

Abstract
Defects in several different connexins have been associated with several different diseases. The most common of these is deafness, where a few mutations in connexin (Cx) 26 have been found to contribute to over 50% of the incidence of non-syndromic deafness in different human populations. Other mutations in Cx26 or Cx30 have also been associated with various skin phenotypes linked to deafness (PPK, BPS, VS, KID, etc.). The large array of disease mutants offer unique opportunities to gain insights into the underlying function of gap junction proteins and their channels in the normal and pathogenic physiology of the cochlea and epidermis. This review focuses on those mutants where the impact on channel function has been assessed, and correlated with the disease phenotype, or organ function in knock-out mouse models. These approaches have provided evidence supporting a role of gap junctions and hemichannels in K+ removal and recycling in the ear, as well as possible roles for nutrient passage, in the cochlea. In contrast, increases in hemichannel opening leading to increased cell death, was associated with several KID skin disease/hearing mutants. In addition to providing clues for therapeutic strategies, these findings allow us to better understand the specific functions of connexin channels that are important for normal tissue function.
Keywords: connexin 26, connexin 30, deafness, skin disease, Ca++ waves, K+ recycling, hemichannels
1. INTRODUCTION
As the sole mediators of the direct exchange of ions, signaling molecules (cAMP, IP3, etc), energy sources (ATP, GTP), reducing/oxidizing agents (glutathione) and nutrients (glucose, amino acids) between cells, it is not surprising that gap junctions have been implicated in the homeostatic and integrative function of most tissues. However, unlike other membrane channels and transporters, where the nature of the signals involved are well established, it has been hard to define the specific underlying mechanisms of this integration of cell behavior, as the identification of the relevant gap junction permeants has proven challenging for these relatively non-specific channels. Nonetheless, extensive evidence from several in vitro and mouse studies have shown that different connexin isotypes cannot substitute for one another and create normal tissue functions, demonstrating that not all connexins are created equal. The picture becomes even more complex with the growing recognition that at least some connexin proteins can also form open hemichannels on the plasma membrane under certain physiological conditions (reduced extracellular Ca++, depolarizing membrane potentials and membrane stretch – [1-3]). This would provide one of the few known routes for the exchange of larger molecules between the cytoplasm and extracellular space. In fact, they have been implicated in ATP release associated with Ca2+ wave propagation [4], nicotinamide-adenine dinucleotide (NAD+) release to allow access to CD38 and conversion to cADP ribose [5], and PGE2 release to regulate bone formation [6]. In addition, the cytoplasmic domains of several connexins, particularly Cx43, have been shown to interact with a number of important structural and signaling molecules (reviewed in Ref. [7]), suggesting additional pathways by which they may be able to influence cell behavior.
Given the broad spectrum of roles connexins can play in integration of cell behavior, it is not surprising that mutations associated with several members of the connexin family have been associated with a diverse array of diseases affecting many tissues (neuronal myelin, eye (lens) and ear (cochlea) structure and function, connective tissue, cardiac function, etc. (reviewed in Ref. [8]). These diseases not only challenge us to seek the means to alleviate their symptoms, they also provide a unique window into understanding the specific functions of connexin proteins in different tissues. Nature has provided us a host of mutations that affect different aspects of channel functions. When these are associated with specific patient phenotypes, and accompanying mechanistic studies in mice engineered to express the defective proteins, we take major steps towards defining their mechanism of action in specific tissues. This becomes increasingly important in the case of gap junctions, as most of the powerful genetic model systems (e.g. Drosophila melanogaster and C. elegans), which have provided invaluable insights into function in other gene families, are of limited use in the study of the connexin gene family, as gap junctions in most invertebrates are composed of a topologically similar, but unrelated family called innexins. The relatives of innexins in vertebrates, a 3 gene family called the pannexins, appear not to form gap junctions, but either form surface hemichannels or play as yet undefined roles in intracellular compartments [9, 10]
Perhaps the most widely studied example of connexin associated disease has been the role of Cx26 (GJB2), and to a lesser extent the other β-connexins Cx30 (GJB6), 30.3 (GJB4) and 31(GJB3), in hearing loss. This is in large part due to the sheer frequency of these mutations, five of which can account for large fractions of all cases of prelingual, non-syndromic deafness in different human populations. In addition to these primarily recessive mutants associated with non-syndromic deafness, other mutations with dominant inheritance patterns have been associated with combined deafness and skin disease phenotypes, and yet others cause problems only in the skin. This constellation of mutations, affecting only 3-4 closely related proteins, provides a unique set of “controls” that help in dissecting out which functions of connexins are critical in the two tissues. There have been several excellent reviews on this topic over the last few years [11-13]. Thus, rather than providing an exhaustive listing of all cases of connexin associated deafness and skin disease, we will focus on mutations in each of the above disease types where we have some information on the functional consequences, and attempt to summarize what this has taught us about the likely roles of gap junctions in maintaining normal cochlear and skin functions.
2. CONNEXIN PHENOTYPES AFFECTING THE EAR AND EPIDERMIS
2.1. Cx26 mutations and Non-Syndromic Hearing Loss (NSHL)
1 in every 1000 children could suffer from congenital hearing loss [14], which can be either syndromic or non-syndromic. Non-syndromic hearing loss (NSHL) is characterized by sensorineural hearing loss in the absence of other symptoms, while syndromic hearing loss affects other organ systems, primarily the skin in the case of connexins. The loci linked to non-syndromic hearing loss could be categorized into dominant (DFNA), recessive (DFNB), X-linked (NFDX), Y-linked (NFDY).
Over 80 loci have been linked to nonsyndromic hearing loss, covering a large spectrum of molecules critical for the normal function of the ear [15]. However, mutations in GJB2 (encoding Cx26) account for half of all congenital and autosomal recessive non-syndromic hearing loss (DFNB1) in every tested population [16-24]. To date, there have been over 150 mutations found in the GJB2 gene. Among these mutations, deletion mutations which cause frameshift and/or truncation of the protein at an early stop codon are the most frequent. Several mutations are present with high prevalence, but with different frequencies in different populations. 35delG [17-22, 24], 167delT [23, 25], 235delC [16, 26], R143W [27] and W24X [28, 29] mutations are, respectively, prevalent in Caucasian, Jewish, east Asian, Ghanan, and Indian/Romani gypsy populations. These mutations have carrier frequencies between 1 and 4%, and contribute to between 30 and >80% of the cases of congenital sensorineural deafness in each of these populations. The high frequency of these mutations has been variously attributed to founder effects, or structural features of the genome (e.g. a string of repeated Gs in the case of 35delG) that increase the likelihood of mutation. However, another possibility that has been proposed is that these mutations could confer some selective advantage [13]. 35delG and R75W carriers have been reported to have a thicker epidermis [30, 31], with the latter heterozygotes also showing higher sweat salinity. These phenotypes may confer a higher resistance to injury or microbial infection. This is also consistent with an observation that Cx26 wt, but not the R75W mutant, rendered HeLa cells more subject to invasion by the gastrointestinal pathogen, S. flexneri [32].
Although the most frequently occurring NSHL mutations produce severely truncated proteins due to frameshift or missense mutations, almost 80% of the described deafness mutations are actually single amino acid changes or deletions. As summarized in [12], these mutations have been found in all domains of Cx26. However, the significance of this is unclear without any information on the functional effects of these mutants. Fortunately, the last few years have seen increasing numbers of these mutations characterized by expression in exogenous systems like Xenopus oocytes or transfected mammalian cell lines. Table I summarize all of the NSHL (in black) and syndromic deafness (red) associated mutations that have been functionally tested in this fashion. Some variability in results have been reported by different labs, perhaps because of different expression systems, different modes of characterization or perhaps even variation in expression levels. In these cases, we have reported the result which shows the higher level of function (e.g. formation of junctional plaques over no gap junction formation).
Table 1.
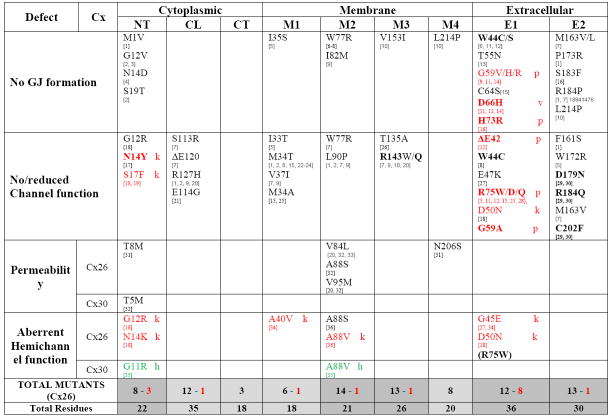
|
Black = Non-syndromic Hearing Loss (NSHL)
 = Syndromic Hearing Loss: K = Keratitis-Icthyosis deafness syndrome (KID); p = PalmoPlanta Keratoderma (PPK); V = Vohwinkel Syndome (VS)
= Syndromic Hearing Loss: K = Keratitis-Icthyosis deafness syndrome (KID); p = PalmoPlanta Keratoderma (PPK); V = Vohwinkel Syndome (VS)
 = hidrotic ectodermal dysplasia (HED)/no hearing loss
= hidrotic ectodermal dysplasia (HED)/no hearing loss
(…) = hemichannels are functional, but not necessarily enhanced compared to wt (gap junctions non-functional)
BOLD – Dominant inhibitory effect on other connexins
NT – amino-terminal domain (20 aa); CL – cytoplasmic loop (38 aa); CT – C-terminal domain (17 aa);
M1 – M4: transmembrane domains 1 through 4 (~20 aa each, except M3 which is ~26 aa);
E1 – E2: extracellular loops 1 (~35 aa) and 2 (~28 aa).
[1] E. Thonnissen, R. Rabionet, M.L. Arbones, X. Estivill, K. Willecke, T. Ott, Human connexin26 (GJB2) deafness mutations affect the function of gap junction channels at different levels of protein expression, Human genetics 111 (2002) 190-197.
[2] P. D’Andrea, V. Veronesi, M. Bicego, S. Melchionda, L. Zelante, E. Di Iorio, R. Bruzzone, P. Gasparini, Hearing loss: frequency and functional studies of the most common connexin26 alleles, Biochemical and biophysical research communications 296 (2002) 685-691.
[3] P.E. Purnick, D.C. Benjamin, V.K. Verselis, T.A. Bargiello, T.L. Dowd, Structure of the amino terminus of a gap junction protein, Archives of biochemistry and biophysics 381 (2000) 181-190.
[4] B. Haack, K. Schmalisch, M. Palmada, C. Bohmer, N. Kohlschmidt, A. Keilmann, U. Zechner, A. Limberger, S. Beckert, H.P. Zenner, F. Lang, S. Kupka, Deficient membrane integration of the novel p.N14D-GJB2 mutant associated with non-syndromic hearing impairment, Human mutation 27 (2006) 1158-1159.
[5] R.S. Mani, A. Ganapathy, R. Jalvi, C.R. Srikumari Srisailapathy, V. Malhotra, S. Chadha, A. Agarwal, A. Ramesh, R.R. Rangasayee, A. Anand, Functional consequences of novel connexin 26 mutations associated with hereditary hearing loss, Eur J Hum Genet 17 (2009) 502-509.
[6] R. Bruzzone, D. Gomes, E. Denoyelle, N. Duval, J. Perea, V. Veronesi, D. Weil, C. Petit, M.M. Gabellec, P. D’Andrea, T.W. White, Functional analysis of a dominant mutation of human connexin26 associated with nonsyndromic deafness, Cell communication & adhesion 8 (2001) 425-431.
[7] R. Bruzzone, V. Veronesi, D. Gomes, M. Bicego, N. Duval, S. Marlin, C. Petit, P. D’Andrea, T.W. White, Loss-of-function and residual channel activity of connexin26 mutations associated with non-syndromic deafness, FEBS letters 533 (2003) 79-88.
[8] P.E. Martin, S.L. Coleman, S.O. Casalotti, A. Forge, W.H. Evans, Properties of connexin26 gap junctional proteins derived from mutations associated with non-syndromal heriditary deafness, Human molecular genetics 8 (1999) 2369-2376.
[9] M. Palmada, K. Schmalisch, C. Bohmer, N. Schug, M. Pfister, F. Lang, N. Blin, Loss of function mutations of the GJB2 gene detected in patients with DFNB1-associated hearing impairment, Neurobiology of disease 22 (2006) 112-118.
[10] G. Mese, E. Londin, R. Mui, P.R. Brink, T.W. White, Altered gating properties of functional Cx26 mutants associated with recessive non-syndromic hearing loss, Human genetics 115 (2004) 191-199.
