motif is also displayed in biologically active natural products.9 We recently described a stereoselective synthesis of trans-2,5-disubstituted pyrrolidine derivatives via Pd-catalyzed alkene aminoarylation reactions of cyclic carbamate substrates such as 10.10 These transformations may proceed via transition state 11, in which ring strain is minimized during the key alkene aminopalladation step (Scheme 4), to provide bicyclic products 12. These products can be converted to pyrrolidines 13 using standard transformations.
Scheme 4.

Alkene aminoarylation strategy for synthesis of trans-2,5-disubstituted pyrrolidines
Substrates 10 were generated in enantiopure form in a few steps from either Boc-protected threonine or serine. A catalyst composed of [(allyl)PdCl]2 and RuPhos provided optimal results, and delivered the desired oxazolidin-2-one products 12 in good yield with >20:1 diastereoselectivity (Scheme 5).
Scheme 5.

Pd-catalyzed aminoarylation reactions of oxazolidin-2-ones
The bicyclic products of the aminoarylation reactions were transformed to the desired trans-2,5-disubstituted pyrrolidines via either hydrolysis with NaOH or reduction with LiAlH4 (Scheme 6). Both methods provided good yields and afforded products 13a–b with no erosion of stereochemical purity.
Scheme 6.

Conversion of Pd-catalyzed products 12a to functionalized trans-2,5-disubstituted pyrrolidines 13a–b
2.1.2 Transformations of Aryl Chloride Electrophiles
The use of aryl chloride electrophiles in cross-coupling reactions is often desirable due to their low cost relative to aryl iodides or bromides. However, the slow rate of oxidative addition of aryl chlorides to Pd(0) necessitates the use of electron-rich ligands, which are known to slow rates of C–C bond forming reductive elimination.11 In preliminary studies, use of electron-rich ligands for Pd-catalyzed alkene aminoarylation reactions of 14 led to the formation of regioisomeric side products 17 (Scheme 7), which are generated via competing β-hydride elimination side reactions that occur when reductive elimination from intermediate 15 is relatively slow. Work by Buchwald and Hartwig has illustrated that bulky electron-rich phosphine ligands can both facilitate oxidative addition of aryl chlorides and promote reductive elimination.12 As such, we explored the use of these ligands for alkene aminoarylation reactions.13
Scheme 7.

Formation of pyrrolidine regioisomers
After optimization we found that Buchwald’s S-Phos ligand14 provided good results for the coupling of a range of aryl chlorides with γ-N-(Boc-amino)alkenes, affording pyrrolidine products with excellent regio- and diastereoselectivity (Scheme 8). Efforts to use this ligand for analogous transformations of γ-N-(arylamino)alkenes led to competing N-arylation of the substrates. However, a catalyst composed of Pd2(dba)3 and PCy2Ph proved to be useful for these latter transformations. The desired pyrrolidine products were generated in good yield, and only relatively small quantities of regioisomeric side-products (ca. 8–10%) were formed.
Scheme 8.

Use of aryl chloride electrophiles in Pd-catalyzed alkene aminoarylation reactions
2.1.3 Synthesis of Hexahydro-3H-pyrrolizin-3-ones
A new synthesis of pyrrolidine-fused lactams (hexahydro-3H-pyrrolizin-3-ones) via Pd-catalyzed alkene aminoarylation reactions was recently reported by Cacchi et. al.15 These products are potentially valuable precursors to substituted pyrrolizidines, which are displayed in many biologically active compounds. As shown in Scheme 9, treatment of lactam substrate 18 with an aryl halide, Cs2CO3 and a Pd2(dba)3/X-Phos catalyst led to the stereoselective formation of products 19 in good yield and with > 20:1 diastereoselectivity. In addition, reactions of enantiomerically enriched substrates proceeded without loss of optical purity. The stereochemical outcome of these reactions is likely due to aminopalladation through a transition state similar to 11 shown above in Scheme 4.
Scheme 9.

