

Abstract
Background
The occurrence of a congenital heart defect has long been thought to have a multifactorial basis, but the evidence is indirect. Complex trait analysis could provide a more nuanced understanding of congenital heart disease.
Methods and Results
We assessed the role of genetic and environmental factors on the incidence of ventricular septal defects caused by a heterozygous Nkx2-5 knockout mutation. We phenotyped >3100 hearts from a second generation intercross of the inbred mouse strains C57BL/6 and FVB/N. Genetic linkage analysis mapped loci with LOD scores of 5-7 on chromosomes 6, 8 and 10 that influence the susceptibility to membranous VSD in Nkx2-5+/- animals. The chromosome 6 locus overlaps one for muscular VSD susceptibility. Multiple logistic regression analysis for environmental variables revealed that maternal age is correlated with the risk of membranous and muscular VSD in Nkx2-5+/- but not wild-type animals. The maternal age effect is unrelated to aneuploidy or a genetic polymorphism in the affected individuals. The risk of a VSD is not only complex but dynamic. Whereas the effect of genetic modifiers on risk remains constant, the effect of maternal aging increases over time.
Conclusions
Enumerable factors contribute to the presentation of a congenital heart defect. The factors that modify rather than cause congenital heart disease substantially affect risk in predisposed individuals. Their characterization in a mouse model offers the potential to narrow the search space in human studies and to develop alternative strategies for prevention.
Keywords: genetic variation, heart defects, congenital, Nkx2-5, genetic modifier, maternal age
Introduction
Based upon clinical observations and animal models, Nora elegantly reasoned in 1968 that multiple genetic and environmental factors contribute to the pathogenesis of a congenital heart defect in an individual. He proposed a multifactorial inheritance model in which a set of genes contributes to a threshold liability to a heart defect and interactions with environmental factors lead to the expression of a phenotype. Nora articulated the possibility that complex trait analysis could help to elucidate the contributing factors and lead to strategies that decrease risk either by replacing or inhibiting a factor.1 Most basic research in the ensuing decades has focused on the simpler problem of defining the monogenic causes of congenital heart disease. Many genes have been discovered, but a plausible strategy to prevent heart defects remains difficult to fathom.2, 3 Complex trait analysis may be worthy of reappraisal in light of knowledge and methods that were unavailable four decades ago.
Complex trait analysis of pediatric cardiology patients, however, would be massively challenging. Genome-wide association studies commonly require tens of thousands of individuals to achieve statistical significance. Population genetic heterogeneity, environmental interactions, and numerous other factors limit the power to detect loci that have small effect for common diseases like type 2 diabetes or traits like height. The analysis of congenital heart disease would likely be even more difficult because of its infrequency, the widely varying presentations, and the heterogeneity of factors that cause disease or modify risk. A mouse model could circumvent these limitations. Experimental findings could then help to narrow the search space for clinical studies based upon the premise that the same molecular genetic pathways are deployed in mouse and human cardiac development.4, 5
Thus we crossed two inbred mouse strains to investigate the effect of genetic background on phenotype in Nkx2-5 haploinsufficient mutants. Human NKX2-5 mutations cause pleiotropic congenital heart defects.6-8 Nkx2-5+/- mice in the inbred C57Bl/6 strain background have a high incidence of similar heart defects, whereas Nkx2-5+/- mice from an outcross to the FVB/N strain (F1) have a substantially reduced incidence. Defects recur in the Nkx2-5+/- F2 progeny of the F1 X F1 intercross. Comparison of the incidences of defects such as atrial and ventricular septal defects (ASD, VSD) and common atrioventricular canal in multiple crosses revealed the profound influence of modifier genes on the mutant phenotype but not wildtype. Each type of heart defect likely has its own set of genetic modifiers, but some genes may influence the development of more than one defect.9
The Nkx2-5+/- F2 population retains manageable complexity within circumscribed boundaries of genetic and environmental variation. The population frequency of each of the two alleles at all polymorphic loci is one half, which eliminates rare polymorphisms. The F1 parents are housed under uniform conditions, leaving parental age and litter size as the only environmental variables that are not held constant during embryonic development. The multifactorial basis of an occurrence of a congenital heart defect seems undisputable. Whether the factors can be systematically delineated is unknown, however, and cannot be known short of direct investigation in a large experiment. Given prior evidence suggesting that genetic factors contribute to specific defects, we focused upon membranous and muscular VSDs in the Nkx2-5+/- F2 population. VSDs are common and easily phenotyped as a binary trait, features that facilitate the analysis of an immensely complex biological system.
Methods
Mouse breeding, collection, and phenotyping
Nkx2-5+/- males in the C57Bl/6 background and wild-type FVB/N females were crossed to produce F1 hybrids. The C57Bl/6 and FVB/N inbred strains were obtained from Charles River Laboratories. Nkx2-5+/- F1 animals were intercrossed to generate F2 progeny. Genomic DNA was isolated from every animal by phenol-chloroform extraction. The Nkx2-5 knockout allele was generated and genotyped as previously described.10 Animals were housed in the same room with a 12 hour day-night cycle and fed the same chow. The experiments were approved by the animal studies committee at Washington University School of Medicine.
Newborn F2 pups (N > 5000) were collected and euthanized within hours of birth. The hearts were dissected and prepared for histology. Neonatal hearts were completely sectioned in the frontal plane. The sections of >3100 Nkx2-5+/- hearts were examined for defects, as previously described.9
Demographic and phenotypic information for each pup was entered into a database., The date of birth, the size of the litter, the parents’ identities and their dates of birth, Nkx2-5 genotype and cardiac diagnoses were recorded.
SNP Genotyping
SNP markers were chosen for the 19 autosomes at an average density of 15-20 cM (N = 120) from the Mouse Genome Informatics database. The density of markers was increased to ~5 cM around mapped loci. SNPs were genotyped on the X (N =3) and Y (N = 1) chromosomes for mapping the X chromosome and gender assignment. Multiplexed SNP genotyping was performed on a Sequenom MassARRAY system. Samples were analyzed on 96-well plates that included two controls of each parental strain and an F1 hybrid. The failure rate for SNP genotyping was 2.3 ± 2.6% (SD).