[11] N.K. Marziano, S.O. Casalotti, A.E. Portelli, D.L. Becker, A. Forge, Mutations in the gene for connexin 26 (GJB2) that cause hearing loss have a dominant negative effect on connexin 30, Human molecular genetics 12 (2003) 805-812.
[12] F. Rouan, T.W. White, N. Brown, A.M. Taylor, T.W. Lucke, D.L. Paul, C.S. Munro, J. Uitto, M.B. Hodgins, G. Richard, trans-dominant inhibition of connexin-43 by mutant connexin-26: implications for dominant connexin disorders affecting epidermal differentiation, J.Cell Sci. 114 (2001) 2105-2113.
[13] S. Melchionda, M. Bicego, E. Marciano, A. Franze, M. Morgutti, G. Bortone, L. Zelante, M. Carella, P. D’Andrea, Functional characterization of a novel Cx26 (T55N) mutation associated to non-syndromic hearing loss, Biochemical and biophysical research communications 337 (2005) 799-805.
[14] T. Thomas, D. Telford, D.W. Laird, Functional domain mapping and selective trans-dominant effects exhibited by Cx26 disease-causing mutations, J.Biol.Chem. 279 (2004) 19157-19168.
[15] A. Oshima, T. Doi, K. Mitsuoka, S. Maeda, Y. Fujiyoshi, Roles of Met-34, Cys-64, and Arg-75 in the assembly of human connexin 26. Implication for key amino acid residues for channel formation and function, The Journal of biological chemistry 278 (2003) 1807-1816.
[16] E.A. de Zwart-Storm, H. Hamm, J. Stoevesandt, P.M. Steijlen, P.E. Martin, M. van Geel, M.A. van Steensel, A novel missense mutation in GJB2 disturbs gap junction protein transport and causes focal palmoplantar keratoderma with deafness, Journal of medical genetics 45 (2008) 161-166.
[17] K. Arita, M. Akiyama, T. Aizawa, Y. Umetsu, I. Segawa, M. Goto, D. Sawamura, M. Demura, K. Kawano, H. Shimizu, A novel N14Y mutation in Connexin26 in keratitis-ichthyosis-deafness syndrome: analyses of altered gap junctional communication and molecular structure of N terminus of mutated Connexin26, The American journal of pathology 169 (2006) 416-423.
[18] J.R. Lee, A.M. Derosa, T.W. White, Connexin mutations causing skin disease and deafness increase hemichannel activity and cell death when expressed in Xenopus oocytes, J.Invest Dermatol. 129 (2009) 870-878.
[19] G. Richard, F. Rouan, C.E. Willoughby, N. Brown, P. Chung, M. Ryynanen, E.W. Jabs, S.J. Bale, J.J. DiGiovanna, J. Uitto, L. Russell, Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome, American journal of human genetics 70 (2002) 1341-1348.
[20] H.L. Wang, W.T. Chang, A.H. Li, T.H. Yeh, C.Y. Wu, M.S. Chen, P.C. Huang, Functional analysis of connexin-26 mutants associated with hereditary recessive deafness, Journal of neurochemistry 84 (2003) 735-742.
[21] Y.H. Choung, S.K. Moon, H.J. Park, Functional study of GJB2 in hereditary hearing loss, The Laryngoscope 112 (2002) 1667-1671.
[22] M. Bicego, M. Beltramello, S. Melchionda, M. Carella, V. Piazza, L. Zelante, F.F. Bukauskas, E. Arslan, E. Cama, S. Pantano, R. Bruzzone, P. D’Andrea, F. Mammano, Pathogenetic role of the deafness-related M34T mutation of Cx26, Human molecular genetics 15 (2006) 2569-2587.
[23] I.M. Skerrett, W.L. Di, E.M. Kasperek, D.P. Kelsell, B.J. Nicholson, Aberrant gating, but a normal expression pattern, underlies the recessive phenotype of the deafness mutant Connexin26M34T, Faseb J 18 (2004) 860-862.
[24] T.W. White, M.R. Deans, D.P. Kelsell, D.L. Paul, Connexin mutations in deafness, Nature 394 (1998) 630-631.
[25] V. Piazza, M. Beltramello, M. Menniti, E. Colao, P. Malatesta, R. Argento, G. Chiarella, L.V. Gallo, M. Catalano, N. Perrotti, F. Mammano, E. Cassandro, Functional analysis of R75Q mutation in the gene coding for Connexin 26 identified in a family with nonsyndromic hearing loss, Clinical genetics 68 (2005) 161-166.
[26] D.L. Beahm, A. Oshima, G.M. Gaietta, G.M. Hand, A.E. Smock, S.N. Zucker, M.M. Toloue, A. Chandrasekhar, B.J. Nicholson, G.E. Sosinsky, Mutation of a conserved threonine in the third transmembrane helix of alpha- and beta-connexins creates a dominant-negative closed gap junction channel, The Journal of biological chemistry 281 (2006) 7994-8009.
[27] B.C. Stong, Q. Chang, S. Ahmad, X. Lin, A novel mechanism for connexin 26 mutation linked deafness: cell death caused by leaky gap junction hemichannels, The Laryngoscope 116 (2006) 2205-2210.
[28] Y. Chen, Y. Deng, X. Bao, L. Reuss, G.A. Altenberg, Mechanism of the defect in gap-junctional communication by expression of a connexin 26 mutant associated with dominant deafness, Faseb J 19 (2005) 1516-1518.
[29] S.W. Yum, J. Zhang, S.S. Scherer, Dominant connexin26 mutants associated with human hearing loss have trans-dominant effects on connexin30, Neurobiology of disease 38 (2010) 226-236.
[30] J. Zhang, S.S. Scherer, S.W. Yum, Dominant Cx26 mutants associated with hearing loss have dominant-negative effects on wild type Cx26, Molecular and cellular neurosciences 47 (2011) 71-78.
[31] G. Mese, V. Valiunas, P.R. Brink, T.W. White, Connexin26 deafness associated mutations show altered permeability to large cationic molecules, Am.J.Physiol Cell Physiol 295 (2008) C966-C974.
[32] Y. Zhang, W. Tang, S. Ahmad, J.A. Sipp, P. Chen, X. Lin, Gap junction-mediated intercellular biochemical coupling in cochlear supporting cells is required for normal cochlear functions, Proc.Natl.Acad.Sci.U.S.A 102 (2005) 15201-15206.
[33] M. Beltramello, V. Piazza, F.F. Bukauskas, T. Pozzan, F. Mammano, Impaired permeability to Ins(1,4,5)P3 in a mutant connexin underlies recessive hereditary deafness, Nat.Cell Biol. 7 (2005) 63-69.
[34] D.A. Gerido, A.M. DeRosa, G. Richard, T.W. White, Aberrant hemichannel properties of Cx26 mutations causing skin disease and deafness, Am.J.Physiol Cell Physiol 293 (2007) C337-C345.
[35] G.M. Essenfelder, R. Bruzzone, J. Lamartine, A. Charollais, C. Blanchet-Bardon, M.T. Barbe, P. Meda, G. Waksman, Connexin30 mutations responsible for hidrotic ectodermal dysplasia cause abnormal hemichannel activity, Human molecular genetics 13 (2004) 1703-1714.
[36] Xu, J. and Nicholson, B.J. - unpublished observations
Based on frequency of mutated residues (shown at the bottom of Table I), M2 shows the highest density of mutations associated with NSHL (67%), followed by M3 (50%), E2 (43%), M4 (40%), NT, CL (36%), M1 and E1 (33%). When the frequency of mutagenesis leading to all forms of deafness, including syndromic forms, is considered, 5 domains show higher than 50% mutation rates (NT, M2, M3, E1 and E2). While still relatively broad, this selective concentration of mutations is likely to have functional significance that could then cast light on the roles played by gap junctions in the ear.
Not surprisingly, the majority of mutants cause generalized folding problems that result in failure in trafficking of Cx26 to the cell surface, or are permissive for the formation of gap junction plaques, but prevent opening of the intercellular channels. As might be expected of mutations that affect folding or conformational changes in a membrane protein, all four transmembrane domains can be affected, although M4 appears to be less important for channel function, consistent with much of the literature. With the exception of the N-terminal domain, which has been clearly implicated in the structure of the pore mouth [33, 34], the cytoplasmic domains do not appear to contribute to the folding and trafficking of the protein, although the cytoplasmic loop region can affect channel opening. Perhaps most striking is the number of mutations in the extracellular domains that can influence both channel function and trafficking. The former is not too surprising, given the importance of these regions in the docking of two hemichannels to form a functional intercellular channel. Most of these mutants, with a few notable exceptions discussed below, have not been tested for their function as hemichannels. However, it is surprising the degree to which these domains also appear to be critical to the correct folding and trafficking of the protein to the cell surface. This may be due to failure of these mutants to form the correct disulfide bonding pattern, which often serves as a measure of “quality control” in the folding of many proteins.
Of greatest interest in terms of shedding light on the role of gap junctions in the ear, are the mutants that affect specific connexin functions. Among NSHL associated mutants, five show selective changes in the permeability of the channels, where ionic conductance and even permeability to anionic fluorescent tracers is maintained, but permeability to larger molecules [cationic tracers (T8M and N206S - [35]) and inositol 1,4,5-trisphosphate (IP3) (V84L, A88S, V95M –[36, 37]] is compromised. The one Cx30 mutant associated with NSHL that has been characterized also appears to be in this category, causing a reduction in IP3 permeability [37]. With one exception, all of these mutations are in the NT and M2 domains. This is consistent with the implication of these domains in the pore in recent structural models [33], although the mutations in M2 all line up on a face of the helix that in the structure face away from the pore. However, the X-ray structure probably represents that of a hemichannel, since the crystals were generated from connexins purified under conditions that typically yield hexamers. By contrast, in situ mapping of the pore domain of intact gap junction channels (Cx32 and 50) indicates that these residues do face the pore in gap junctions ([38], Toloue and Nicholson, unpublished results). These results suggest that passage between cells of specific larger metabolites, rather than ions, is critical to normal hearing. The significance of this to the current models of gap junction function in the ear is discussed below.
2.2. Cx26 mutations and syndromic deafness
GJB2 mutations have also been linked to syndromic hearing loss, which is accompanied by a variety of mild to severe skin disorders. The skin syndromes have been categorized into several types according to their symptoms, including Vohwinkel syndrome (VS), Bart–Pumphrey syndrome (BPS), palmoplanta keratoderma (PPK), keratitis–ichthyosis deafness syndrome (KID), and hystrix-like icthyosis deafness syndrome (HID). The first three of these skin diseases (VS, BPS and PPK) seem to represent a constellation of similar phenotypes, involving palmoplanta hyperkeratosis (thickening of the palms and soles of the feet). VS also includes notable constriction bands around the digits that can lead to auto-amputation, and BPS includes nail thickening and brittleness. KID and HID are similar to one another, but distinct from the other three syndromes, in that skin lesions are not restricted to any one region, and they include hair loss (eyebrows and eyelashes) and icthyosis that causes disruption of the dermal barrier and increased susceptibility to infection that can be lethal. KID also involves extensive keratitis that in severe cases can lead to blindness.
In contrast to mutations associated with NSHL, all mutations causing syndromic deafness are missense mutations and are inherited in a dominant manner [39-42]. Since none of the complete loss of protein mutants associated with NSHL cause skin problems, and Cx26 conditional knockout and Cx30-null mice also exhibit no skin phenotype, it is likely that connexins in skin have redundant functions, so that lack of one type of connexin would not cause skin disorders [43, 44]. Since the connexin alleles causing syndromic hearing loss are dominant, it is plausible to speculate that these mutations cause disease through either a transdominant effect on other connexin proteins expressed in the skin, or through a specific gain-of-function that is not suppressed by any of the other wt connexins expressed in the skin, of which there are nine.
Consistent with the clustering of the different skin syndromes into similar groupings, Cx26 mutations which cause VS-BPS-PPK syndromes show significant overlap, and all have been found to have a transdominant effect on other connexins expressed in the epidermis. These mutations include ΔE42 (PPK), G59A (PPK), D66H (VS) and R75W (PPK) [45, 46]. Of these, G59S is also associated with both VS and BPS [47]. N54 is also associated with BPS or PPK, depending on whether it is mutated to a K or H, respectively [48, 49]. As summarized in Table I, all of these mutations that have been characterized are loss of function mutations with problems in trafficking, or the formation of functional gap junction channels.