Synthesis of hexahydro-3H-pyrrolizin-3-ones
The transformations were effective with a range of aryl bromides, chlorides, and triflates as coupling partners. Moreover, the use of Cs2CO3 as base allowed for tolerance of functional groups such as esters, aldehydes, and nitro groups, which are not compatible with stronger bases such as NaOtBu.5 However, the scope of these reactions with respect to lactam size or substitution pattern was not explored.
2.1.4 Enantioselective Synthesis of Monosubstituted Pyrrolidines
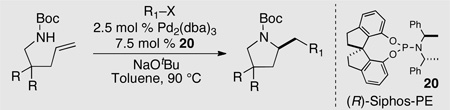
A number of interesting biologically active molecules contain monosubstituted pyrrolidine cores.16 As such, the development of enantioselective transformations for the construction of these units is of considerable significance. Our group recently reported an asymmetric alkene aminoarylation reaction for the synthesis of enantioenriched pyrrolidines.17 During our studies we discovered that the monodentate phosphoramidite ligand (R)-Siphos-PE (20),18 gave the best asymmetric induction (Table 1). In contrast, little to no stereocontrol was observed with chiral bidentate ligands. This effect may be due to a requirement for aminopalladation via a monophosphine palladium complex.6
Table 1.
Enantioselective synthesis of substituted pyrrolidines
 | |||||
|---|---|---|---|---|---|
| Entry | R | R1 | X | ee | Yield |
| 1 | H | 2-naphthyl | Br | 82% | 78% |
| 2 | H | (Z)-β-styryl | Br | 94% | 61% |
| 3 | H | Br | 82% | 61% | |
| 4 | Me | C6H4-m-CF3 | I | 91% | 71% |
| 5 | Me | C6H4-p-NMe2 | Br | 92% | 70% |
The optimized conditions allowed for the synthesis of numerous enantioenriched 2-(arylmethyl)pyrrolidines in good yield and ee (Table 1). Electron-rich, -poor, and -neutral aryl bromides and iodides were suitable coupling partners for the alkene aminoarylation reactions, and the highest enantioselectivities were obtained with alkenyl bromide electrophiles.
This method was applied towards a concise enantioselective synthesis of the natural product (−)-tylophorine 22 (Scheme 10).19 Pyrrolidine 21 was generated in 69% yield (88% ee) via enantioselective aminoarylation. This intermediate was converted to 22 in two steps and near quantitative yield.
Scheme 10.

Synthesis of (−)-tylophorine
2.1.5 Asymmetric Total Synthesis of (+)-Aphanorphine
The utility of enantioselective Pd-catalyzed alkene aminoarylation has also been illustrated through our recent enantioconvergent synthesis of the benzomorphan alkaloid (+)-aphanorphine (23).20,21 Our approach to this target involved two key transformations: (1) an intramolecular Friedel-Crafts alkylation;22 and (2) an asymmetric Pd-catalyzed alkene aminoarylation reaction (Scheme 11).17 A key element of our strategy involved the conversion of racemic substrate 25 to a pair of enantioenriched diastereomers 24 with the same absolute configuration at C2 via a catalyst-controlled reaction.23 This mixture was then converted to a single enantiopure product via intramolecular Friedel-Crafts arylation.
Scheme 11.

Strategy for enantioconvergent synthesis of (+)-aphanorphine
Substrate 25 was synthesized in 3 steps from commercially available N-Boc-1-amino-2-propanol in 81% overall yield. The asymmetric alkene aminoarylation reaction between 25 and 4-bromoanisole (26) generated a 1:1 mixture of pyrrolidines 24a–b in 75% yield (Scheme 12).
Scheme 12.

Asymmetric alkene aminoarylation of 25
The enantioselectivity of the alkene aminoarylation reaction was determined to be 81% through the conversion of 24a–b to known intermediate 28 via Friedel-Crafts alkylation (Scheme 13).22 To complete the total synthesis of (+)-aphanorphine, 28 was transformed to 29 via cleavage of the tosyl group and N-methylation. Finally, O-demethylation with BBr324 afforded (+)-aphanorphine in 63% yield. This route provided (+)-aphanorphine in 10 steps and 13% overall yield from commercially available materials.
Scheme 13.

Synthesis of (+)-aphanorphine
2.2 Pd(II)-Catalyzed Arene C–H Activation/Alkene Aminoarylation
Michael and coworkers have developed a new cascade arene solvent C–H activation/alkene aminoarylation reaction that takes advantage of the unique reactivity of high oxidation state Pd(IV) complexes.25 This method was used to generate a number of different 2-(arylmethyl)pyrrolidines (Scheme 14). Related transformations were also applied towards the construction of other 5-, 6-, and 7-membered nitrogen heterocycles in good to excellent yield. High regioselectivity favoring generation of para-substituted products was observed.
Scheme 14.