Genetic Linkage Analyses
Genetic linkage analyses were conducted using R/QTL to map loci that have an independent or main effect on membranous or muscular VSD susceptibility in one-way scans.11 Of the Nkx2-5+/- F2 pups SNP genotyped, 233 had a membranous VSD, 80 a muscular VSD, and 284 a structurally normal heart. Interval mapping using the Expectation-Maximization algorithm on a binary model yielded LOD scores representing the probability of linkage at each SNP marker and imputed genotypes at 5 cM intervals between markers. Permutation analysis was performed on the entire dataset or selected chromosomes to determine genome-wide or sub-genomic significance thresholds, respectively. A minimum of 2000 permutations were tested; performing up to 10000 did not appreciably affect genome-wide thresholds.11 The power to detect a main effect locus was calculated using R/qtlDesign.12
A two-way scan was conducted to identify additive or epistatic interactions between locus pairs. Each SNP and imputed marker was tested in combination with every other marker for correlation with phenotype. As a locus can have a main or epistatic effect or both, the data were fit to models that include zero, one, or two main effect loci with or without an epistatic interaction between two loci. LOD scores were calculated for each model, i.e., full (two main effect loci, one interaction between them), additive (two loci, no interaction), interactive (no locus, one interaction), Fv1 (one locus, one interaction) and Av1 (one locus, no interaction). Significant interactions are determined by comparison of models and their significance thresholds. Genome-wide significance thresholds were determined by permutation analysis.11
Genetic linkage analysis for membranous VSD modifier loci was performed with maternal age as an additive or interactive covariate using R/QTL.11 Additive covariate analysis accounts for potential residual variance, whereas the interactive covariate analysis detects gene by covariate specific interactions.
Statistical analyses of environmental modifiers
Multiple logistic regression analyses using the R statistical software package were performed to determine effect of three independent variables, i.e., litter size and maternal and paternal age, on the dichotomous dependent variable, i.e., VSD present or normal. The membranous and muscular VSD phenotypes were analyzed separately. Parental age odd ratios are expressed with age in terms of a 30-day month. Statistical significance was defined by P < 0.05.
A multiple logistic regression model was developed to portray the combinatorial effects of genetic and environmental modifiers on membranous VSD risk. The four independent risk factors were the genotypes at the chromosomes 6, 8 and 10 membranous VSD modifier loci and maternal age. The model was based upon a cohort of 2437 Nkx2-5+/- F2 animals that had a membranous VSD or normal heart. As not all normal animals are SNP genotyped, their genotypes are deemed “missing at random”.13 The missing genotypes were imputed based upon the distribution in the 284 normal animals genotyped for linkage analysis. Multiple logistic regression on the imputed or known genotypes at the three modifier loci and the maternal age for each F2 pup yielded an estimate of the odds ratio for each risk factor in the model. Fifty iterations of the regression analysis with multiple, independent imputations for each ungenotyped normal animal were performed to establish confidence intervals.13, 14
Copy Number Variation Analysis
Chromosomal aneuploidy and copy number variation (CNV) in the Nkx2-5+/- C57Bl/6 X FVB/N F2 population was evaluated using the SNP allele ratio (SAR) algorithm in the R.SQNM software application. The method plots the amounts of PCR product for the two alleles of a SNP assay against each other across the entire genotyped population. The distributions of the three genotypes, i.e., homozygous for either parental allele or heterozygous, are then determined. A genotype that falls outside the distributions is flagged as an outlier or potential CNV.15 Mixtures of FVB/N and C57Bl/6 genomic DNA in 1:2, 1:3, 2:1 and 3:1 ratios served as positive controls.
Results
Multiple modifier loci influence susceptibility to membranous VSD in Nkx2-5+/- animals
To map the genetic modifiers of VSD susceptibility in Nkx2-5 mutants, we phenotyped >3100 Nkx2-5+/- F2 progeny of the C57Bl/6 X FVB/N F1 X F1 intercross. The incidences of specific malformations were previously reported for about one half of the animals phenotyped for this study.9 We mapped modifier loci for membranous and muscular VSDs separately, based upon evidence that they have distinct sets of genetic modifiers.9 Nkx2-5+/- animals that have structurally normal hearts served as the control population in the linkage analyses.
Genetic linkage analysis for membranous VSD modifier loci revealed three peaks on chromosomes 6 (peak at 104.6 Mb), 8 (60.2 Mb) and 10 (9 Mb) with LOD scores of 5-7 (Fig. 1A). There was no association with gender. The appearance of smaller peaks telomeric to the maximum peak on chromosome 10 suggests the possibility of two or more separate loci. Based on the large sample size and marker density, the power of detecting a locus that has a LOD score of 3.5 is 97%. Any additional loci, should they exist, likely have a small effect.
Figure 1.
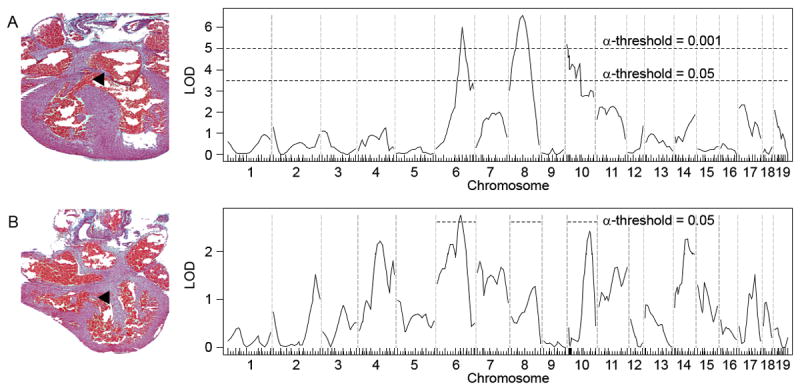
Genetic linkage analyses for loci that modify membranous and muscular VSD susceptibility in Nkx2-5+/- animals from the C57Bl/6 X FVB/N F2 population. An example of each VSD type is shown. (A) At least three significant membranous VSD modifier loci exist on chromosomes 6, 8 and 10. Genome-wide significance thresholds are indicated by the dotted lines (α = 0.001 and 0.05). (B) Genetic linkage analysis for muscular VSD modifier loci reveals a significant overlap of the chromosome 6 peak with a membranous VSD locus. The significance thresholds shown were determined by permutation of genotypes on chromosomes 6, 8 and 10, which contain the membranous VSD modifier loci. N = 233 membranous VSDs, 80 muscular VSDs, and 284 structurally normal hearts.