Of the Cx26 mutations causing the more severe HID-KID syndromes that have been tested for function, with the exception of S17F, have all been found to increase hemichannel activity (Table I). These mutations include G12R, N14K, A40V, G45E, D50N [50-52] and A88V (Xu and Nicholson, unpublished results). Transgenic expression of Cx26G45E in mice correlated well with human KID pathology. Electrical recordings showed increased hemichannel current [53]. Although all of the mutations which lead to HID-KID cause increased hemichannel opening, they seem to exert their effects through different mechanisms. Cx26A40V alters hemichannel sensitivity to extracellular Ca2+, while Cx26G45E, which was located in the pore using cysteine accessibility (SCAM) studies, increases the hemichannel permeability to Ca2+ [54]. Our unpublished data demonstrates that A88V enhances hemichannel surface expression without affecting other channel properties, thus also causing larger hemichannel currents. Interestingly, other mutations at this site (A88S) that increase hemichannel currents by affecting voltage gating, but not surface expression, result only in hearing defects, as the voltage gating of hemichannels is not a major mode of regulation in the skin. Given that, in almost all exogenous expression systems, these mutants result in compromised cell health due to constitutively leaky hemichannels, it seems likely that cell death is a major source of the etiology of the KID-HID spectrum of syndromes.
2.3 Other Connexin mutations in deafness and skin disease – Cx30, 30.3 and 31
Mutations in three other beta connexin genes, GJB3 (Cx31), GJB4 (Cx30.3) and GJB6 (Cx30) have also been associated with various forms of NSHL (Cx31 E183K, delI141, I141V and delD66; Cx30 T5M)[55-57], syndromic deafness associated with Erythrokeratoderma Variablis (EKV) (Cx31 G12D, R42P and C86S; Cx30.3 F137L)[58, 59] and a form of skin disease without accompanying hearing problems, Hidrotic Ectodermal Dysplasia (HED) (Cx30 G11R and A88V)[60]. EKV presents with hyperkeratotic plaques and variable areas of erythema, while HED (also called Clouston syndrome) involves hair loss, nail hypoplasia, hyper pigmentation and palmoplanta hyperkeratosis. The vast majority of the mutations cause loss of function, with failure of the protein to traffic to the surface. In the cases associated with EKV, this is severe enough that the accumulation of unfolded protein can lead to cell death [61]. In the NSHL cases, whether recessive or dominant, there was typically no compromise to cell health, nor major loss of intercellular coupling. As shown in Table I, the HED associated mutants, like those in Cx26 that caused KID syndrome, caused an increase in hemichannel activity that likely results in increased cell death ([62], Xu and Nicholson, unpublished results).
3. FUNCTION OF CONNEXINS IN THE EAR
3.1 Connexin distribution in the ear
RNA expression for a number of connexins has been detected in the cochlea (Cxs26, 29, 30, 31, 43, 30.2, 37 and 46 – [63, 64]), however, immunocytochemistry has only confirmed expression in a few of these cases. Cx26 and Cx30 are the most abundantly expressed, primarily in the supporting cells, fibrocytes in spiral limbus and spiral ligament, and stria vascularis of both humans and rodents [65-67], but not in the inner or outer hair cells or the marginal cells of the stria vascularis [63, 66-68]. Cx29 is found primarily in myelinating glial cells [69], although it has been detected in low levels in the stria vascularis [70]. Reports of Cx31 expression have been varied, but it has been reported to have some overlapping expression with Cx26 and 30 in fibrocytes of the spiral ligament and limbus [71] and below the spiral prominence [68]. Cx32, 43 and 45 are all expressed in the cochlea earlier in development, but expression is either lost (Cx32 – [72]) or redistributed to the expected locations (bone for Cx43 ([73]), capillaries for Cx45.
In terms of the major connexins, Cx26 and Cx30 show extensive co-localization immunocytochemically [63, 74, 75]. Their co-immunoprecipitation, both from cochlea tissue and exogenously expressing cell lines, also supports the existence of Cx26/Cx30 heteromeric channels [63, 74-76]. At the prenatal stage, staining is concentrated to narrower bands of cells in the spiral limbus and spiral ligament, but absent in the supporting cells [75]. After birth, connexin expression gradually increases in the supporting cells of the cochlea [75, 77]. Prior to P8, immunostaining indicates that only Cx26 is expressed in the Claudius cells, Hensen’s cells and inner sulcus cells, although both Cx30 and Cx26 are distributed in the outer sulcus regions [78]. The most dramatic change postnatally is that Cx30 then replaces Cx26 in the supporting cells surrounding the outer hair cells (Dieters’ and Hensen’s cells) between P0 and P12 [75, 77]. The function of gap junction channels is also very dynamic during development, as suggested by dye transfer studies [77].
3.2 Functional Properties of the major cochlear connexins (Cx26 and 30)
While Cx26 and Cx30 share high sequence homology, they have been found to have quite different permeability properties. Cx26 gap junction channels are readily permeable to the negatively charged dyes, Lucifer yellow, calcein, and carboxyfluorescein, but Cx30 gap junction channels are largely impermeable to these molecules, even when comparisons are made between cells with very similar electrical coupling. In contrast to the anionic dyes, both connexin gap junctions are equally permeable to positively charged dyes like ethidium bromide or propidium iodide [76, 79, 80]. Cx26 was also found to mediate Ca++ wave propagation between cells more efficiently than Cx30 [36]. Based on their extensive co-localization in the cochlea, Cx26 and 30 have also been co-expressed in exogenous systems, yielding single channel conductances and gating behavior not observed between cells expressing only one connexin [76]. This strongly indicates the existence of heteromeric channels, consistent with co-immunoprecipitation of these two connexins in the ear [63, 75, 76]. In terms of permeability to dyes, these heteromeric channels appear to be dominated by Cx26 [75, 76], and show even faster Ca++ wave propagation than cells coupled by Cx26 alone [75]. Thus, it seems possible that heteromeric channels can gain unique permeability properties not shown by either homomeric channel. In situ, it is difficult to predict what proportions of homomeric and heteromeric channels exist, although this is likely to vary between different parts of the cochlea where the relative levels of the two connexins differ, such as Dieters’ cells in adult mice, which primarily express Cx30.
3.3 Insights from mouse genetic models
To understand the physiological functions of the gap junctional network in the cochlea, and how defects in it cause deafness, mouse genetic models have proven invaluable tools. However, the mouse genetic models only reproduce the most profound and simplified symptoms seen in the human patients carrying connexin mutations, thus caution in interpreting them needs to be exercised in light of differences that exist in mouse and human physiology, and how (and when) connexin function was disrupted. In particular, the range of clinical phenotypes resulting from Cx26 mutations is generally much larger than has been reported to date in mice, ranging from pre-lingual to progressive and late onset deafness of varying severity. Such variations have even been seen between siblings with the same Cx26 mutation [81], indicating that some of this variation is due to the diversity of genetic background present in the human population, but not mimicked in lab mouse models.
Several mice lines have been developed to study Cx26 function, but since germline Cx26 knockout is lethal in mice due to its requirement for placental function [82], two conditional Cx26 knockout mice were developed. One is a spatially specific knock out, which used the Otog promoter to specifically delete Cx26 in the epithelial cells. The other is a temporal knockout, which utilized a ligand inducible recombinase to delete Cx26 after E19 [83, 84]. Both mice lines display moderate hearing loss and hair cell degeneration, but the cell death in the latter line is more rapid and extensive. Cell degeneration of spiral ganglion neurons was also observed in the latter line. Cell degeneration in the former line starts from the inner hair cells, while in the latter line it starts form the outer hair cells. The endolymphatic potential (EP) of these mice is minimally affected at P12, when the organ of Corti development is complete and the EP reaches mature levels [85], but drops to one-third wt levels in the adult, with a 50% loss in K+ concentration in this compartment. The significance of this for the function of the ear will be discussed below. Another mouse genetic model of deafness utilized the knock-in of a dominant disease mutation, Cx26R75W. Due to its well characterized dominant-negative effect on gap junction channel function, transgenic expression of the mutant protein [43], or its transient transfection into the ear of adult mice [86], should serve to ablate all endogenous gap junction function. These mice show quite different phenotypes, with the former showing severe hearing loss and outer hair cell loss, and the latter rather minor hearing effects and no loss of hair cells, suggesting that expression of the mutant connexin over the lifetime of the mouse contributes to disease. Other effects on SG neuron degeneration in these models are likely due to their broader expression patterns in the cochlea, compared to the knock-outs, which were targeted to the epithelial system. There are also reports that expression of this mutant can lead to arrested development of the organ of Corti, [87], an effect also reported in some Cx26 conditional knock-outs [88, 89]. This suggests that cx26 signaling may also be important to different aspects of development of this part of the ear.
Cx30 null mice were generated by deletion of the Cx30 coding region [44]. These mice show significant hearing loss, accompanied by hair cell loss, and failure to develop an EP, even though K+ levels are elevated in the endolymph. Hair cell degeneration occurs after P18 and the outer hair cells are damaged more severely. Cx30T5M knock-in mice were also generated due to the interest that this mutation specifically impairs biochemical coupling of the cells, while leaving electrical coupling intact [90]. These mice display mild deafness, but nonetheless show a significant increase of hearing threshold.
Interpretation of these knock-out studies are complicated by the findings that loss of either connexin results in a decrease in the expression level of the other, apparently through a PLC-NF-κB signaling mechanism, although it is not clear how Cx26 and Cx30 expression could affect this pathway [91]. The reciprocal regulation of Cx26 and Cx30 could also be due to their direct interaction, which might stabilize them and extend their half-life on the plasma membrane. Thus, it was unclear if any of these knock-out phenotypes should be interpreted in terms of loss of the properties of a specific connexin (or specific heteromeric forms), or just generalized reductions in coupling. To investigate this, two recent studies tested the effect of replacing one connexin with another. Most instructive was the finding that deafness caused by loss of Cx30 could be corrected by over-expression of Cx26 through a BAC chromosome, thereby re-establishing the original coupling levels, but now with only one connexin isotype [92]. In contrast, BAC expression of Cx30 failed to rescue the morphological or hearing phenotype of Cx26 null mice [78], demonstrating that Cx26 channels are necessary, while Cx30 channels are not sufficient, for normal hearing. It has been suggested that the earlier expression of Cx26 than Cx30 in the supporting cells (Cx30 is first seen at P6) could be the reason why Cx26 is indispensible, and its knock-out causes more severe phenotypes and earlier cell degeneration than seen in Cx30-null mice. Alternatively, the more restrictive permeability of Cx30 channels to larger anions may limit its ability to replace Cx26. In this regard, it has recently been shown that replacement of Cx26 with Cx32 in a knock-out mouse, results in a normal hearing phenotype [93]. Thus, while Cx26 has unique properties that it apparently does not share with Cx30, they are sufficiently mimicked by Cx32 to maintain ear function. Intriguingly, Cx26 and Cx30 share much closer sequence homology than Cx26 has with Cx32, demonstrating that the properties that are important to preserve are likely dictated by a small part of the connexin structure. These results suggest that both net levels of expression of connexins, as well as their specific properties, are critical for the auditory function of the ear.
3.4 Proposed models for gap junction function in the ear – insights from functional deficits of connexin deafness mutants
Cx26 and Cx30 form two gap junction networks in the cochlea: the epithelial cell system and the connective tissue cell system. Epithelial cells include supporting cells from interdental cells medially to the outer sulcus cells laterally. Medially, the epithelial GJ system connects inner hair cells to the endolymph though interdental cells or the extracellular space of the spiral limbus. Laterally, this system connects the basolateral space of outer hair cells to the spiral ligament extracellular space. The GJ system in the connective tissue then connects the extracellular space of spiral ligament to the intrastrial space (Fig. 1A). As most of the GJB2 and GJB6 mutations disrupt gap junction coupling, it is apparent that this network of intercellular communication is important for the normal function of the cochlea. The recessive nature of most connexin deafness mutants indicates that they do not affect the expression or function of other connexins, even the product of the wt allele in a heterozygote. This would indicate that loss of any one connexin is sufficient to induce disease, and there is little “redundancy” in the cochlea, in contrast to skin disease discussed below where most mutants are dominant
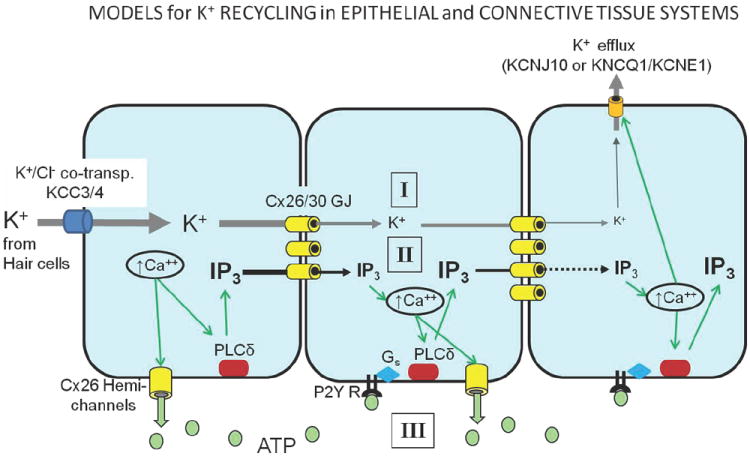
Figure 1.