Solvent C–H activation/alkene aminoarylation
In contrast to Pd(0/II)-catalyzed alkene aminoarylations involving aryl halide electrophiles, which proceed via syn-aminopalladation processes, the Pd(II/IV)-catalyzed reactions involve anti-aminopalladation pathways. As shown in Scheme 15, anti-aminopalladation of substrate 30 affords Pd(II)-alkyl species 32. Subsequent oxidation to Pd(IV) with NFBS followed by arene C–H activation affords intermediate 34, which undergoes C–C bond forming reductive elimination to yield the product 31. It appears that the preference for anti-aminopalladation may result from the slightly acidic reaction conditions, which suppress formation of Pd-amido complexes that are required for syn-aminopalladation to occur.
Scheme 15.

Catalytic cycle
3 Synthesis of Other 5-Membered Heterocycles via Pd-Catalyzed Alkene Aminoarylation
3.1 Synthesis of Pyrazolidines
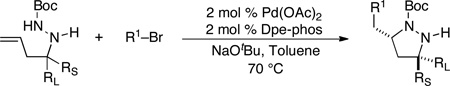
Substituted pyrazolidines are valuable intermediates in complex molecule synthesis, as the N–N bond can be reductively cleaved to yield 1,3-diamine derivatives. We recently reported a strategy for the stereoselective synthesis of 3,5-disubstituted pyrazolidines in which manipulation of allylic strain through choice of nitrogen protecting group was used to dictate the stereochemical outcome of these transformations (Scheme 16).26 Substrates lacking a protecting group on the internal nitrogen atom were converted to cis-3,5-disubstituted products 37 via cyclization through transition state 36, in which R1 is equatorial (Scheme 16 and Table 3). In contrast, reactions of substrates bearing N2-Boc or aryl groups were transformed to trans-3,5-disubstituted products 39 by way of transition state 38, in which A(1,3) strain is minimized via axial orientation of R1 (Scheme 16 and Table 2). Conversion of the products to substituted 1,3-diamines was accomplished by SmI2-mediated reductive cleavage of the N–N bond.
Scheme 16.

Pyrazolidine stereochemistry
Table 3.
Synthesis of cis-3,5-disubstituted pyrazolidines
 | |||||
|---|---|---|---|---|---|
| Entry | RS | RL | R1 | Yield | dr (crude dr) |
| 1 | H | C3H7 | p-ClPh | 70 | >20:1 (7:1) |
| 2 | Ph | m-MePh | 66 | 13:1 (10:1) | |
| 3 | Me | Ph | p-PhPh | 83 | 6:1 (6:1) |
Table 2.
Synthesis of trans-3,5-disubstituted pyrazolidines
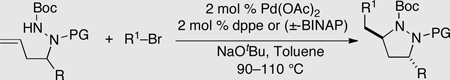 | |||||
|---|---|---|---|---|---|
| Entry | PG | R | R1 | Yield | dr (crude dr) |
| 1 | Ph | Ph | p-PhC(O)Ph | 74 | 20:1 (11:1) |
| 2 | PMP | C3H7 | p-CNPh | 63 | >20:1 (>20:1) |
| 3 | Boc | Ph | 2-naphthyl | 55 | >20:1(>20:1) |
| 4 | Boc | C3H7 | m-CF3Ph | 81 | >20:1(>20:1) |
3.2 Synthesis of Isoxazolidines
Substituted isoxazolidines are displayed in a number of biologically active compounds, and serve as synthetically useful precursors to substituted 1,3-amino alcohols. Although construction of these compounds is often accomplished via 1,3-dipolar cycloaddition reactions, the synthesis of 3,5-disubstituted isoxazolidines via this route often suffers from modest diastereoselectivity. We have developed an alternative approach to the stereoselective generation of cis-3,5-disubstituted isoxazolidines via Pd-catalyzed alkene aminoarylation reactions of O-butenyl hydroxylamine derivatives.27
The substrates for the aminoarylation reactions were prepared in three steps from readily available homoallylic alcohols. A catalyst composed of Pd2(dba)3/P(tBu3) provided good results, and the disubstituted isoxazolidine products were generated in moderate to good yield with diastereoselectivities that were typically around 20:1. Both aryl and alkenyl halides were suitable electrophiles, and related substrates bearing a substituent at the allylic position were converted to trans-4,5-disubstituted isoxazolidines with good yield and stereocontrol (Scheme 17).
Scheme 17.