Previously, backcrosses of the Nkx2-5+/- C57Bl/6 X FVB/N F1 animals to the parental strains indicated that each strain carries either a protective or susceptibility allele at individual modifier loci.9 Effect plots at the three membranous VSD modifier loci demonstrate this. C57Bl/6 and FVB/N bear the recessive, susceptibility alleles at the chromosomes 6 and 8 loci, respectively (Fig. 2). A single copy of the dominant, protective allele negates the effect of the susceptibility allele at either locus. In contrast, the chromosome 10 locus bears a semi-dominant pattern of inheritance. C57Bl/6 and FVB/N respectively carry the susceptibility and protective alleles, and heterozygosity is associated with intermediate risk.
Figure 2.
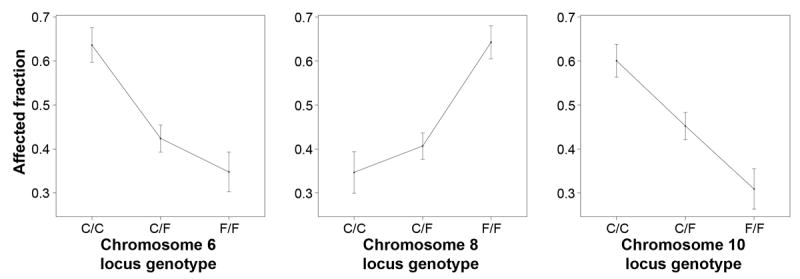
Effect plots demonstrate the patterns of inheritance at membranous VSD modifier loci mapped in the C57Bl/6 X FVB/N F2 population. The fraction of Nkx2-5+/- animals genotyped in the linkage analysis that have a membranous VSD is plotted with respect to the SNP genotype at the locus peak. C/C, C57Bl/6 homozygote; C/F, C57Bl/6-FVB/N heterozygote; F/F, FVB/N homozygote.
Genetic differences between muscular and membranous VSDs
Mutations of genes like Nkx2-5 cause pleiotropic defects presumably because the genes affect diverse cardiac developmental pathways. Modifier genes make the pathways more or less susceptible to the causative mutation. To determine whether the pathways can be distinguished for anatomically distinct defects in the ventricular septum, we mapped muscular VSD modifier loci. Linkage analysis yielded no muscular VSD modifier locus surpassing the genome-wide significance threshold probably in part because of the smaller sample size. (The incidence of muscular VSD is one third that of membranous VSD in the C57Bl/6 X FVB/N F2 population.) Again, no association with gender was observed.
Nevertheless, comparison of the muscular and membranous VSD linkage plots indicates that the two defects have distinct sets of modifiers except perhaps for a locus on chromosome 6 (Fig. 1B). When only the three chromosomes that contain membranous VSD loci are considered in a subgenome-wide scan, the muscular VSD chromosome 6 locus centered at 94.6 Mb significantly overlaps with the membranous VSD locus at 104.6 Mb. C57Bl/6 carries the susceptibility allele at this locus for both muscular and membranous VSD. Whether the same gene on chromosome 6 affects both membranous and muscular VSD susceptibility, however, remains to be determined. The sample size is sufficiently large such that there is a 97% likelihood of detecting a locus on chromosomes 6, 8 or 10 that has a LOD score of 2. Therefore, the membranous VSD modifier loci on chromosomes 8 and 10 either do not affect muscular VSD susceptibility or have very small effects.
Epistatic interactions between membranous VSD modifier loci are not detected
The genes that underlie the VSD modifier loci interact epistatically with the Nkx2-5 mutant allele; they have no apparent effect on the wild type. Epistatic interactions between modifier genes would add an additional layer of complexity to an already complex trait. In the case of membranous VSD, segregation analysis of the Nkx2-5+/- animals in the C57Bl/6 background and C57Bl/6 X FVB/N F1 and F2 crosses suggested that no epistatic interaction exists between the modifiers.9 Consistent with this prediction, a two-way genome scan between all potential locus pairs revealed no significant epistatic interaction (Fig. 3). A more limited scan to detect loci that interact with the chromosomes 6, 8 or 10 loci also found none, although each locus has additive effects with the others on membranous VSD susceptibility (Fig. 3). The smaller number of muscular VSDs precludes a two-way genome scan for epistasis, which segregation analysis suggests may exist for Nkx2-5+/- animals in the C57Bl/6 X FVB/N backgrounds.9
Figure 3.
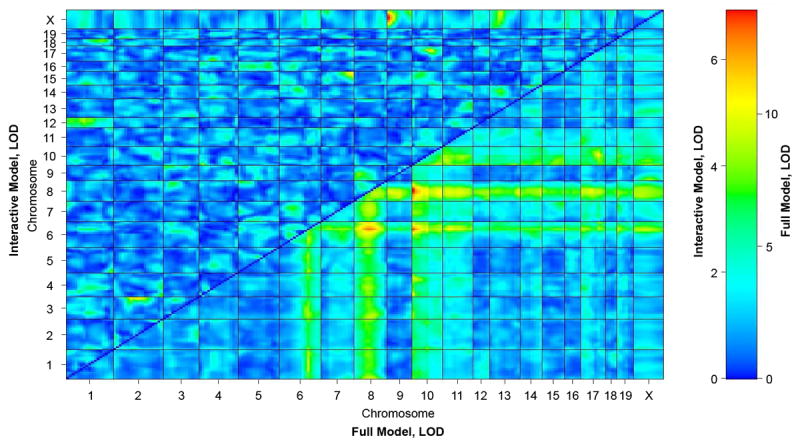
A two-way scan reveals additive but no epistatic interactions between membranous VSD modifier loci. Data are fit to models that assume the presence of zero, one, or two main effect loci with or without epistatic interaction. For an epistatic interaction to be significant, a model that includes the interaction term must fit the data better than one that does not. The full model includes two main effect loci and an interaction between them. The interactive model includes an interaction between two loci but no main effect of either. The LOD score of each marker pair is plotted for the interactive and full models above and below the diagonal, respectively. Significant epistatic interactions must satisfy the conditions that (1) Full LOD > 8.51 and (2) Interactive LOD > 6.1 or Fv1 LOD > 6.69; Fv1 LOD scores, which represent a model of a locus that has a main effect and an interaction with another locus that has no main effect, are not shown.