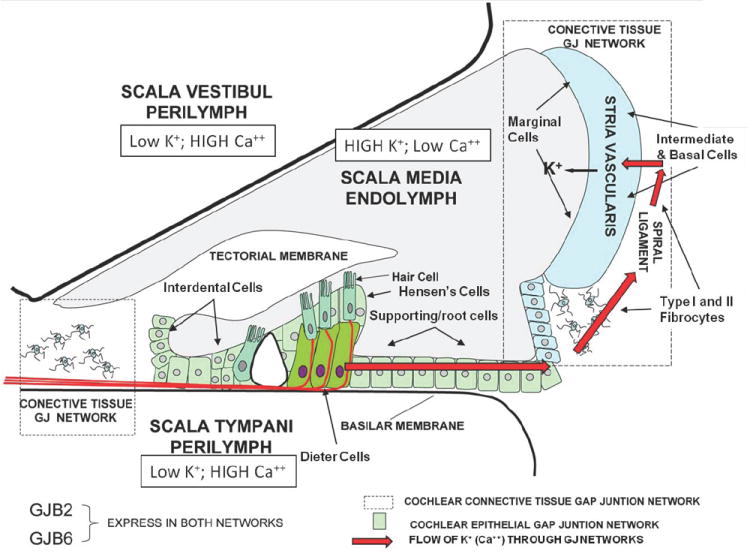
A. Diagram of the cochlea and its various compartments, showing the proposed return of K+ (possibly mediated by a propagated Ca++ wave) from the hair cells to the endolymph (red arrows) through the epithelial (green color) and connective tissue (dotted box) gap junction networks. The former is comprised of Dieters’ cells and supporting cells, while the latter is comprised of types I and II fibrocytes in the spiral ligament and basal and intermediate cells of the stria vascularis. Ultimately, released K+ is taken up by the marginal cells of the stria vascularis and released into the endolymph, thus maintaining the elevated K+ levels (and high resting potential of ~+100mV) in this compartment. This is essential for amplifying the responses of hair cells to sound, by increasing the driving potential for K+ flux across the hair cell membrane when it is activated.
- passive K+ flux through gap junctions. This seems unlikely, as it would decay over a relatively short distance
- a regenerative Ca++ wave that is propagated by IP3 flux between cells. IP3 is regenerated in each cell by IP3 induced Ca++ release from intracellular stores that activates phospholipase C (PLC). Ultimately, in the stria vascularis, K+ is released into the intrastrial space or the endolymph through KCNJ10 (Kir4.1) or PIP2 regulated (KNCQ1/KCNE1) K+ channels.
- A regenerative Ca++ wave that is propagated extracellularly by Ca++ activated release of ATP through connexin hemichannels which then activates P2Y receptors in adjacent cells. These in turn can activate PLCδ, which will generate IP3 and re-initiate the Ca++ response.
However, the fact that gap junction channels are permeable to a wide spectrum of molecules makes it difficult to define their exact functions in any system, including the ear. Some clues, however, have been provided by disease mutants that have caused more specific loss of selective functions of the protein, as summarized in Table I. The insights that these, along with mouse studies described above, have provided into connexin function in the cochlea are summarized below in the context of some of the models that guide current thinking.
3.4.1 K+ recycling
The apical side of hair cells and supporting cells face the endolymph, which contains a low concentration of Ca++ (20μM), but high concentrations of K+ (>100mM), that can be maintained when the endolymph and perilymph are electrically isolated from on another via tight junctions between the basal cells of the stria. This occurs in mammalian systems, and results in a positive endolymphatic potential (EP) of 100-150mV. Sound stimulation opens mechanosensitive channels to allow K+ to enter the hair cell from the endolymph. Since the cytoplasm of the hair cell has a typical resting potential of -80mV, the electromotive force driving the K+ current is substantially higher than seen in most neurons where the extracellular environment is at ground, thus amplifying the inward K+ current across the membrane. The receptor potential generated causes neurotransmitter release from hair cells, as well as K+ release into interstitial space, possibly though KCNQ4 channels. This excess extracellular K+ is then absorbed by the supporting cells through the KCC4 K+/Cl- co-transporter [94-96]. Mutations in KCNQ4 or KCC4 are known to cause deafness due to hair cell degeneration. Since cell loss only occurs after hearing onset in conditional GJB2, and KCC4 knockout animals, it has been proposed that high local extracellular K+ levels are toxic to hair cells by depolarizing their membrane potential, and producing hyperexcitability in the cells. This is a well established phenomenon in other neuronal populations, and the gap junction network connecting astrocytes has been implicated in the buffering of this extracellular K+. This is also likely to be true in the supporting and epithelial cells of the cochlea, and its failure would lead to hair cell loss after they become active.
However, in the ear this uptake and intercellular distribution of K+ may have another role. The EP described above is first generated several days prior to the onset of hearing, and reaches adult levels at the onset of hearing at P12, K+ from the stria intermediate cells is secreted into intrastrial space by KCNJ10 (Kir4.1) [97, 98], from where marginal cells absorb it by a Na+/K+/2Cl- co-transporter, NKCC1, and secrete it into the endolymph by the PIP2 regulated K+ channel (KNCQ1/KCNE1 - reviewed in Ref. [99]). The K+ secretion from the stria intermediate cells is thought to be the end product of “recycling” of K+ initially released from hair cells, through the epithelial and connective tissue gap junction networks [100, 101]. Gap junction coupling between fibrocytes is established as early as P2. Coupling between fibrocytes and basal, as well as intermediate cells, in the stria vascularis, is established by P7 in mammals [102] and is required for completion of the K+ recycling “circuit” by bypassing the extracellular tight junction barrier between the stria and the perilymphatic space of the spiral ligament. Thus, gap junction systems in the cochlea may be responsible for both K+ clearance from the hair cells, and EP formation. While the former is generally accepted, several lines of evidence have been used argue against a role for the GJ network in the epithelia cells contributing to EP formation. Firstly, Cx30-null mice do not develop normal EP, but maintain electrical coupling between supporting cells [44, 103]. Cx30T5M transgenic mice, mimicking a form of NSHL (Table I), also show impaired hearing, but normal electrical coupling between supporting cells as measured by double electrode patch clamp [90]. Secondly, knockout of Cx26 in epithelial cells, or transgenic expression of the dominant deafness mutant, Cx26R75W, do not impair initial EP development, but still cause deafness [43, 83], and Cx26 mutants like V84L, which, like Cx30T5M, show normal electrical coupling, still show hearing loss. Thirdly, knockout of KCC4, which is the transporter in the supporting cells to siphon K+ secreted from hair cells into epithelial cells, does not show the collapse of Reissner’s membrane, which is always seen in animals with impaired K+ secretion from stria vascularis, but still show hair cell degeneration [94].
The problem with these conclusions is that they often presuppose that the K+ recycling in the cochlea might occur through direct passive diffusion of K+ ions. However, this is unlikely, as such diffusion would be unlikely to propagate over any distance unless it were part of a regenerating system. Such a system could be provided through intercellular Ca++ waves known to occur in the cochlea (Fig. 1B). In this model, an initial Ca++ response generated by depolarization from initial K+ uptake, would generate an IP3 response through stimulation of PLCδ. The IP3 could then pass through gap junctions to a neighboring cell, where it would, in turn, activate Ca++ release from intracellular stores, and initiate the whole process again. Ultimately, this Ca++ response can also trigger opening of Ca++ sensitive K+ channels at the apical surface of root cells into the extracellular space of the spiral ligament (end of the endothelial cell propagation) or from the connective tissue system into the intrastrial space where it can be taken up by the marginal cells. An alternative, extracellular route also could involve connexins. The same initial Ca++ signal could induce opening of hemichannels, resulting in ATP release from the cell. Diffusion of this ATP to the neighboring cell would activate P2Y receptors, which would then generate an IP3 signal through G-protein mediated activation of PLC, producing a Ca++ spike that would then regenerate the same cycle in that cell. Thus, both extracellular and intercellular Ca++ waves are both mediated by connexins and could provide the means of propagating a signal over a long distance that could effectively shuttle K+ from its release from hair cells back to the endolymph. Both mechanisms of Ca++ wave propagation, which is proposed to mediate this regenerative K+ flow, have been demonstrated in the immature cochlea [104].
These models explain many of the observations above that had seemed contradictory to the K+ recycling model. While several of the knock-outs (Cx30 or Cx26) had appeared to leave electrical coupling intact, this is difficult to quantify with the complex connectivity of cells seen in situ. Furthermore, the above model would indicate that it is not ion flow that is relevant, but that of larger molecules like IP3 and ATP, the flux of which is likely to be more sensitive to a reduction in channel number than electrical coupling. This would also explain why mutants such as Cx26V84L, A88S and V95M as well as Cx30T5M, all of which selectively ablate IP3, but not ionic, permeability cause deafness. It also likely explains why Cx30 cannot replace loss of Cx26, while Cx26 can replace loss of Cx30 in the transgenic mice experiments described above, since Cx30 homomeric channels have been shown to be much less permeable to large anions, including IP3, than Cx26 homomeric channels, or heteromeric channels of Cx26 and 30 [36]. The Cx26R75W mutation was shown to still allow development of the EP, even in the absence of gap junction channels [43]. However, its ability to form hemichannels remains intact [105], so that it is possible that at least the K+ recycling function could be preserved through the extracellular route of ATP release. Thus, in the light of these regenerative models of Ca++ wave mediated K+ recycling, many of the observations from mouse mutants and human disease mutations are consistent with at least one of the critical roles of connexins in the cochlea being to remove K+ from the vicinity of the hair cells and return it to the endolymph.
3.4.2 Metabolite transfer
Aside from their role in propagation of Ca++ waves, gap junctions have also been shown to pass non-signaling permeants, including nutrients, such as amino acids and glucose. Since the organ of Corti is an avascular organ, gap junction channels in supporting cells and connective tissue cells may be critical for the energy supply of the cochlea, in a manner similar to that proposed for the eye lens [106]. Consistent with this, Cx30-null mice were found to have impaired permeability to 2-NPDG, an analog to D-glucose, and to display increased ROS production [103]. This is consistent with the contention that decreases in coupling that go undetected electrically, could still impair larger metabolite/nutrient transfer to the point of compromising cell health. Finally, it should not be forgotten that non-canonical permeants larger than 1kDa, including short peptides [107], siRNA [108] and miRNA [109, 110] have also been shown to pass through gap junctions. Such signaling, which has not been investigated in the case of the cochlea, may be critical for cochlear homeostasis, and contribute to the variations seen in deafness phenotypes between different connexin mutants.
3.4.3 A Role for Hemichannels?
So far, there have been no reports specifically indicating loss of hemichannel function involved in the etiology of connexin linked congenital deafness. If there are connexin mutations which cause diseases due to loss of hemichannel function, one of the reasons why they may have not been identified is possibly because mutations which impair hemichannel functions would very possibly compromise the gap junction channel function as well. However, several deafness mutants have been identified that show normal gap junction function, but enhanced hemichannel function (Table I). As noted above, hemichannels likely represent one of the major mechanisms for generation of propagatable Ca++ waves in the cochlea though release of ATP. ATP release from the Kolliger organ through hemichannels has also been suggested to be responsible for spontaneous hair cell activity before hearing onset [111]. The existence of a hemichannel population was supported by immunostaining of the apical surface of supporting cells by antibodies targeting the extracellular loops of Cx26 [112]. It is also worth noting that, while the endolymph has very high K+ levels, its Ca2+ levels are very low, around at ~20 μM (Wangemann and Schacht, 1996), which would facilitate hemichannel opening. Therefore, the hemichannel functions in the cochlea warrant closer attention.
Of course, other than changes in nutrient and signal transfer, a likely consequence of significant increases in hemichannel activity in the cochlea is increased cell death. Hemichannel opening is strictly regulated by the cell through a combination of membrane potential, high extracellular Ca++ and limited expression of unopposed hemichannels at the cell surface. This is presumably because widespread opening of large, relatively non-selective pores on the cell surface would compromise cell health. Given that hair cells do not express Cx26 or 30, it is unlikely that mutants of these genes with enhanced hemichannel function contribute directly to the hair cell death seen in patients and mice models. However, they could compromise the functional integrity, or even cause cell death [83]. in the supporting parts of the cochlea, which would then lead indirectly to hair cell death. Cell death can also arise through accumulation of unfolded proteins in the cell that can lead to a pathogenic unfolded protein response. This may be the underlying cause of deafness with several connexin mutations which show no evidence of enhanced hemichannel activity, but instead accumulate expressed protein intracellularly (e.g. Cx31 G12D – [61]).