Synthesis of isoxazolidines
The high diastereoselectivity in these transformations has been ascribed to cyclization through transition state 40 in which the R1 group is in an equatorial position and the carbamate moiety is in a pseudoaxial position and oriented at a 90° angle with respect to the C3–O bond This orientation of the carbamate minimizes electronic repulsion of the neighboring heteroatoms,27 and contrasts to the preferred orientation of carbamate moieties in related pyrrolidine-forming reactions (e.g., Scheme 3).
4 Aminoarylation Reactions For the Synthesis of 3-, 6-, and 7-Membered Heterocycles
4.1 Synthesis of Aziridines
The synthesis of aziridines via Pd-catalyzed alkene aminoarylation was recently reported by Yorimitsu and Oshima.28 A catalyst composed of Pd2(dba)3 and S-Phos was effective for the coupling of allylic amines with aryl and alkenyl halides to yield a variety of substituted aziridines (Table 4). However, the presence of two large groups at C1 appears to be necessary, as substrates bearing small C1 groups (e.g. methyl, entry 4) were transformed in modest yield due to competing substrate N-arylation.
Table 4.
Synthesis of aziridines
 | |||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Ar-X | Yield | dr |
| 1 | Ph | Ph | p-CF3C6H4Cl | 99 | -- |
| 2 | Me | Ph | PhBr | 90 | >99:1 |
| 3 | Me | tBu | PhBr | 42 | 59:41 |
| 4 | Me | Me | PhBr | 66 | -- |
The stereochemical outcome of these reactions is believed to arise from cyclization through transition state 41, in which the larger C1 substituent is in an axial orientation to avoid developing A(1,3)-strain with the N-aryl group. The transformation of a substrate bearing a very large tert-butyl group proceeded with low (59:41) diastereoselectivity (entry 3), which may be due to unfavorable steric interactions between the tert-butyl group and either the alkene or a ligand on the metal. Deuterium labeling experiments support a mechanism similar to that described above for the formation of pyrrolidines (Scheme 2), in which syn-aminopalladation of the alkene plays a key role.
4.2 Synthesis of Morpholines
The morpholine scaffold is found in several natural products and bioactive molecules29 and is often appended onto pharmaceutical lead compounds to improve pharmacokinetic properties. Our group has developed a new approach to the synthesis of substituted morpholines via Pd-catalyzed alkene aminoarylation reactions of O-allyl-1,2-aminoalcohol derivatives.30 The requisite substrates for these transformations were prepared in 3 steps from commercially available enantiopure amino acids. The alkene aminoarylation reactions proceeded smoothly using conditions similar to those previously employed by our group in a related synthesis of cis-2,6-disubstituted piperazines.31
A variety of cis-3,5-disubstituted morpholines were generated in good yield and with excellent stereocontrol (Scheme 18). The high diastereoselectivities (>20:1 dr) observed in these reactions are believed to be the result of syn-aminopalladation via boat-like transition state 42. This method was also useful for the diastereoselective construction of fused bicyclic morpholines. However, efforts to generate 2,3- or 2,5-disubstituted morpholines resulted in formation of products with low diastereoselectivity (ca. 2:1 dr).
Scheme 18.

Synthesis of morpholines
4.3 Synthesis of Saturated 1,4-Benzodiazepines
Benzodiazepines are considered “privileged structures” in medicinal chemistry due to their wide range of therapeutic activities.32 We recently illustrated the use of alkene aminoarylation reactions for the preparation of saturated 1,4-benzodiazepines, which provides a new entry into an important class of compounds and demonstrates the potential for 7-membered ring formation via this strategy.33
Substrates 43 were synthesized in 4 steps from methyl-2-aminobenzoate. A catalyst composed of PdCl2(MeCN)2 and PPh2Cy was effective for the conversion of a variety of substrates bearing different N-aryl groups into saturated 1,4-benzodiazepine products 44 in good yield (Scheme 19). The reactions were effective with numerous aryl halide coupling partners, and substrates bearing a substituent at the allylic position were converted to cis-2,3-disubstituted products with excellent diastereoselectivity. This stereochemical outcome likely results from syn-aminopalladation via boat-like transition state 45, in which the R group is oriented in an equatorial position and the Pd–N bond is eclipsed with the alkene.
Scheme 19.

Synthesis of saturated 1,4-benzodiazepines
A related synthesis of 1,4-benzodiazepin-5-ones 47 was accomplished via Pd-catalyzed alkene aminoarylation reactions of amide substrates 46. Transformations of 46 were most effective when P(p-F-C6H4)3 was used as the ligand (Scheme 20).
Scheme 20.

Synthesis of saturated 1,4-benzodiazepin-5-ones
5 Pd-Catalyzed Synthesis of Fused-, Bridged-, and Spiro-Polycyclic Heterocycles
5.1 Cascade Alkene Difunctionalization Reactions
Pyrrolizidines and their benzo-fused analogs are displayed in a broad array of biologically active compounds and natural products. We recently developed a concise synthesis of benzo-fused pyrrolizidines 51 from aryl halides and N-allyl-2-allylaniline 48 via a cascade alkene aminopalladation/carbopalladation/reductive elimination sequence (Scheme 21). The cascade reaction transforms simple substrates into complex products through formation of three bonds, two stereocenters, and two rings in a single step. However, to achieve this transformation it was necessary to find a catalyst that promotes carbopalladation from intermediate 49 in preference to competing reductive elimination.
Scheme 21.