Maternal age affects the susceptibility to VSD in Nkx2-5+/- animals
Epidemiologists parse naturally heterogeneous environments to discover factors that are associated with but do not necessarily cause disease. In contrast, variation is intentionally eliminated in laboratory experiments. Certain variables in our experiment, however, could not be easily held constant. Even though the parents were housed under identical conditions, three environmental factors – maternal age, paternal age and litter size – vary among gestating F2 pups. The effects of the three variables were examined in multiple logistic regression analyses on membranous and muscular VSD risk.
To our surprise, we discovered a significant relationship between maternal age and VSD risk. Older mothers produce Nkx2-5+/- offspring that have a higher risk of either a membranous or muscular VSD (P = 1.7 × 10-4 and 8.5 × 10-4, respectively; Table 1). The increase in risk per month of maternal aging is small, but the cumulative risk over the reproductive lifetime of a mother becomes sizable, as plotted for membranous VSD (Fig. 4). Paternal age and litter size had marginally significant negative correlations with the incidence of muscular and membranous VSD, respectively (Table 1). None of the variables had an effect in Nkx2-5 wild-type animals. Maternal age is thus a modifying but not causative factor for VSD in our experimental model. Maternal age could reflect another variable more directly related to risk, such as the number of preceding litters. This possibility cannot be excluded because maternal age and parity are strongly correlated. The effect of either paternal age or litter size on VSD risk is inconsistent or marginal.
Table 1.
Multiple logistic regression analysis reveals a significant association between maternal age and the incidence of membranous and muscular VSD in Nkx2-5+/- animals from the C57Bl/6 X FVB/N F2 population. Each month of maternal aging is associated with a small increase in risk. The effect of paternal age or litter size is of questionable or marginal significance
| Factor | Membranous VSD | Muscular VSD | ||||
|---|---|---|---|---|---|---|
| OR | 95% CI | P | OR | 95% CI | P | |
| Maternal age, months | 1.11 | 1.05-1.17 | 1.7 × 10-4 | 1.22 | 1.09-1.38 | 8.5 × 10-4 |
| Paternal age, months | 0.98 | 0.94-1.03 | NS | 0.89 | 0.79-0.99 | 0.043 |
| Litter size, N | 0.948 | 0.880-1.000 | 0.051 | 1.036 | 0.932-1.147 | NS |
OR, odds ratio. CI, confidence interval. NS, not significant.
Figure 4.
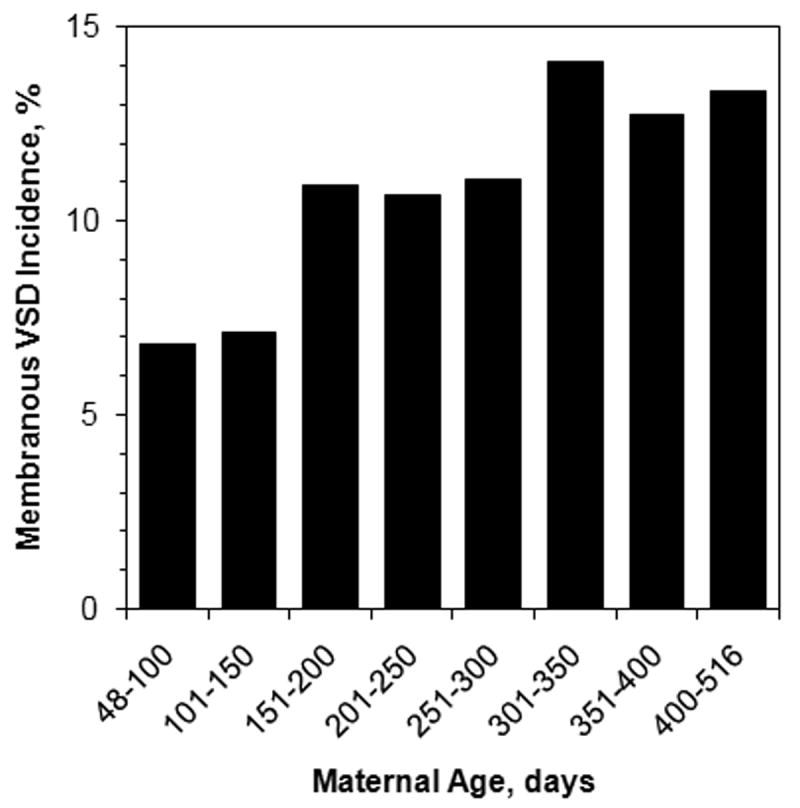
The incidence of membranous VSD in Nkx2-5+/- animals from the C57Bl/6 X FVB/N intercross is significantly correlated with the age of the mother, as determined by multiple logistic regression analysis (P = 1.7 × 10-4).
A biologic factor related to maternal aging must influence cardiac development in Nkx2-5 mutant embryos. In addition, some embryos could be more or less genetically susceptible to the factor. To evaluate this possibility, linkage analyses were performed with maternal age as an additive or interactive covariate, which accounts for the effect of maternal age independent of or dependent upon the genotype at a locus. No significant interaction was detected between maternal age and embryo genotype at any locus in the genome (data not shown). This is consistent with the similar distribution of genotypes at all modifier loci regardless of maternal age (Fig. S1). Thus, while a maternal age-dependent factor must interact with a gene product in the embryo to influence cardiac development, the interaction appears independent of any genetic polymorphism in an F2 embryo, including ones in modifier loci.