3.4.4 Relative roles of Cx26, 30, 30.3 and 31
There are many fewer mutations found on GJB6, 4 and 3 associated with deafness. Most of the GJB6 mutants associated with NSHL are deletion mutants, which likely lead to premature termination of the protein [113-115], and provide little insight into their functional importance. These deletions are typically homozygous, compound heterozygous or trans-heterozygous with a Cx26 recessive mutation, suggesting that deafness may only result when not only Cx30 function is lost, but also Cx26. Larger deletion mutations, which truncate GJB6 (Cx30), may also affect the transcription level of the neighboring Cx26 gene in an allele specific manner, should they also affect the cis-regulatory elements of the Cx26 gene [116, 117]. These observations are all consistent with mouse models which indicate that increase in Cx26 expression can alleviate the deafness phenotype of Cx30 knock-out mice [92].
The exceptions to these NSHL associated deletion mutations are Cx30T5M [57] and Cx30A40V [118]. Only Cx30T5M has been functionally characterized, being shown to have reduced permeability to IP3 [37], similar to several of the NSHL causing Cx26 mutations (Table I). Two other point mutations of Cx30, one in the NT (G11R) and the other in M2 (A88V), cause HED, a skin disease with aspects of both PPK (hyperkeratosis of palms and feet) as well KID (hair loss), but with no hearing phenotype. Both mutants cause increased hemichannel opening, as do several of the mutations of Cx26 associated with KID syndrome. However, since these Cx26 mutations do show a hearing phenotype, this suggests that either Cx30 hemichannels may be differentially regulated in the ear, and/or that similar mutations in Cx26 and 30 (e.g. A88V) have different effects in channel function in the two isotypes.
Mutations in GJB4 (Cx30.3) and GJB3 (Cx31) associated with both NSHL (Cx31 - E183K, ΔI141 and I141V) and syndromic deafness linked to EKV (Cx30.3F137L and Cx31 G12D, R42P and C86S) or peripheral neuropathy (Cx31ΔD66) [55, 56, 58, 59, 119, 120] fail to produce functional channels, with the proteins typically accumulating in the cytoplasm. Thus, they cast little light on the functional roles of these connexins in the ear or skin. It is notable, however, that the mutants associated with EKV accumulate sufficiently in the cytosol in exogenously expressed systems to lead to cell death.
4. FUNCTION OF CONNEXINS IN THE SKIN
As noted above, most of the mutations associated with NSHL are inherited recessively, while those which also cause skin disease are dominant, even when the mutations are in the same regions of the protein, and cause similar disruption in trafficking of the protein to the cell surface or formation of functional gap junction channels (first two rows, Table I). This suggests that, unlike the ear where at least Cx26 seems essential for normal function, there may be redundancy in the skin. Thus, loss of any one connexin is not deleterious, unless it also affects other connexins with which it interacts. Consistent with this conclusion is the frequency of mutations that cause dominant effects on co-expressed connexins (bold type in Table I), which is much higher among syndromic than non-syndromic deafness mutations of Cx26 (5 of 8 compared to 5 of 38 sites characterized, respectively, in first 2 rows of Table I). Such a conclusion is not surprising, given the much higher abundance of different connexins with overlapping expression patterns in the skin, where Cx26, 30, 30.3, 31, 31.1, 37. 40 and 43 are broadly distributed, with differing patterns during development [121-124].
The specific functions of gap junctions in the skin remain largely obscure to date. They have been implicated in the formation of communication compartments among keratinocytes that could correlate with epidermal proliferative units during development [125], potentially explaining the hyperkeratosis seen in many connexin associated skin phenotypes. However, this is likely to be complex, as the upregulation of Cx26 expression, not its loss, is associated with skin thickening in response to retinoids [126], and thickening of mouse epidermis in transgenic mice [127]. Connexin regulation during wound healing has also been extensively studied, with elevation of Cx26 and 30, and reduction in Cx31.1 and 43, occurring successively following wounding [128-130]. The functional relevance of this has been demonstrated using topical Cx43 knockdown, which results in significant enhancement of wound healing rates in several model systems [124, 125, 131, 132].
As discussed above for the NSHL mutations, the greatest insight into the specific functions of Cx26 or other connexins in the proliferative or wound healing responses of skin would be provided by mutations that selectively change specific properties of the channels. Notably, none of the mutants of either Cx26 or Cx30 that change the permeability properties of gap junction channels cause any skin problems (third row of Table I). However, before concluding that metabolite transfer through gap junctions is not critical for skin function, it is possible that the effects of these mutants could be masked, as none of them have been shown to have dominant effects on other connexins with which they are co-expressed.
In contrast, another manner in which a connexin disease mutant could have dominant inheritance, other than dominant negative effects on wt connexins, is if the mutation induces a gain of function. Thus, it is not surprising that almost all of the mutations of Cx26 that cause an increase in hemichannel activity, cause syndromic deafness (fourth row of Table I), mostly associated with the more severe KID syndrome. Even the two mutations of Cx30 that cause increased hemichannels opening (G11R and A88V - [60, 133]) cause skin disease, which in this case is not associated with deafness (HED or Clouston’s syndrome). This disease has features of KID (keratosis and involvement of hair follicles), but also of the other cluster of syndromes, PPK (thickening of the epidermis on the palms and feet) and BPS (nail thickening). While the specific effects of increased hemichannel opening in keratinocytes in situ has not been directly assessed, it seems likely that this would lead to increased cell death, as it does in the ear [62]. The one case of a mutant that increases hemichannels function, but does not cause skin disease (Cx26A88S), causes a loss of hemichannel voltage gating, which is likely an important regulator in the ear, but not keratinocytes, which have very low membrane potentials (Xu and Nicholson, personal observations).
5. PROSPECTS FOR THE FUTURE
While we do not have all of the answers as to how gap junctions or hemichannels contribute to the normal function of the ear or skin, the functional analyses of disease associated mutants summarized above has stimulated a new era of discovery in this area. As more mouse models are developed, and functional analyses of disease mutants in exogenous systems expand to include detailed permeability analyses for metabolites and signaling molecules, we will certainly gain a greater understanding of how gap junction networks serve to integrate cochlear and epidermal functions. Even structural information on these channels will prove relevant once we reach the resolution where we can make predictions as to the functional effects of different mutations on both hemichannel and gap junction conformations of these channels. The insights we have already gained from this provide a compelling argument for the integration of the most basic molecular studies of gap junction properties with in vivo analyses of mutant phenotypes, as only then are we likely to truly understand the molecular basis of organ function. Such insights will also be essential to developing truly effective therapeutic strategies.
HIGHLIGHTS.
Disease mutants provide insights into gap junction function in ear/skin function.
Selective loss of IP3 permeability in NSHL, implicates Ca++ waves in normal hearing
Increased hemichannel activity causes cell death associated with dominant skin disease.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCE LIST
- 1.Bao L, Sachs F, Dahl G. Connexins are mechanosensitive. American journal of physiology. 2004;287:C1389–1395. doi: 10.1152/ajpcell.00220.2004. [DOI] [PubMed] [Google Scholar]
- 2.Ebihara L, Steiner E. Properties of a nonjunctional current expressed from a rat connexin46 cDNA in Xenopus oocytes. J Gen Physiol. 1993;102:59–74. doi: 10.1085/jgp.102.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paul DL, Ebihara L, Takemoto LJ, Swenson KI, Goodenough DA. Connexin46, a novel lens gap junction protein, induces voltage-gated currents in nonjunctional plasma membrane of Xenopus oocytes. J Cell Biol. 1991;115:1077–1089. doi: 10.1083/jcb.115.4.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stout CE, Costantin JL, Naus CC, Charles AC. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J Biol Chem. 2002;277:10482–10488. doi: 10.1074/jbc.M109902200. [DOI] [PubMed] [Google Scholar]
- 5.Bruzzone S, Franco L, Guida L, Zocchi E, Contini P, Bisso A, Usai C, De Flora A. A self-restricted CD38-connexin 43 cross-talk affects NAD+ and cyclic ADP-ribose metabolism and regulates intracellular calcium in 3T3 fibroblasts. J Biol Chem. 2001;276:48300–48308. doi: 10.1074/jbc.M107308200. [DOI] [PubMed] [Google Scholar]
- 6.Cherian PP, Siller-Jackson AJ, Gu S, Wang X, Bonewald LF, Sprague E, Jiang JX. Mechanical strain opens connexin 43 hemichannels in osteocytes: a novel mechanism for the release of prostaglandin. Molecular biology of the cell. 2005;16:3100–3106. doi: 10.1091/mbc.E04-10-0912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herve JC, Bourmeyster N, Sarrouilhe D, Duffy HS. Gap junctional complexes: from partners to functions. Progress in biophysics and molecular biology. 2007;94:29–65. doi: 10.1016/j.pbiomolbio.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 8.Pfenniger A, Wohlwend A, Kwak BR. Mutations in connexin genes and disease. European journal of clinical investigation. 2011;41:103–116. doi: 10.1111/j.1365-2362.2010.02378.x. [DOI] [PubMed] [Google Scholar]
- 9.D’Hondt C, Ponsaerts R, De Smedt H, Vinken M, De Vuyst E, De Bock M, Wang N, Rogiers V, Leybaert L, Himpens B, Bultynck G. Pannexin channels in ATP release and beyond: an unexpected rendezvous at the endoplasmic reticulum. Cell Signal. 2011;23:305–316. doi: 10.1016/j.cellsig.2010.07.018. [DOI] [PubMed] [Google Scholar]
- 10.Sosinsky GE, Boassa D, Dermietzel R, Duffy HS, Laird DW, MacVicar B, Naus CC, Penuela S, Scemes E, Spray DC, Thompson RJ, Zhao HB, Dahl G. Pannexin channels are not gap junction hemichannels. Channels (Austin) 2011;5:193–197. doi: 10.4161/chan.5.3.15765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoang Dinh E, Ahmad S, Chang Q, Tang W, Stong B, Lin X. Diverse deafness mechanisms of connexin mutations revealed by studies using in vitro approaches and mouse models. Brain research. 2009;1277:52–69. doi: 10.1016/j.brainres.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee JR, White TW. Connexin-26 mutations in deafness and skin disease. Expert reviews in molecular medicine. 2009;11:e35. doi: 10.1017/S1462399409001276. [DOI] [PubMed] [Google Scholar]
- 13.Scott CA, Kelsell DP. Key functions for gap junctions in skin and hearing. Biochem J. 2011;438:245–254. doi: 10.1042/BJ20110278. [DOI] [PubMed] [Google Scholar]