Cascade reaction sequence
Our studies revealed that ligand 52 was the most effective at promoting the cascade reaction. Several substrates bearing different substitution patterns underwent the cascade reaction to afford the desired products in good yield and diastereoselectivity (Scheme 22).34 This method was also used for the conversion of N-allyl-2-(but-3-enyl)aniline to the analogous benzo-fused indolizidine derivatives.
Scheme 22.

Synthesis of benzo-fused pyrrolizidines and indolizidines
Interestingly, when substrate 53 was subjected to the optimized conditions, rearranged product 55 was observed (Scheme 23). Deuterium labeling studies suggest that the aminopalladation/carbopalladation cascade provides Pd-alkyl intermediate 54, which then undergoes an unusual 1,3-Pd shift prior to reductive elimination.35
Scheme 23.

Formation of 55 via 1,3-palladium migration
5.2 Tandem N-Arylation/Alkene Aminoarylation Reactions
The transformations described above provide an efficient means for generating benzo-fused pyrrolizidine and indolizidine derivatives bearing C2 arylmethyl groups. In contrast, the synthesis of related compounds 58 bearing arylmethyl groups at C3 was accomplished through use of intramolecular N-arylation/intermolecular alkene aminoarylation reactions of substrates 56 (Scheme 24).36 The chemoselectivity of these cascade reactions was controlled by the high reactivity of aryl bromides vs. aryl chlorides, which allowed the intramolecular N-arylation of the aryl bromide to occur prior to the intermolecular alkene aminoarylation reaction with the less reactive aryl chloride.
Scheme 24.

Cascade N-arylation/alkene aminoarylation
The use of electron rich phosphine ligands was essential in performing both transformations with a single catalyst.37 The bis-phosphine Cy4Dpe-Phos provided good results with electron-neutral or electron-rich aryl chloride coupling partners (Scheme 25). However, when electron-poor aryl chlorides were employed, optimal results were obtained with a Pd2(dba)3/PCy3. The transformations proceeded with moderate to good diastereoselectivity, and also provided access to tricyclic benzo-fused morpholines and pyrazolidines (albeit in modest yield).
Scheme 25.

Synthesis of fused tricyclic heterocycles
5.3 Intramolecular Alkene Aminoarylation Reactions
5.3.1 Synthesis of Tropane Derivatives
The tropane scaffold is displayed in a large number of natural products and pharmaceutically relevant molecules. We have recently shown that tropane derivatives 60 can be constructed through intramolecular alkene aminoarylation reactions of substrates 59 (Scheme 26).38 Enantiomerically enriched substrates (92–99% ee) were synthesized in 4 steps; control of absolute configuration was achieved via addition of a homoallylic Grignard reagent to a chiral N-tert-butanesulfinyl imine.
Scheme 26.

Synthesis of tropane derivatives
Monodentate phosphine ligands proved superior to bidentate ligands in the intramolecular reactions, and PCy3 provided optimal results in most cases. The scope of the intramolecular carboamination was broad, and a number of enantiomerically enriched benzo-fused tropanes were synthesized in good yield. The reactions were effective with a variety of aryl, alkenyl, and heteroaryl halides. Moreover, substrates with substituted alkenes were cleanly converted into benzo-fused tropanes bearing either quaternary stereocenters or two adjacent stereocenters.
The utility of the intramolecular aminoarylation reactions were demonstrated through a concise synthesis of NMDA antagonist 63 (Scheme 27). Substrate 61 was prepared in 3 steps (99% ee), and intramolecular alkene aminoarylation of 61 using S-Phos as ligand afforded 62 in 71% yield. Removal of the PMP group with aqueous CAN then provided enantiopure 63 in good yield.
Scheme 27.

Synthesis of NMDA antagonist 63
5.3.2 Synthesis of Spirooxindoles
Spirooxindoles are displayed in a variety of natural products and have also been investigated as potential pharmaceuticals or agrochemicals. Zhu and coworkers have recently employed alkene aminoarylation reactions of anilides bearing pendant alkenes (64) for the generation of spirooxindoles 65 (Scheme 28).39 Substrates bearing a substituent (R1) adjacent to the nitrogen atom were stereoselectively transformed to products bearing two stereocenters. In contrast to most Pd-catalyzed alkene aminoarylation reactions, these transformations proceed via an anti-aminopalladation mechanism and endocyclization of the amino group onto the pendant alkene is observed. The regiochemistry of these reactions may be controlled by the electronic bias of the substrate alkene group.
Scheme 28.