The genotypes at the chromosomes 6, 8 and 10 modifier loci and maternal age are independent variables that affect the incidence of membranous VSD caused by Nkx2-5 mutation. To illustrate in concrete terms how the set of modifying factors influence risk, we developed a multiple logistic regression model that estimates the incidence of membranous VSD as a function of various combinations of susceptibility genotypes at the three modifier loci and maternal age (Fig. 5). Spanning an order of magnitude, the incidences from low- to high-risk groups strikingly depict the influence of modifying factors among individuals who carry the identical disease-causing mutation. As indicated by the odds ratios, the incremental risk associated with one month of maternal aging is smaller than the effect of a susceptibility genotype at any one of the three membranous VSD modifier loci (Fig. 5). The maternal age risk accrues over time, however, and can surpass the risk at any genetic locus, which remains constant. The number of months of maternal aging that would equate with the incremental risk of a susceptibility genotype is about 4, 8, and 6 for the chromosomes 6, 8 and 10 loci, respectively. For perspective, the mean age of each pup’s mother in this study is 195 ± 98 days (SD) with a range of 48-516 days.
Fig. 5.
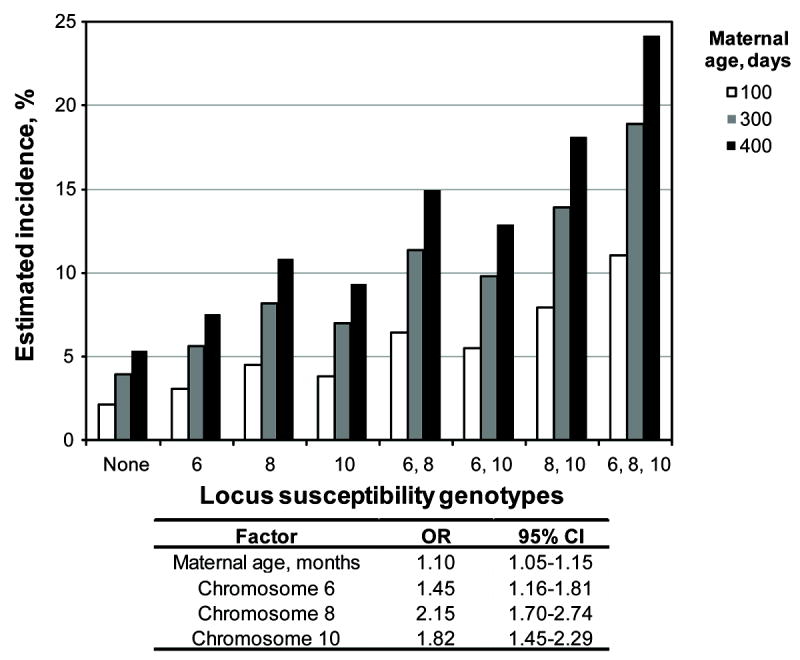
A multiple logistic regression model estimates the incidence of membranous VSD in the Nkx2-5+/- F2 population as a function of bearing a susceptibility genotype at none, one or more of the chromosomes 6, 8, and 10 loci and maternal age. The incremental risk associated with one month of maternal aging is small relative to a susceptibility genotype at any locus, but the cumulative risk associated with maternal aging can surpass any genetic effect, which remains constant. OR, odds ratio. CI, confidence interval.
Chromosomal aneuploidy is not the basis of the maternal age effect
Chromosomal aneuploidy is a well-known phenomenon associated with oocyte aging that can cause developmental defects. We hence looked for copy number variation at a SNP or set of contiguous SNPs by measuring the ratios of the amount of PCR product for each SNP allele obtained during genotyping. None of ~250 Nkx2-5+/- F2 animals, whether normal or affected, or 2 F1 parental controls showed a deviation from the expected ratio for a heterozygous (1 C57Bl/6 allele:1 FVB/N allele) or homozygous (2 C57Bl/6 or FVB/N alleles) at any SNP marker, let alone set of SNPs on a chromosome. In contrast, control samples in which C57Bl/6 and FVB/N genomic DNA were mixed in unequal ratios to simulate aneuploidy were readily identified (Fig. 6). On average, 6.3 ± 0.7 of 8 controls (i.e., two each at 2:1, 3:1, 1:2 and 1:3 ratios of C57Bl/6 and FVB/N DNA) were flagged as potential CNVs at any particular SNP. Thus, chromosomal aneuploidy or acquired copy number variants that span SNP markers, i.e., ~20 cM or more, cannot account for the maternal age effect.
Figure 6.
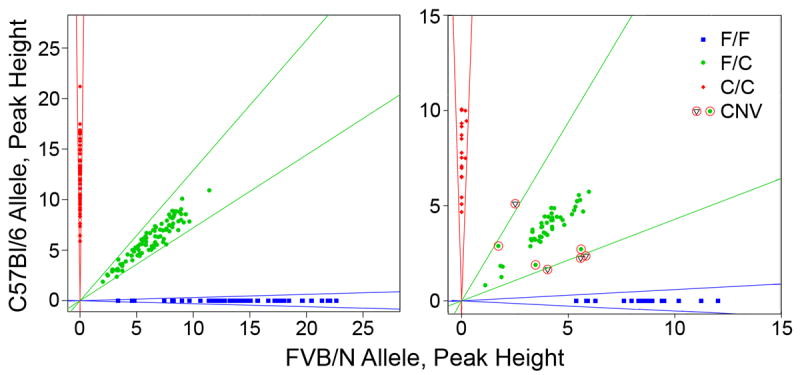
Chromosomal aneuploidy or CNVs cannot account for the increased incidence of VSD in Nkx2-5+/- animals born to older mothers. Representative plots of the PCR products for the two alleles of a SNP genotyping assay are shown. In a typical 96-well assay (left panel), no animal is flagged as having a SNP CNV. In contrast (right panel), control samples that contain unequal mixtures of parental DNA in 2:1 or 3:1 ratios are consistently flagged as potential CNVs, based on a SNP allele ratio that falls below a defined clustering strength (circled circles) or lies outside the Gaussian distribution cluster (circled triangles) for heterozygotes (C57Bl/6:FVB/N genotype, C/F) or homozygotes (C57Bl/6 or FVB/N, C/C or F/F).