- 14.Cohen MMJ, Gorlin RJ. Epidemiology, etiology, and genetic patterns. In: Gorlin RJ, Toriello HV, Cohen MM Jr, editors. Hereditary hearing loss and its syndromes. Vol. 28. Oxford monographs on medical genetics; 1995. pp. 9–21. [Google Scholar]
- 15.Dror AA, Avraham KB. Hearing impairment: a panoply of genes and functions. Neuron. 2010;68:293–308. doi: 10.1016/j.neuron.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 16.Abe S, Usami S, Shinkawa H, Kelley PM, Kimberling WJ. Prevalent connexin 26 gene (GJB2) mutations in Japanese. J Med Genet. 2000;37:41–43. doi: 10.1136/jmg.37.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Denoyelle F, Marlin S, Weil D, Moatti L, Chauvin P, Garabedian EN, Petit C. Clinical features of the prevalent form of childhood deafness, DFNB1, due to a connexin-26 gene defect: implications for genetic counselling. Lancet. 1999;353:1298–1303. doi: 10.1016/S0140-6736(98)11071-1. [DOI] [PubMed] [Google Scholar]
- 18.Estivill X, Fortina P, Surrey S, Rabionet R, Melchionda S, D’Agruma L, Mansfield E, Rappaport E, Govea N, Mila M, Zelante L, Gasparini P. Connexin-26 mutations in sporadic and inherited sensorineural deafness. Lancet. 1998;351:394–398. doi: 10.1016/S0140-6736(97)11124-2. [DOI] [PubMed] [Google Scholar]
- 19.Gabriel H, Kupsch P, Sudendey J, Winterhager E, Jahnke K, Lautermann J. Mutations in the connexin26/GJB2 gene are the most common event in non-syndromic hearing loss among the German population. Hum Mutat. 2001;17:521–522. doi: 10.1002/humu.1138. [DOI] [PubMed] [Google Scholar]
- 20.Lench N, Houseman M, Newton V, Van CG, Mueller R. Connexin-26 mutations in sporadic non-syndromal sensorineural deafness. Lancet. 1998;351:415. doi: 10.1016/s0140-6736(98)24006-2. [DOI] [PubMed] [Google Scholar]
- 21.Loffler J, Nekahm D, Hirst-Stadlmann A, Gunther B, Menzel HJ, Utermann G, Janecke AR. Sensorineural hearing loss and the incidence of Cx26 mutations in Austria. Eur J Hum Genet. 2001;9:226–230. doi: 10.1038/sj.ejhg.5200607. [DOI] [PubMed] [Google Scholar]
- 22.Murgia A, Orzan E, Polli R, Martella M, Vinanzi C, Leonardi E, Arslan E, Zacchello F. Cx26 deafness: mutation analysis and clinical variability. J Med Genet. 1999;36:829–832. [PMC free article] [PubMed] [Google Scholar]
- 23.Sobe T, Vreugde S, Shahin H, Berlin M, Davis N, Kanaan M, Yaron Y, Orr-Urtreger A, Frydman M, Shohat M, Avraham KB. The prevalence and expression of inherited connexin 26 mutations associated with nonsyndromic hearing loss in the Israeli population. Hum Genet. 2000;106:50–57. doi: 10.1007/s004390051009. [DOI] [PubMed] [Google Scholar]
- 24.Wilcox SA, Saunders K, Osborn AH, Arnold A, Wunderlich J, Kelly T, Collins V, Wilcox LJ, Kinlay Gardner RJ, Kamarinos M, Cone-Wesson B, Williamson R, Dahl HH. High frequency hearing loss correlated with mutations in the GJB2 gene. Hum Genet. 2000;106:399–405. doi: 10.1007/s004390000273. [DOI] [PubMed] [Google Scholar]
- 25.Morell RJ, Kim HJ, Hood LJ, Goforth L, Friderici K, Fisher R, Van Camp G, Berlin CI, Oddoux C, Ostrer H, Keats B, Friedman TB. Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. N Engl J Med. 1998;339:1500–1505. doi: 10.1056/NEJM199811193392103. [DOI] [PubMed] [Google Scholar]
- 26.Dai P, Yu F, Han B, Yuan Y, Li Q, Wang G, Liu X, He J, Huang D, Kang D, Zhang X, Yuan H, Schmitt E, Han D, Wong LJ. The prevalence of the 235delC GJB2 mutation in a Chinese deaf population. Genet Med. 2007;9:283–289. doi: 10.1097/gim.0b013e31804d2371. [DOI] [PubMed] [Google Scholar]
- 27.Brobby GW, Muller-Myhsok B, Horstmann RD. Connexin 26 R143W mutation associated with recessive nonsyndromic sensorineural deafness in Africa. N Engl J Med. 1998;338:548–550. doi: 10.1056/NEJM199802193380813. [DOI] [PubMed] [Google Scholar]
- 28.Alvarez A, del Castillo I, Villamar M, Aguirre LA, Gonzalez-Neira A, Lopez-Nevot A, Moreno-Pelayo MA, Moreno F. High prevalence of the W24X mutation in the gene encoding connexin-26 (GJB2) in Spanish Romani (gypsies) with autosomal recessive non-syndromic hearing loss. Am J Med Genet A. 2005;137A:255–258. doi: 10.1002/ajmg.a.30884. [DOI] [PubMed] [Google Scholar]
- 29.Joseph AY, Rasool TJ. High frequency of connexin26 (GJB2) mutations associated with nonsyndromic hearing loss in the population of Kerala, India. Int J Pediatr Otorhinolaryngol. 2009;73:437–443. doi: 10.1016/j.ijporl.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 30.Meyer CG, Amedofu GK, Brandner JM, Pohland D, Timmann C, Horstmann RD. Selection for deafness? Nat Med. 2002;8:1332–1333. doi: 10.1038/nm1202-1332. [DOI] [PubMed] [Google Scholar]
- 31.D’Adamo P, Guerci VI, Fabretto A, Faletra F, Grasso DL, Ronfani L, Montico M, Morgutti M, Guastalla P, Gasparini P. Does epidermal thickening explain GJB2 high carrier frequency and heterozygote advantage? Eur J Hum Genet. 2009;17:284–286. doi: 10.1038/ejhg.2008.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Man YK, Trolove C, Tattersall D, Thomas AC, Papakonstantinopoulou A, Patel D, Scott C, Chong J, Jagger DJ, O’Toole EA, Navsaria H, Curtis MA, Kelsell DP. A deafness-associated mutant human connexin 26 improves the epithelial barrier in vitro. J Membr Biol. 2007;218:29–37. doi: 10.1007/s00232-007-9025-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maeda S, Nakagawa S, Suga M, Yamashita E, Oshima A, Fujiyoshi Y, Tsukihara T. Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature. 2009;458:597–602. doi: 10.1038/nature07869. [DOI] [PubMed] [Google Scholar]
- 34.Oshima A, Tani K, Hiroaki Y, Fujiyoshi Y, Sosinsky GE. Three-dimensional structure of a human connexin26 gap junction channel reveals a plug in the vestibule. Proc Natl Acad Sci U S A. 2007;104:10034–10039. doi: 10.1073/pnas.0703704104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mese G, Valiunas V, Brink PR, White TW. Connexin26 deafness associated mutations show altered permeability to large cationic molecules. Am J Physiol Cell Physiol. 2008;295:C966–C974. doi: 10.1152/ajpcell.00008.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beltramello M, Piazza V, Bukauskas FF, Pozzan T, Mammano F. Impaired permeability to Ins(1,4,5)P3 in a mutant connexin underlies recessive hereditary deafness. Nat Cell Biol. 2005;7:63–69. doi: 10.1038/ncb1205. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Y, Tang W, Ahmad S, Sipp JA, Chen P, Lin X. Gap junction-mediated intercellular biochemical coupling in cochlear supporting cells is required for normal cochlear functions. Proc Natl Acad Sci U S A. 2005;102:15201–15206. doi: 10.1073/pnas.0501859102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skerrett IM, Aronowitz J, Shin JH, Cymes G, Kasperek E, Cao FL, Nicholson BJ. Identification of amino acid residues lining the pore of a gap junction channel. J Cell Biol. 2002;159:349–360. doi: 10.1083/jcb.200207060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heathcote K, Syrris P, Carter ND, Patton MA. A connexin 26 mutation causes a syndrome of sensorineural hearing loss and palmoplantar hyperkeratosis (MIM 148350) Journal of medical genetics. 2000;37:50–51. doi: 10.1136/jmg.37.1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maestrini E, Korge BP, Ocana-Sierra J, Calzolari E, Cambiaghi S, Scudder PM, Hovnanian A, Monaco AP, Munro CS. A missense mutation in connexin26, D66H, causes mutilating keratoderma with sensorineural deafness (Vohwinkel’s syndrome) in three unrelated families. Human molecular genetics. 1999;8:1237–1243. doi: 10.1093/hmg/8.7.1237. [DOI] [PubMed] [Google Scholar]
- 41.Richard G, Rouan F, Willoughby CE, Brown N, Chung P, Ryynanen M, Jabs EW, Bale SJ, DiGiovanna JJ, Uitto J, Russell L. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. American journal of human genetics. 2002;70:1341–1348. doi: 10.1086/339986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uyguner O, Tukel T, Baykal C, Eris H, Emiroglu M, Hafiz G, Ghanbari A, Baserer N, Yuksel-Apak M, Wollnik B. The novel R75Q mutation in the GJB2 gene causes autosomal dominant hearing loss and palmoplantar keratoderma in a Turkish family. Clinical genetics. 2002;62:306–309. doi: 10.1034/j.1399-0004.2002.620409.x. [DOI] [PubMed] [Google Scholar]
- 43.Kudo T, Kure S, Ikeda K, Xia AP, Katori Y, Suzuki M, Kojima K, Ichinohe A, Suzuki Y, Aoki Y, Kobayashi T, Matsubara Y. Transgenic expression of a dominant-negative connexin26 causes degeneration of the organ of Corti and non-syndromic deafness. Hum Mol Genet. 2003;12:995–1004. doi: 10.1093/hmg/ddg116. [DOI] [PubMed] [Google Scholar]
- 44.Teubner B, Michel V, Pesch J, Lautermann J, Cohen-Salmon M, Sohl G, Jahnke K, Winterhager E, Herberhold C, Hardelin JP, Petit C, Willecke K. Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Hum Mol Genet. 2003;12:13–21. doi: 10.1093/hmg/ddg001. [DOI] [PubMed] [Google Scholar]
- 45.Rouan F, White TW, Brown N, Taylor AM, Lucke TW, Paul DL, Munro CS, Uitto J, Hodgins MB, Richard G. trans-dominant inhibition of connexin-43 by mutant connexin-26: implications for dominant connexin disorders affecting epidermal differentiation. J Cell Sci. 2001;114:2105–2113. doi: 10.1242/jcs.114.11.2105. [DOI] [PubMed] [Google Scholar]
- 46.Thomas T, Telford D, Laird DW. Functional domain mapping and selective trans-dominant effects exhibited by Cx26 disease-causing mutations. J Biol Chem. 2004;279:19157–19168. doi: 10.1074/jbc.M314117200. [DOI] [PubMed] [Google Scholar]
- 47.Alexandrino F, Sartorato EL, Marques-de-Faria AP, Steiner CE. G59S mutation in the GJB2 (connexin 26) gene in a patient with Bart-Pumphrey syndrome. Am J Med Genet A. 2005;136:282–284. doi: 10.1002/ajmg.a.30822. [DOI] [PubMed] [Google Scholar]
- 48.Akiyama M, Sakai K, Arita K, Nomura Y, Ito K, Kodama K, McMillan JR, Kobayashi K, Sawamura D, Shimizu H. A novel GJB2 mutation p.Asn54His in a patient with palmoplantar keratoderma, sensorineural hearing loss and knuckle pads. The Journal of investigative dermatology. 2007;127:1540–1543. doi: 10.1038/sj.jid.5700711. [DOI] [PubMed] [Google Scholar]
- 49.Richard G, Brown N, Ishida-Yamamoto A, Krol A. Expanding the phenotypic spectrum of Cx26 disorders: Bart-Pumphrey syndrome is caused by a novel missense mutation in GJB2. The Journal of investigative dermatology. 2004;123:856–863. doi: 10.1111/j.0022-202X.2004.23470.x. [DOI] [PubMed] [Google Scholar]
- 50.Gerido DA, DeRosa AM, Richard G, White TW. Aberrant hemichannel properties of Cx26 mutations causing skin disease and deafness. Am J Physiol Cell Physiol. 2007;293:C337–C345. doi: 10.1152/ajpcell.00626.2006. [DOI] [PubMed] [Google Scholar]
- 51.Lee JR, Derosa AM, White TW. Connexin mutations causing skin disease and deafness increase hemichannel activity and cell death when expressed in Xenopus oocytes. J Invest Dermatol. 2009;129:870–878. doi: 10.1038/jid.2008.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stong BC, Chang Q, Ahmad S, Lin X. A novel mechanism for connexin 26 mutation linked deafness: cell death caused by leaky gap junction hemichannels. Laryngoscope. 2006;116:2205–2210. doi: 10.1097/01.mlg.0000241944.77192.d2. [DOI] [PubMed] [Google Scholar]