Synthesis of spirooxindoles
5.3.3 Cascade C–H Functionalization/Intramolecular Alkene Aminoarylation Reactions
Two research groups have recently reported intramolecular alkene aminoarylation reactions that effect C–H functionalization of the arene and do not require the presence of a halogenated electrophile. Zhu has illustrated that substrates 66 are transformed to spirooxindoles 65 when treated with PdCl2 in the presence of excess PhI(OAc)2 (Scheme 29).40 Although the need for halogenated substrates is eliminated when these conditions are employed, chemical yields and diastereoselectivities are modest.
Scheme 29.

Synthesis of spirooxindoles via C–H functionalization
In independent studies Yang has illustrated that benzamide derivatives bearing pendant alkenes (67) are transformed to fused polycyclic heterocycles 68 via intramolecular alkene aminoarylation (Scheme 30).41 Substituents on the arene moiety are well-tolerated, and disubstituted alkene substrates are stereoselectively transformed into heterocyclic products with net syn-addition to the double bond. Mechanistic studies indicate the reactions likely proceed by way of syn-aminopalladation of the alkene followed by arene C–H functionalization.
Scheme 30.

Synthesis of fused polycyclic heterocycles
Conclusions
The transformations above illustrate the utility of Pd-catalyzed alkene aminoarylation reactions for the stereocontrolled construction of a broad array of nitrogen heterocycles. Although there has been considerable progress in this field, there is also great potential for future developments. In the coming years it seems likely that increasingly complex structures will become accessible using this approach, and further work on catalyst development will likely lead to improved scope, stereocontrol, and efficiency.
Acknowledgment
The authors acknowledge the NIH-NIGMS (GM017650 and GM098314) for financial support of this work. DMS thanks the ACS Division of Organic Chemistry for a graduate fellowship and the University of Michigan for a Rackham predoctoral fellowship.
Biographies

John P. Wolfe was born in Greeley, CO, and received his B.A. degree from the University of Colorado, Boulder in 1994. As an undergraduate he conducted research in the labs of Professor Gary A. Molander. He received his Ph.D. degree in 1999 from the Massachusetts Institute of Technology under the guidance of Professor Stephen L. Buchwald. Following the completion of his Ph.D. studies, he spent three years as an NIH postdoctoral fellow in the lab of Professor Larry E. Overman at the University of California, Irvine. He joined the faculty at the University of Michigan in July, 2002, where he is currently an Associate Professor of Chemistry. Professor Wolfe’s current research is directed towards the development of new palladium-catalyzed reactions for the stereoselective synthesis of heterocycles, and new reactions of enediolate nucleophiles for enantioselective synthesis of functionalized tertiary alcohols. His research accomplishments have been recognized with several awards, including the Dreyfus New Faculty Award (2002), the Research Corporation Innovation Award (2002), the 3M Untenured Faculty Award (2003–2005), the Amgen Young Investigator Award (2004), the Lilly Grantee Award (2005), the Camille Dreyfus Teacher-Scholar Award (2006), and the GlaxoSmithKline Scholar Award (2008–2009).