Discussion
The notion that congenital heart disease has a multifactorial basis blurs two distinct hypotheses. The commonly considered hypothesis supposes the existence of many causes of congenital heart disease in a population. The multiple-etiology hypothesis is undeniable. The second hypothesis supposes that genetic and environmental factors in addition to the main cause determine the manifestation of congenital heart disease in an individual. The complex trait hypothesis is almost certainly true but receives less attention. The present work directly approaches congenital heart disease as a complex trait. The analysis is simplified by holding the etiology constant, focusing upon ventricular septal defects, and using a mouse model in which the population frequency of either of the two alleles at any polymorphic locus is one half. Despite the anatomic simplicity of VSDs, the analyses reveal substantial complexity, including the interactions of genes and environment with Nkx2-5. The results suggest how gaps in knowledge of pathogenesis may be filled and provide a more nuanced picture of the multifactorial basis of a heart defect.
Despite being one of the first genetic mutations discovered to cause congenital heart disease, only a couple details are known about how an Nkx2-5 mutation might lead to a defect. First, Nkx2-5 represses Bmp2 signaling to regulate proliferation in the second heart field and morphogenesis of the outflow tract.16 Second, a physical interaction between Whsc1 (Wolf-Hirschhorn syndrome candidate 1) and Nkx2-5 could contribute to some of the defects associated with mutations of either gene.17 Contrast this with how well the mechanisms that lead from mutations of Tbx1 to conotruncal defects like tetralogy of Fallot are understood. Key insights into secondary heart field development undoubtedly helped.18 Similarly helpful insights for Nkx2-5 and the pathogenesis of simple VSDs are lacking. The three highly significant membranous VSD modifier loci hence offer footholds into genetic interactions with Nkx2-5 that affect the development of the ventricular septum. The chromosome 6 locus contains no known cardiac developmental genes. The chromosome 8 locus contains two known genes. The locus is centered within 1 Mb of Hand2, also known as dHand. Nkx2-5-/-/dHand-/- mutant embryos completely lack a ventricle, whereas the double heterozygotes are viable.19 Tll1, a transcriptional target of Nkx2-5, is located within 6 Mb of the peak.20 Tll1-/- embryos have abnormal or underdeveloped muscular ventricular septae.21 The chromosome 10 locus contains two genes, Cited2 and Hey2, 8 and 21 Mb away from the peak. Mutations of Cited2 and Hey2 cause abnormal cardiac development in man and mouse models.22-24 A Mouse Genome Informatics query revealed no non-synonymous polymorphism between C57Bl/6 and FVB/N in any of the known cardiac developmental genes. The mostly circumstantial evidence for them as potential modifiers should raise skepticism that any actually underlies a locus.
An unbiased genetic strategy thus seems best suited to discover genes not currently suspected to be involved in cardiac development. The number of undiscovered genes is unknown, but linkage analysis can offer a minimum estimate for individual cardiac phenotypes. In the case of membranous VSD, the three modifier loci mapped in the C57Bl/6 X FVB/N intercross largely account for the low incidence previously reported in Nkx2-5+/- F1 animals, as illustrated by the low risk groups in Fig. 6. Different polymorphic loci would likely be mapped in other genetic strain backgrounds. The total number of genetic modifiers for all defects could be large, given that undetected loci clearly exist for muscular VSD in the C57Bl/6 X FVB/N intercross.
Whatever the VSD modifier genes are, their polymorphisms render specific developmental pathways more or less sensitive to the effect of Nkx2-5 mutation. The genes must comprise functional modules, sets of genes related by a biological function or mutant phenotype, that are involved in ventricular septal development.25 The modules are poorly defined, but one may intuit that they serve pathways related to myocardial growth, tissue remodeling or fusion of endocardial cushions between the inflow and outflow tracts.26 Some modules may be deployed in the development of a single cardiac structure, whereas others may be used in several. For example, a chromosome 6 locus may represent a module important for the growth and integrity of the ventricular septum, as the susceptibility genotype appears to increase the risk of both membranous and muscular VSD in Nkx2-5+/- animals. On the other hand, loci on chromosomes 8 and 10 may subserve modules specific to closure of the membranous ventricular septum but not the integrity of the muscular septum. The elucidation of the modifier genes and their functional modules should clarify mechanisms of normal development and pathogenesis, as has been the case for diseases classified as ciliopathies, channelopathies, and cohesinopathies.25 Identification of modifier genes via the mapping of advanced intercross lines or combined crosses, high-throughput sequencing of embryonic cardiac mRNA, or other complementary methods will help to define the functional modules relevant to congenital heart disease. Once a modifier gene is identified, the inheritance patterns of the alleles, e.g., dominant, recessive or co-dominant as defined by the linkage studies, can help to dissect functional mechanisms.
The present analyses have features of genetic and epidemiologic studies. We expected that genetic loci would be mapped but were truly surprised to discover that maternal age affects the risk of membranous and muscular VSD. The higher incidence of either VSD in Nkx2-5+/- pups from older mothers indicates a genuine biological basis for the commonly reported correlation between maternal age and the risk of congenital heart disease that is independent of any chromosomal abnormality in humans.27-33 The result is not consistently observed, however, highlighting the difficulty of establishing the truth of epidemiologic findings by epidemiology alone.32, 34, 35 Variation in a genetic predisposition to congenital heart disease between the surveyed populations could contribute to the inconsistent correlation, just as the effect of maternal age is observed in Nkx2-5+/- but not wild-type pups.
The possibility that the maternal age effect may have the same basis in mice and humans should motivate investigation of the mechanistic basis. No genetic factor in the offspring, such as chromosomal aneuploidy or polymorphic locus, was found as an explanation. The biologic basis of the effect could reside in the oocyte or the uterine milieu of the developing embryo. In either case the “experimental epidemiologic” findings lend themselves to testable hypotheses. For example, an ovarian transfer experiment between young and old mouse mothers could localize the maternal age factor. The experiment could never be done in humans, but the results would constrain hypotheses regarding genetic or epigenetic alterations in the offspring or humoral factors in the mother that could be examined in a clinical setting.