- 53.Mese G, Sellitto C, Li L, Wang HZ, Valiunas V, Richard G, Brink PR, White TW. The Cx26-G45E mutation displays increased hemichannel activity in a mouse model of the lethal form of keratitis-ichthyosis-deafness syndrome. Mol Biol Cell. 2011;22:4776–4786. doi: 10.1091/mbc.E11-09-0778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanchez HA, Mese G, Srinivas M, White TW, Verselis VK. Differentially altered Ca2+ regulation and Ca2+ permeability in Cx26 hemichannels formed by the A40V and G45E mutations that cause keratitis ichthyosis deafness syndrome. J Gen Physiol. 2010;136:47–62. doi: 10.1085/jgp.201010433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xia JH, Liu CY, Tang BS, Pan Q, Huang L, Dai HP, Zhang BR, Xie W, Hu DX, Zheng D, Shi XL, Wang DA, Xia K, Yu KP, Liao XD, Feng Y, Yang YF, Xiao JY, Xie DH, Huang JZ. Mutations in the gene encoding gap junction protein beta-3 associated with autosomal dominant hearing impairment. Nature genetics. 1998;20:370–373. doi: 10.1038/3845. [DOI] [PubMed] [Google Scholar]
- 56.Liu XZ, Xia XJ, Xu LR, Pandya A, Liang CY, Blanton SH, Brown SD, Steel KP, Nance WE. Mutations in connexin31 underlie recessive as well as dominant non-syndromic hearing loss. Human molecular genetics. 2000;9:63–67. doi: 10.1093/hmg/9.1.63. [DOI] [PubMed] [Google Scholar]
- 57.Grifa A, Wagner CA, D’Ambrosio L, Melchionda S, Bernardi F, Lopez-Bigas N, Rabionet R, Arbones M, Monica MD, Estivill X, Zelante L, Lang F, Gasparini P. Mutations in GJB6 cause nonsyndromic autosomal dominant deafness at DFNA3 locus. Nat Genet. 1999;23:16–18. doi: 10.1038/12612. [DOI] [PubMed] [Google Scholar]
- 58.Richard G, Smith LE, Bailey RA, Itin P, Hohl D, Epstein EH, Jr, DiGiovanna JJ, Compton JG, Bale SJ. Mutations in the human connexin gene GJB3 cause erythrokeratodermia variabilis. Nature genetics. 1998;20:366–369. doi: 10.1038/3840. [DOI] [PubMed] [Google Scholar]
- 59.Wilgoss A, Leigh IM, Barnes MR, Dopping-Hepenstal P, Eady RA, Walter JM, Kennedy CT, Kelsell DP. Identification of a novel mutation R42P in the gap junction protein beta-3 associated with autosomal dominant erythrokeratoderma variabilis. The Journal of investigative dermatology. 1999;113:1119–1122. doi: 10.1046/j.1523-1747.1999.00792.x. [DOI] [PubMed] [Google Scholar]
- 60.Lamartine J, Munhoz Essenfelder G, Kibar Z, Lanneluc I, Callouet E, Laoudj D, Lemaitre G, Hand C, Hayflick SJ, Zonana J, Antonarakis S, Radhakrishna U, Kelsell DP, Christianson AL, Pitaval A, Der Kaloustian V, Fraser C, Blanchet-Bardon C, Rouleau GA, Waksman G. Mutations in GJB6 cause hidrotic ectodermal dysplasia. Nature genetics. 2000;26:142–144. doi: 10.1038/79851. [DOI] [PubMed] [Google Scholar]
- 61.Tattersall D, Scott CA, Gray C, Zicha D, Kelsell DP. EKV mutant connexin 31 associated cell death is mediated by ER stress. Human molecular genetics. 2009;18:4734–4745. doi: 10.1093/hmg/ddp436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Essenfelder GM, Bruzzone R, Lamartine J, Charollais A, Blanchet-Bardon C, Barbe MT, Meda P, Waksman G. Connexin30 mutations responsible for hidrotic ectodermal dysplasia cause abnormal hemichannel activity. Human molecular genetics. 2004;13:1703–1714. doi: 10.1093/hmg/ddh191. [DOI] [PubMed] [Google Scholar]
- 63.Ahmad S, Chen S, Sun J, Lin X. Connexins 26 and 30 are co-assembled to form gap junctions in the cochlea of mice. Biochem Biophys Res Commun. 2003;307:362–368. doi: 10.1016/s0006-291x(03)01166-5. [DOI] [PubMed] [Google Scholar]
- 64.Buniello A, Montanaro D, Volinia S, Gasparini P, Marigo V. An expression atlas of connexin genes in the mouse. Genomics. 2004;83:812–820. doi: 10.1016/j.ygeno.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 65.Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, Mueller RF, Leigh IM. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature. 1997;387:80–83. doi: 10.1038/387080a0. [DOI] [PubMed] [Google Scholar]
- 66.Kikuchi T, Kimura RS, Paul DL, Adams JC. Gap junctions in the rat cochlea: immunohistochemical and ultrastructural analysis. Anat Embryol (Berl) 1995;191:101–118. doi: 10.1007/BF00186783. [DOI] [PubMed] [Google Scholar]
- 67.Lautermann J, ten Cate WJ, Altenhoff P, Grummer R, Traub O, Frank H, Jahnke K, Winterhager E. Expression of the gap-junction connexins 26 and 30 in the rat cochlea. Cell Tissue Res. 1998;294:415–420. doi: 10.1007/s004410051192. [DOI] [PubMed] [Google Scholar]
- 68.Forge A, Becker D, Casalotti S, Edwards J, Marziano N, Nevill G. Gap junctions in the inner ear: comparison of distribution patterns in different vertebrates and assessement of connexin composition in mammals. J Comp Neurol. 2003;467:207–231. doi: 10.1002/cne.10916. [DOI] [PubMed] [Google Scholar]
- 69.Eiberger J, Kibschull M, Strenzke N, Schober A, Bussow H, Wessig C, Djahed S, Reucher H, Koch DA, Lautermann J, Moser T, Winterhager E, Willecke K. Expression pattern and functional characterization of connexin29 in transgenic mice. Glia. 2006;53:601–611. doi: 10.1002/glia.20315. [DOI] [PubMed] [Google Scholar]
- 70.Tang W, Zhang Y, Chang Q, Ahmad S, Dahlke I, Yi H, Chen P, Paul DL, Lin X. Connexin29 is highly expressed in cochlear Schwann cells, and it is required for the normal development and function of the auditory nerve of mice. J Neurosci. 2006;26:1991–1999. doi: 10.1523/JNEUROSCI.5055-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xia AP, Ikeda K, Katori Y, Oshima T, Kikuchi T, Takasaka T. Expression of connexin 31 in the developing mouse cochlea. Neuroreport. 2000;11:2449–2453. doi: 10.1097/00001756-200008030-00022. [DOI] [PubMed] [Google Scholar]
- 72.Lopez-Bigas N, Arbones ML, Estivill X, Simonneau L. Expression profiles of the connexin genes, Gjb1 and Gjb3, in the developing mouse cochlea. Gene Expr Patterns. 2002;2:113–117. doi: 10.1016/s0925-4773(02)00299-x. [DOI] [PubMed] [Google Scholar]
- 73.Cohen-Salmon M, Maxeiner S, Kruger O, Theis M, Willecke K, Petit C. Expression of the connexin43- and connexin45-encoding genes in the developing and mature mouse inner ear. Cell and tissue research. 2004;316:15–22. doi: 10.1007/s00441-004-0861-2. [DOI] [PubMed] [Google Scholar]
- 74.Forge A, Marziano NK, Casalotti SO, Becker DL, Jagger D. The inner ear contains heteromeric channels composed of cx26 and cx30 and deafness-related mutations in cx26 have a dominant negative effect on cx30. Cell Commun Adhes. 2003;10:341–346. doi: 10.1080/cac.10.4-6.341.346. [DOI] [PubMed] [Google Scholar]
- 75.Sun J, Ahmad S, Chen S, Tang W, Zhang Y, Chen P, Lin X. Cochlear gap junctions coassembled from Cx26 and 30 show faster intercellular Ca2+ signaling than homomeric counterparts. Am J Physiol Cell Physiol. 2005;288:C613–C623. doi: 10.1152/ajpcell.00341.2004. [DOI] [PubMed] [Google Scholar]
- 76.Yum SW, Zhang J, Valiunas V, Kanaporis G, Brink PR, White TW, Scherer SS. Human connexin26 and connexin30 form functional heteromeric and heterotypic channels. Am J Physiol Cell Physiol. 2007;293:C1032–C1048. doi: 10.1152/ajpcell.00011.2007. [DOI] [PubMed] [Google Scholar]
- 77.Jagger DJ, Forge A. Compartmentalized and signal-selective gap junctional coupling in the hearing cochlea. J Neurosci. 2006;26:1260–1268. doi: 10.1523/JNEUROSCI.4278-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Qu Y, Tang W, Zhou B, Ahmad S, Chang Q, Li X, Lin X. Early developmental expression of connexin26 in the cochlea contributes to its dominate functional role in the cochlear gap junctions. Biochem Biophys Res Commun. 2011 doi: 10.1016/j.bbrc.2011.11.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Beltramello M, Bicego M, Piazza V, Ciubotaru CD, Mammano F, D’Andrea P. Permeability and gating properties of human connexins 26 and 30 expressed in HeLa cells. Biochem Biophys Res Commun. 2003;305:1024–1033. doi: 10.1016/s0006-291x(03)00868-4. [DOI] [PubMed] [Google Scholar]
- 80.Manthey D, Banach K, Desplantez T, Lee CG, Kozak CA, Traub O, Weingart R, Willecke K. Intracellular domains of mouse connexin26 and -30 affect diffusional and electrical properties of gap junction channels. J Membr Biol. 2001;181:137–148. doi: 10.1007/s00232-001-0017-1. [DOI] [PubMed] [Google Scholar]
- 81.Marlin S, Feldmann D, Blons H, Loundon N, Rouillon I, Albert S, Chauvin P, Garabedian EN, Couderc R, Odent S, Joannard A, Schmerber S, Delobel B, Leman J, Journel H, Catros H, Lemarechal C, Dollfus H, Eliot MM, Delaunoy JL, David A, Calais C, Drouin-Garraud V, Obstoy MF, Goizet C, Duriez F, Fellmann F, Helias J, Vigneron J, Montaut B, Matin-Coignard D, Faivre L, Baumann C, Lewin P, Petit C, Denoyelle F. GJB2 and GJB6 mutations: genotypic and phenotypic correlations in a large cohort of hearing-impaired patients. Archives of otolaryngology--head & neck surgery. 2005;131:481–487. doi: 10.1001/archotol.131.6.481. [DOI] [PubMed] [Google Scholar]
- 82.Gabriel HD, Jung D, Butzler C, Temme A, Traub O, Winterhager E, Willecke K. Transplacental uptake of glucose is decreased in embryonic lethal connexin26-deficient mice. The Journal of cell biology. 1998;140:1453–1461. doi: 10.1083/jcb.140.6.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cohen-Salmon M, Ott T, Michel V, Hardelin JP, Perfettini I, Eybalin M, Wu T, Marcus DC, Wangemann P, Willecke K, Petit C. Targeted ablation of connexin26 in the inner ear epithelial gap junction network causes hearing impairment and cell death. Curr Biol. 2002;12:1106–1111. doi: 10.1016/s0960-9822(02)00904-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sun Y, Tang W, Chang Q, Wang Y, Kong W, Lin X. Connexin30 null and conditional connexin26 null mice display distinct pattern and time course of cellular degeneration in the cochlea. J Comp Neurol. 2009;516:569–579. doi: 10.1002/cne.22117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Steel KP, Barkway C. Another role for melanocytes: their importance for normal stria vascularis development in the mammalian inner ear. Development (Cambridge, England) 1989;107:453–463. doi: 10.1242/dev.107.3.453. [DOI] [PubMed] [Google Scholar]
- 86.Maeda Y, Fukushima K, Kawasaki A, Nishizaki K, Smith RJ. Cochlear expression of a dominant-negative GJB2R75W construct delivered through the round window membrane in mice. Neurosci Res. 2007;58:250–254. doi: 10.1016/j.neures.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 87.Minekawa A, Abe T, Inoshita A, Iizuka T, Kakehata S, Narui Y, Koike T, Kamiya K, Okamura HO, Shinkawa H, Ikeda K. Cochlear outer hair cells in a dominant-negative connexin26 mutant mouse preserve non-linear capacitance in spite of impaired distortion product otoacoustic emission. Neuroscience. 2009;164:1312–1319. doi: 10.1016/j.neuroscience.2009.08.043. [DOI] [PubMed] [Google Scholar]
- 88.Qu Y, Tang W, Zhou B, Ahmad S, Chang Q, Li X, Lin X. Early developmental expression of connexin26 in the cochlea contributes to its dominate functional role in the cochlear gap junctions. Biochemical and biophysical research communications. 417:245–250. doi: 10.1016/j.bbrc.2011.11.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang Y, Chang Q, Tang W, Sun Y, Zhou B, Li H, Lin X. Targeted connexin26 ablation arrests postnatal development of the organ of Corti. Biochemical and biophysical research communications. 2009;385:33–37. doi: 10.1016/j.bbrc.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schutz M, Scimemi P, Majumder P, De Siati RD, Crispino G, Rodriguez L, Bortolozzi M, Santarelli R, Seydel A, Sonntag S, Ingham N, Steel KP, Willecke K, Mammano F. The human deafness-associated connexin 30 T5M mutation causes mild hearing loss and reduces biochemical coupling among cochlear non-sensory cells in knock-in mice. Hum Mol Genet. 2010;19:4759–4773. doi: 10.1093/hmg/ddq402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ortolano S, Di PG, Crispino G, Anselmi F, Mammano F, Chiorini JA. Coordinated control of connexin 26 and connexin 30 at the regulatory and functional level in the inner ear. Proc Natl Acad Sci U S A. 2008;105:18776–18781. doi: 10.1073/pnas.0800831105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ahmad S, Tang W, Chang Q, Qu Y, Hibshman J, Li Y, Sohl G, Willecke K, Chen P, Lin X. Restoration of connexin26 protein level in the cochlea completely rescues hearing in a mouse model of human connexin30-linked deafness. Proc Natl Acad Sci U S A. 2007;104:1337–1341. doi: 10.1073/pnas.0606855104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Degen J, Schutz M, Dicke N, Strenzke N, Jokwitz M, Moser T, Willecke K. Connexin32 can restore hearing in connexin26 deficient mice. European journal of cell biology. 2011;90:817–824. doi: 10.1016/j.ejcb.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 94.Boettger T, Hubner CA, Maier H, Rust MB, Beck FX, Jentsch TJ. Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4. Nature. 2002;416:874–878. doi: 10.1038/416874a. [DOI] [PubMed] [Google Scholar]