Danielle M. Schultz was born in 1984 in Minocqua, WI. She received her B.S. in Chemistry and Cell and Molecular Biology in 2007 at the University of Wisconsin-La Crosse, where she synthesized novel serotonin agonists under the supervision of Professor Aaron Monte. She is currently in the fifth year of her Ph.D. studies in Professor John P. Wolfe’s lab. Her research has been focused on Pd-catalyzed alkene difunctionalization reactions for the synthesis of fused and bridging heterocycles.
References
- 1.For prior reviews, see: Wolfe JP. Synlett. 2008:2913. Wolfe JP. Eur. J. Org. Chem. 2007:571. Kotov V, Scarborough CC, Stahl SS. Inorg. Chem. 2007;46:1910. doi: 10.1021/ic061997v. McDonald RI, Liu G, Stahl SS. Chem. Rev. 2011;111:2981. doi: 10.1021/cr100371y.
- 2.For Cu-catalyzed reactions, see: Chemler SR. Org. Biomol. Chem. 2009;7:3009. doi: 10.1039/B907743J. Chemler SR. J. Organomet. Chem. 2011;696:150. doi: 10.1016/j.jorganchem.2010.08.041.
- 3.For Au-catalyzed reactions, see: Zhang G, Cui L, Wang Y, Zhang L. J. Am. Chem. Soc. 2010;132:1474. doi: 10.1021/ja909555d. Brenzovich WE, Jr, Benitez D, Lackner AD, Shunatona HP, Tkatchouk E, Goddard WA, III, Toste FD. Angew. Chem. Int. Ed. 2010;49:5519. doi: 10.1002/anie.201002739. Mankad NP, Toste FD. J. Am. Chem. Soc. 2010;132:12859. doi: 10.1021/ja106257n. Tkatchouk E, Mankad NP, Benitez D, Goddard WA, III, Toste FD. J. Am. Chem. Soc. 2011;133:14293. doi: 10.1021/ja2012627.
- 4.Ney JE, Wolfe JP. Angew. Chem. Int. Ed. 2004;43:3605. doi: 10.1002/anie.200460060. [DOI] [PubMed] [Google Scholar]
- 5.Bertrand MB, Neukom JD, Wolfe JP. J. Org. Chem. 2008;73:8851. doi: 10.1021/jo801631v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.The syn-migratory insertion of unactivated alkenes into Pd–N bonds had not been documented prior to our experiments in this area. See: Neukom JD, Perch NS, Wolfe JP. J. Am. Chem. Soc. 2010;132:6276. doi: 10.1021/ja9102259. Hanley PS, Markovic D, Hartwig JF. J. Am. Chem. Soc. 2010;132:6302. doi: 10.1021/ja102172m. Neukom JD, Perch NS, Wolfe JP. Organometallics. 2011;30:1269. Hanley PS, Hartwig JF. J. Am. Chem. Soc. 2011;133:15661. doi: 10.1021/ja205722f.
- 7.Jepsen TH, Larsen M, Nielsen MB. Tetrahedron. 2010;66:6133. [Google Scholar]
- 8.(a) Sprott KT, Corey EJ. Org. Lett. 2003;5:2465. doi: 10.1021/ol034706k. [DOI] [PubMed] [Google Scholar]; (b) Frisch K, Landa A, Saaby S, Jorgensen KA. Angew. Chem. Int. Ed. 2005;44:6058. doi: 10.1002/anie.200501900. [DOI] [PubMed] [Google Scholar]
- 9.Asano N, Nash RJ, Molyneux RJ, Fleet GWJ. Tetrahedron: Asymmetry. 2000;11:1645. [Google Scholar]
- 10.Lemen GS, Wolfe JP. Org. Lett. 2010;12:2322. doi: 10.1021/ol1006828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Littke AF, Fu GC. Angew. Chem. Int. Ed. 2002;41:4176. doi: 10.1002/1521-3773(20021115)41:22<4176::AID-ANIE4176>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 12.(a) Surry DS, Buchwald SL. Angew. Chem. Int. Ed. 2008;47:6338. doi: 10.1002/anie.200800497. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hartwig JF. Inorg. Chem. 2007;46:1936. doi: 10.1021/ic061926w. [DOI] [PubMed] [Google Scholar]
- 13.Rosen BR, Ney JE, Wolfe JP. J. Org. Chem. 2010;75:2756. doi: 10.1021/jo100344k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walker SD, Barder TE, Martinelli JR, Buchwald SL. Angew. Chem. Int. Ed. 2004;43:1871. doi: 10.1002/anie.200353615. [DOI] [PubMed] [Google Scholar]
- 15.Bagnoli L, Cacchi S, Fabrizi G, Goggiamani A, Scarponi C, Tiecco M. J. Org. Chem. 2010;75:2134. doi: 10.1021/jo1002032. [DOI] [PubMed] [Google Scholar]
- 16.Cole DC, Lennox WJ, Lombardi S, Ellingboe JW, Bernotas RC, Tawa GJ, Mazandarani H, Smith DL, Zhang G, Coupet J, Schechter LE. J. Med. Chem. 2005;48:353. doi: 10.1021/jm049243i. [DOI] [PubMed] [Google Scholar]