Many genetic mutations are known to cause congenital heart disease, but human patients and animal models rarely follow strict Mendelian patterns of inheritance. The present work illuminates the genetic and environmental sources of complexity for cardiac developmental traits. When as few as four factors can have a meaningful impact on the risk of membranous VSD in an Nkx2-5+/- population, modifiers should warrant as much consideration as the causes of congenital heart disease. Even if an embryo’s genotype at a locus or a mother’s age cannot be changed, targeting the affected molecular pathways could in principle reduce the burden of congenital heart disease.
Supplementary Material
Congenital heart disease is a complex trait. The same deleterious mutation can cause presentations ranging from normal to life-threatening because modifying factors also influence phenotype. The modifiers have received less scrutiny than the causes not least because their analysis is far more challenging in heterogeneous populations. Nevertheless, the factors, especially ones that reduce risk, may suggest preventive strategies that a focus on monogenic causes has not. To begin to delineate the modifiers, we studied newborn mouse pups that carry a heterozygous knockout mutation of Nkx2-5. Mutations of this cardiac transcription factor cause pleiotropic heart defects in human and mouse. We enumerated the genetic and environmental factors that modify the risk of ventricular septal defects (VSD) in the progeny of a cross between two inbred strains. Genetic linkage analysis revealed three membranous VSD susceptibility loci on chromosomes 6, 8, and 10. The chromosome 6 locus may also influence muscular VSD susceptibility, but the other two loci do not. Interestingly, we observed that the age of the mother affects the risk of membranous and muscular VSD too. The maternal age effect is unrelated to chromosomal aneuploidy, which corroborates a common but unexplained epidemiologic observation. The effects of the genetic loci and maternal age explain the variation in risk of membranous VSD in this animal model, which spans nearly an order of magnitude between low-and high-risk groups. Characterization of the pathways that underlie modifying factors in a mouse model may help to guide future human studies.
Acknowledgments
We thank James Cheverud, D.C. Rao and David Wilson for their expert advice and support. We appreciate the use of the Morphology Core at the Washington University Digestive Diseases Research Core Center (DDRCC).
Funding Sources: J.B. Winston and C.E. Schulkey were supported by Ruth L. Kirschstein National Research Service Awards from the Developmental Cardiology and Pulmonary Training Program (NIH T32 HL007873). P.Y. Jay was a scholar of the Child Health Research Center of Excellence in Developmental Biology at Washington University School of Medicine (NIH K12-HD001487). This work was supported by the American Heart Association, Children’s Discovery Institute, Children’s Heart Foundation, Hartwell Foundation, March of Dimes (1-FY07-453) and NIH (HL105857) (PYJ). The DDRCC is supported by the NIH (P30 DK52574).
Footnotes
Conflict of Interest Disclosures: None.
References
- 1.Nora JJ. Multifactorial inheritance hypothesis for the etiology of congenital heart diseases. The genetic-environmental interaction. Circulation. 1968;38:604–617. doi: 10.1161/01.cir.38.3.604. [DOI] [PubMed] [Google Scholar]
- 2.Wessels MW, Willems PJ. Genetic factors in non-syndromic congenital heart malformations. Clin Genet. 2010;78:103–123. doi: 10.1111/j.1399-0004.2010.01435.x. [DOI] [PubMed] [Google Scholar]
- 3.Benson DW. Genetic Origins of Pediatric Heart Disease. Pediatr Cardiol. 2009;31:422–429. doi: 10.1007/s00246-009-9607-y. [DOI] [PubMed] [Google Scholar]
- 4.Moon A. Mouse models of congenital cardiovascular disease. Curr Top Dev Biol. 2008;84:171–248. doi: 10.1016/S0070-2153(08)00604-2. [DOI] [PubMed] [Google Scholar]
- 5.Horsthuis T, Christoffels VM, Anderson RH, Moorman AF. Can recent insights into cardiac development improve our understanding of congenitally malformed hearts? Clin Anat. 2009;22:4–20. doi: 10.1002/ca.20723. [DOI] [PubMed] [Google Scholar]
- 6.Benson DW, Silberbach GM, Kavanaugh-McHugh A, Cottrill C, Zhang Y, Riggs S, et al. Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J Clin Invest. 1999;104:1567–1573. doi: 10.1172/JCI8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McElhinney DB, Geiger E, Blinder J, Benson DW, Goldmuntz E. NKX2.5 mutations in patients with congenital heart disease. J Am Coll Cardiol. 2003;42:1650–1655. doi: 10.1016/j.jacc.2003.05.004. [DOI] [PubMed] [Google Scholar]
- 8.Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, et al. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science. 1998;281:108–111. doi: 10.1126/science.281.5373.108. [DOI] [PubMed] [Google Scholar]
- 9.Winston JB, Erlich JM, Green CA, Aluko A, Kaiser KA, Takematsu M, et al. Heterogeneity of genetic modifiers ensures normal cardiac development. Circulation. 2010;121:1313–1321. doi: 10.1161/CIRCULATIONAHA.109.887687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanaka M, Chen Z, Bartunkova S, Yamasaki N, Izumo S. The cardiac homeobox gene Csx/Nkx2.5 lies genetically upstream of multiple genes essential for heart development. Development. 1999;126:1269–1280. doi: 10.1242/dev.126.6.1269. [DOI] [PubMed] [Google Scholar]
- 11.Broman KW, Sen S. A Guide to QTL Mapping with R/qtl. New York: Springer; 2009. [Google Scholar]
- 12.Sen S, Satagopan JM, Broman KW, Churchill GA. R/qtlDesign: inbred line cross experimental design. Mamm Genome. 2007;18:87–93. doi: 10.1007/s00335-006-0090-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He Y. Missing data analysis using multiple imputation: getting to the heart of the matter. Circ Cardiovasc Qual Outcomes. 2010;3:98–105. doi: 10.1161/CIRCOUTCOMES.109.875658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donders AR, van der Heijden GJ, Stijnen T, Moons KG. Review: a gentle introduction to imputation of missing values. J Clin Epidemiol. 2006;59:1087–1091. doi: 10.1016/j.jclinepi.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 15.Shi T. CNV analysis using Sequenom MassARRAY: An R tutorial. 2008 https://www.mysequenom.com/Applications/Application_Updates_Typeraspx?path=r-scripts.