- 95.Kharkovets T, Dedek K, Maier H, Schweizer M, Khimich D, Nouvian R, Vardanyan V, Leuwer R, Moser T, Jentsch TJ. Mice with altered KCNQ4 K+ channels implicate sensory outer hair cells in human progressive deafness. EMBO J. 2006;25:642–652. doi: 10.1038/sj.emboj.7600951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kubisch C, Schroeder BC, Friedrich T, Lutjohann B, El-Amraoui A, Marlin S, Petit C, Jentsch TJ. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell. 1999;96:437–446. doi: 10.1016/s0092-8674(00)80556-5. [DOI] [PubMed] [Google Scholar]
- 97.Marcus DC, Wu T, Wangemann P, Kofuji P. KCNJ10 (Kir4.1) potassium channel knockout abolishes endocochlear potential. American journal of physiology. 2002;282:C403–407. doi: 10.1152/ajpcell.00312.2001. [DOI] [PubMed] [Google Scholar]
- 98.Ando M, Takeuchi S. Immunological identification of an inward rectifier K+ channel (Kir4.1) in the intermediate cell (melanocyte) of the cochlear stria vascularis of gerbils and rats. Cell and tissue research. 1999;298:179–183. doi: 10.1007/s004419900066. [DOI] [PubMed] [Google Scholar]
- 99.Wangemann P. K+ cycling and the endocochlear potential. Hearing research. 2002;165:1–9. doi: 10.1016/s0378-5955(02)00279-4. [DOI] [PubMed] [Google Scholar]
- 100.Spicer SS, Schulte BA. The fine structure of spiral ligament cells relates to ion return to the stria and varies with place-frequency. Hear Res. 1996;100:80–100. doi: 10.1016/0378-5955(96)00106-2. [DOI] [PubMed] [Google Scholar]
- 101.Zhao HB, Kikuchi T, Ngezahayo A, White TW. Gap junctions and cochlear homeostasis. J Membr Biol. 2006;209:177–186. doi: 10.1007/s00232-005-0832-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kelly JJ, Forge A, Jagger DJ. Development of gap junctional intercellular communication within the lateral wall of the rat cochlea. Neuroscience. 2011;180:360–369. doi: 10.1016/j.neuroscience.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 103.Chang Q, Tang W, Ahmad S, Zhou B, Lin X. Gap junction mediated intercellular metabolite transfer in the cochlea is compromised in connexin30 null mice. PLoS One. 2008;3:e4088. doi: 10.1371/journal.pone.0004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Anselmi F, Hernandez VH, Crispino G, Seydel A, Ortolano S, Roper SD, Kessaris N, Richardson W, Rickheit G, Filippov MA, Monyer H, Mammano F. ATP release through connexin hemichannels and gap junction transfer of second messengers propagate Ca2+ signals across the inner ear. Proc Natl Acad Sci U S A. 2008;105:18770–18775. doi: 10.1073/pnas.0800793105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen Y, Deng Y, Bao X, Reuss L, Altenberg GA. Mechanism of the defect in gap-junctional communication by expression of a connexin 26 mutant associated with dominant deafness. Faseb J. 2005;19:1516–1518. doi: 10.1096/fj.04-3491fje. [DOI] [PubMed] [Google Scholar]
- 106.Mathias RT, Kistler J, Donaldson P. The lens circulation. The Journal of membrane biology. 2007;216:1–16. doi: 10.1007/s00232-007-9019-y. [DOI] [PubMed] [Google Scholar]
- 107.Neijssen J, Herberts C, Drijfhout JW, Reits E, Janssen L, Neefjes J. Cross-presentation by intercellular peptide transfer through gap junctions. Nature. 2005;434:83–88. doi: 10.1038/nature03290. [DOI] [PubMed] [Google Scholar]
- 108.Valiunas V, Polosina YY, Miller H, Potapova IA, Valiuniene L, Doronin S, Mathias RT, Robinson RB, Rosen MR, Cohen IS, Brink PR. Connexin-specific cell-to-cell transfer of short interfering RNA by gap junctions. The Journal of physiology. 2005;568:459–468. doi: 10.1113/jphysiol.2005.090985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Katakowski M, Buller B, Wang X, Rogers T, Chopp M. Functional microRNA is transferred between glioma cells. Cancer research. 2011;70:8259–8263. doi: 10.1158/0008-5472.CAN-10-0604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lim PK, Bliss SA, Patel SA, Taborga M, Dave MA, Gregory LA, Greco SJ, Bryan M, Patel PS, Rameshwar P. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer research. 2010;71:1550–1560. doi: 10.1158/0008-5472.CAN-10-2372. [DOI] [PubMed] [Google Scholar]
- 111.Tritsch NX, Yi E, Gale JE, Glowatzki E, Bergles DE. The origin of spontaneous activity in the developing auditory system. Nature. 2007;450:50–55. doi: 10.1038/nature06233. [DOI] [PubMed] [Google Scholar]
- 112.Majumder P, Crispino G, Rodriguez L, Ciubotaru CD, Anselmi F, Piazza V, Bortolozzi M, Mammano F. ATP-mediated cell-cell signaling in the organ of Corti: the role of connexin channels. Purinergic Signal. 2010;6:167–187. doi: 10.1007/s11302-010-9192-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.del CI, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Telleria D, Menendez I, Moreno F. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med. 2002;346:243–249. doi: 10.1056/NEJMoa012052. [DOI] [PubMed] [Google Scholar]
- 114.Lerer I, Sagi M, Ben-Neriah Z, Wang T, Levi H, Abeliovich D. A deletion mutation in GJB6 cooperating with a GJB2 mutation in trans in non-syndromic deafness: A novel founder mutation in Ashkenazi Jews. Hum Mutat. 2001;18:460. doi: 10.1002/humu.1222. [DOI] [PubMed] [Google Scholar]
- 115.Pallares-Ruiz N, Blanchet P, Mondain M, Claustres M, Roux AF. A large deletion including most of GJB6 in recessive non syndromic deafness: a digenic effect? Eur J Hum Genet. 2002;10:72–76. doi: 10.1038/sj.ejhg.5200762. [DOI] [PubMed] [Google Scholar]
- 116.Rodriguez-Paris J, Schrijver I. The digenic hypothesis unraveled: the GJB6 del(GJB6-D13S1830) mutation causes allele-specific loss of GJB2 expression in cis. Biochem Biophys Res Commun. 2009;389:354–359. doi: 10.1016/j.bbrc.2009.08.152. [DOI] [PubMed] [Google Scholar]
- 117.Rodriguez-Paris J, Tamayo ML, Gelvez N, Schrijver I. Allele-specific impairment of GJB2 expression by GJB6 deletion del(GJB6-D13S1854) PLoS One. 2011;6:e21665. doi: 10.1371/journal.pone.0021665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yang JJ, Huang SH, Chou KH, Liao PJ, Su CC, Li SY. Identification of mutations in members of the connexin gene family as a cause of nonsyndromic deafness in Taiwan. Audiology & neurootology. 2007;12:198–208. doi: 10.1159/000099024. [DOI] [PubMed] [Google Scholar]
- 119.Lopez-Bigas N, Olive M, Rabionet R, Ben-David O, Martinez-Matos JA, Bravo O, Banchs I, Volpini V, Gasparini P, Avraham KB, Ferrer I, Arbones ML, Estivill X. Connexin 31 (GJB3) is expressed in the peripheral and auditory nerves and causes neuropathy and hearing impairment. Human molecular genetics. 2001;10:947–952. doi: 10.1093/hmg/10.9.947. [DOI] [PubMed] [Google Scholar]
- 120.Macari F, Landau M, Cousin P, Mevorah B, Brenner S, Panizzon R, Schorderet DF, Hohl D, Huber M. Mutation in the gene for connexin 30.3 in a family with erythrokeratodermia variabilis. American journal of human genetics. 2000;67:1296–1301. doi: 10.1016/s0002-9297(07)62957-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Butterweck A, Elfgang C, Willecke K, Traub O. Differential expression of the gap junction proteins connexin45, -43, -40, -31, and -26 in mouse skin. European journal of cell biology. 1994;65:152–163. [PubMed] [Google Scholar]
- 122.Di WL, Rugg EL, Leigh IM, Kelsell DP. Multiple epidermal connexins are expressed in different keratinocyte subpopulations including connexin 31. The Journal of investigative dermatology. 2001;117:958–964. doi: 10.1046/j.0022-202x.2001.01468.x. [DOI] [PubMed] [Google Scholar]
- 123.Goliger JA, Paul DL. Expression of gap junction proteins Cx26, Cx31.1, Cx37, and Cx43 in developing and mature rat epidermis. Dev Dyn. 1994;200:1–13. doi: 10.1002/aja.1002000102. [DOI] [PubMed] [Google Scholar]
- 124.Wiszniewski L, Limat A, Saurat JH, Meda P, Salomon D. Differential expression of connexins during stratification of human keratinocytes. The Journal of investigative dermatology. 2000;115:278–285. doi: 10.1046/j.1523-1747.2000.00043.x. [DOI] [PubMed] [Google Scholar]
- 125.Potten CS. Cell replacement in epidermis (keratopoiesis) via discrete units of proliferation. International review of cytology. 1981;69:271–318. doi: 10.1016/s0074-7696(08)62326-8. [DOI] [PubMed] [Google Scholar]
- 126.Masgrau-Peya E, Salomon D, Saurat JH, Meda P. In vivo modulation of connexins 43 and 26 of human epidermis by topical retinoic acid treatment. J Histochem Cytochem. 1997;45:1207–1215. doi: 10.1177/002215549704500904. [DOI] [PubMed] [Google Scholar]
- 127.Djalilian AR, McGaughey D, Patel S, Seo EY, Yang C, Cheng J, Tomic M, Sinha S, Ishida-Yamamoto A, Segre JA. Connexin 26 regulates epidermal barrier and wound remodeling and promotes psoriasiform response. The Journal of clinical investigation. 2006;116:1243–1253. doi: 10.1172/JCI27186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Brandner JM, Houdek P, Husing B, Kaiser C, Moll I. Connexins 26, 30, and 43: differences among spontaneous, chronic, and accelerated human wound healing. The Journal of investigative dermatology. 2004;122:1310–1320. doi: 10.1111/j.0022-202X.2004.22529.x. [DOI] [PubMed] [Google Scholar]
- 129.Coutinho P, Qiu C, Frank S, Tamber K, Becker D. Dynamic changes in connexin expression correlate with key events in the wound healing process. Cell biology international. 2003;27:525–541. doi: 10.1016/s1065-6995(03)00077-5. [DOI] [PubMed] [Google Scholar]
- 130.Goliger JA, Paul DL. Wounding alters epidermal connexin expression and gap junction-mediated intercellular communication. Molecular biology of the cell. 1995;6:1491–1501. doi: 10.1091/mbc.6.11.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pollok S, Pfeiffer AC, Lobmann R, Wright CS, Moll I, Martin PE, Brandner JM. Connexin 43 mimetic peptide Gap27 reveals potential differences in the role of Cx43 in wound repair between diabetic and non-diabetic cells. Journal of cellular and molecular medicine. 2011;15:861–873. doi: 10.1111/j.1582-4934.2010.01057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Qiu C, Coutinho P, Frank S, Franke S, Law LY, Martin P, Green CR, Becker DL. Targeting connexin43 expression accelerates the rate of wound repair. Curr Biol. 2003;13:1697–1703. doi: 10.1016/j.cub.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 133.Smith FJ, Morley SM, McLean WH. A novel connexin 30 mutation in Clouston syndrome. The Journal of investigative dermatology. 2002;118:530–532. doi: 10.1046/j.0022-202x.2001.01689.x. [DOI] [PubMed] [Google Scholar]