- 17.Mai DN, Wolfe JP. J. Am. Chem. Soc. 2010;132:12157. doi: 10.1021/ja106989h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo X-X, Xie J-H, Hou G-H, Shi W-J, Wang L-X, Zhou Q-L. Tetrahedron: Asymmetry. 2004;15:2231. [Google Scholar]
- 19.For an analogous synthesis of (±)-tylophorine, see: Rossiter LM, Slater ML, Giessert RE, Sakwa SA, Herr RJ. J. Org. Chem. 2009;74:9554. doi: 10.1021/jo902114u.
- 20.Mai DN, Rosen BR, Wolfe JP. Org. Lett. 2011;13:2932. doi: 10.1021/ol2009895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gulavita N, Hori A, Shimizu Y, Laszlo P, Clardy J. Tetrahedron Lett. 1988;29:4381. [Google Scholar]
- 22.(a) Ma Z, Hu H, Xiong W, Zhai H. Tetrahedron. 2007;63:7523. [Google Scholar]; (b) Zhai H, Luo S, Ye C, Ma Y. J. Org. Chem. 2003;68:8268. doi: 10.1021/jo0348726. [DOI] [PubMed] [Google Scholar]
- 23.Pyrrolidine-forming reactions of substrates bearing homoallylic substituents proceed with low diastereoselectivity. See references 1a–b and 7.
- 24.Fuchs JR, Funk RL. Org. Lett. 2001;3:3923. doi: 10.1021/ol016795b. [DOI] [PubMed] [Google Scholar]
- 25.(a) Rosewall CF, Sibbald PA, Liskin DV, Michael FE. J. Am. Chem. Soc. 2009;131:9488. doi: 10.1021/ja9031659. [DOI] [PubMed] [Google Scholar]; (b) Sibbald PA, Rosewall CF, Swartz RD, Michael FE. J. Am. Chem. Soc. 2009;131:15945. doi: 10.1021/ja906915w. [DOI] [PubMed] [Google Scholar]
- 26.Giampietro NC, Wolfe JP. J. Am. Chem. Soc. 2008;130:12907. doi: 10.1021/ja8050487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lemen GS, Giampietro NC, Hay MB, Wolfe JP. J. Org. Chem. 2009;74:2533. doi: 10.1021/jo8027399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayashi S, Yorimitsu H, Oshima K. Angew. Chem. Int. Ed. 2009;48:7224. doi: 10.1002/anie.200903178. [DOI] [PubMed] [Google Scholar]
- 29.Wijtmans R, Vink MKS, Shoemaker HE, van Delft FL, Blaauw RH, Rutjes FPJT. Synthesis. 2004:641. [Google Scholar]
- 30.Leathen ML, Rosen BR, Wolfe JP. J. Org. Chem. 2009;74:5107. doi: 10.1021/jo9007223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.(a) Nakhla JS, Wolfe JP. Org. Lett. 2007;9:3279. doi: 10.1021/ol071241f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nakhla JS, Schultz DM, Wolfe JP. Tetrahedron. 2009;65:6549. doi: 10.1016/j.tet.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.(a) Ellman JA. Acc. Chem. Res. 1996;29:132. [Google Scholar]; (b) Horton DA, Bourne GT, Smythe ML. Chem. Rev. 2003;103:893. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]
- 33.Neukom JD, Aquino AS, Wolfe JP. Org. Lett. 2011;13:2196. doi: 10.1021/ol200429a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schultz DM, Wolfe JP. Org. Lett. 2010;12:1028. doi: 10.1021/ol100033s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.The migration of palladium from one sp3-hybridized carbon to another has only been observed on one prior occasion. See: Heumann A, Bäckvall JE. Angew. Chem. Int. Ed. Engl. 1985;24:207.
- 36.Lemen GS, Wolfe JP. Org. Lett. 2011;13:3218. doi: 10.1021/ol201123b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.For the synthesis of N-aryl-2-benzylpyrrolidines and indolines via cascade N-arylation/alkene aminoarylation, see: Lira R, Wolfe JP. J. Am. Chem. Soc. 2004;126:13906. doi: 10.1021/ja0460920. Yang Q, Ney JE, Wolfe JP. Org. Lett. 2005;7:2575. doi: 10.1021/ol050647u.
- 38.Schultz DM, Wolfe JP. Org. Lett. 2011;13:2962. doi: 10.1021/ol201051q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jaegli S, Erb W, Retailleau P, Vors J–P, Neuville L, Zhu J. Chem. Eur. J. 2010;16:5863. doi: 10.1002/chem.201000312. [DOI] [PubMed] [Google Scholar]
- 40.Jaegli S, Dufour J, Wei H–L, Piou T, Duan X–H, Vors J–P, Neuville L, Zhu J. Org. Lett. 2010;12:4498. doi: 10.1021/ol101778c. [DOI] [PubMed] [Google Scholar]
- 41.Yip K–T, Yang D. Org. Lett. 2011;13:2134. doi: 10.1021/ol2006083. [DOI] [PubMed] [Google Scholar]