- 16.Prall OW, Menon MK, Solloway MJ, Watanabe Y, Zaffran S, Bajolle F, et al. An Nkx2-5/Bmp2/Smad1 negative feedback loop controls heart progenitor specification and proliferation. Cell. 2007;128:947–959. doi: 10.1016/j.cell.2007.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nimura K, Ura K, Shiratori H, Ikawa M, Okabe M, Schwartz RJ, et al. A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf-Hirschhorn syndrome. Nature. 2009;460:287–291. doi: 10.1038/nature08086. [DOI] [PubMed] [Google Scholar]
- 18.Dyer LA, Kirby ML. The role of secondary heart field in cardiac development. Dev Biol. 2009;336:137–144. doi: 10.1016/j.ydbio.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamagishi H, Yamagishi C, Nakagawa O, Harvey RP, Olson EN, Srivastava D. The combinatorial activities of Nkx2.5 and dHAND are essential for cardiac ventricle formation. Dev Biol. 2001;239:190–203. doi: 10.1006/dbio.2001.0417. [DOI] [PubMed] [Google Scholar]
- 20.Sabirzhanova I, Sabirzhanov B, Bjordahl J, Brandt J, Jay PY, Clark TG. Activation of Tolloid-like 1 gene expression by the cardiac specific homeobox gene Nkx2-5. Dev Growth Differ. 2009;51:403–410. doi: 10.1111/j.1440-169X.2009.01097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark TG, Conway SJ, Scott IC, Labosky PA, Winnier G, Bundy J, et al. The mammalian Tolloid-like 1 gene, Tll1, is necessary for normal septation and positioning of the heart. Development. 1999;126:2631–2642. doi: 10.1242/dev.126.12.2631. [DOI] [PubMed] [Google Scholar]
- 22.Bamforth SD, Braganca J, Eloranta JJ, Murdoch JN, Marques FI, Kranc KR, et al. Cardiac malformations, adrenal agenesis, neural crest defects and exencephaly in mice lacking Cited2, a new Tfap2 co-activator. Nat Genet. 2001;29:469–474. doi: 10.1038/ng768. [DOI] [PubMed] [Google Scholar]
- 23.Sperling S, Grimm CH, Dunkel I, Mebus S, Sperling HP, Ebner A, et al. Identification and functional analysis of CITED2 mutations in patients with congenital heart defects. Hum Mutat. 2005;26:575–582. doi: 10.1002/humu.20262. [DOI] [PubMed] [Google Scholar]
- 24.Sakata Y, Koibuchi N, Xiang F, Youngblood JM, Kamei CN, Chin MT. The spectrum of cardiovascular anomalies in CHF1/Hey2 deficient mice reveals roles in endocardial cushion, myocardial and vascular maturation. J Mol Cell Cardiol. 2006;40:267–273. doi: 10.1016/j.yjmcc.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 25.Zaghloul NA, Katsanis N. Functional modules, mutational load and human genetic disease. Trends Genet. 2010;26:168–176. doi: 10.1016/j.tig.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Webb S, Brown NA, Anderson RH. Formation of the atrioventricular septal structures in the normal mouse. Circ Res. 1998;82:645–656. doi: 10.1161/01.res.82.6.645. [DOI] [PubMed] [Google Scholar]
- 27.Forrester MB, Merz RD. Descriptive epidemiology of selected congenital heart defects, Hawaii, 1986-1999. Paediatr Perinat Epidemiol. 2004;18:415–424. doi: 10.1111/j.1365-3016.2004.00594.x. [DOI] [PubMed] [Google Scholar]
- 28.Hollier LM, Leveno KJ, Kelly MA, McIntire DD, Cunningham FG. Maternal age and malformations in singleton births. Obstet Gynecol. 2000;96:701–706. doi: 10.1016/s0029-7844(00)01019-x. [DOI] [PubMed] [Google Scholar]
- 29.Kidd SA, Lancaster PA, McCredie RM. The incidence of congenital heart defects in the first year of life. J Paediatr Child Health. 1993;29:344–349. doi: 10.1111/j.1440-1754.1993.tb00531.x. [DOI] [PubMed] [Google Scholar]
- 30.Materna-Kiryluk A, Wisniewska K, Badura-Stronka M, Mejnartowicz J, Wieckowska B, Balcar-Boron A, et al. Parental age as a risk factor for isolated congenital malformations in a Polish population. Paediatr Perinat Epidemiol. 2009;23:29–40. doi: 10.1111/j.1365-3016.2008.00979.x. [DOI] [PubMed] [Google Scholar]
- 31.Miller A, Riehle-Colarusso T, Siffel C, Frias JL, Correa A. Maternal age and prevalence of isolated congenital heart defects in an urban area of the United States. Am J Med Genet A. 2011;155A:2137–2145. doi: 10.1002/ajmg.a.34130. [DOI] [PubMed] [Google Scholar]
- 32.Pradat P, Francannet C, Harris JA, Robert E. The epidemiology of cardiovascular defects, part I: a study based on data from three large registries of congenital malformations. Pediatr Cardiol. 2003;24:195–221. doi: 10.1007/s00246-002-9401-6. [DOI] [PubMed] [Google Scholar]
- 33.Reefhuis J, Honein MA. Maternal age and non-chromosomal birth defects, Atlanta--1968-2000: teenager or thirty-something, who is at risk? Birth Defects Res A Clin Mol Teratol. 2004;70:572–579. doi: 10.1002/bdra.20065. [DOI] [PubMed] [Google Scholar]
- 34.Baird PA, Sadovnick AD, Yee IM. Maternal age and birth defects: a population study. Lancet. 1991;337:527–530. doi: 10.1016/0140-6736(91)91306-f. [DOI] [PubMed] [Google Scholar]
- 35.Loane M, Dolk H, Morris JK. Maternal age-specific risk of non-chromosomal anomalies. BJOG. 2009;116:1111–1119. doi: 10.1111/j.1471-0528.2009.02227.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.