

Diverse proteins form amyloid aggregates that may be functional or pathological. Here the authors use the E. coli fiber export system to produce amyloid fibrils composed of heterologous proteins. Several yeast prion proteins and a human huntingtin variant form amyloid fibrils when directed for export via the curli export pathway, whereas nonamyloidogenic proteins do not. This study thus establishes a simple screening platform to identify amyloidogenic proteins.
Keywords: Sup35, amyloid, curli, huntingtin, protein export, yeast prions
Abstract
Diverse proteins are known to be capable of forming amyloid aggregates, self-seeding fibrillar assemblies that may be biologically functional or pathological. Well-known examples include neurodegenerative disease-associated proteins that misfold as amyloid, fungal prion proteins that can transition to a self-propagating amyloid form and certain bacterial proteins that fold as amyloid at the cell surface and promote biofilm formation. To further explore the diversity of amyloidogenic proteins, generally applicable methods for identifying them are critical. Here we describe a cell-based method for generating amyloid aggregates that relies on the natural ability of Escherichia coli cells to elaborate amyloid fibrils at the cell surface. We use several different yeast prion proteins and the human huntingtin protein to show that protein secretion via this specialized export pathway promotes acquisition of the amyloid fold specifically for proteins that have an inherent amyloid-forming propensity. Furthermore, our findings establish the potential of this E. coli-based system to facilitate the implementation of high-throughput screens for identifying amyloidogenic proteins and modulators of amyloid aggregation.
Diverse proteins from all domains of life are capable of forming amyloid aggregates made up of highly ordered β-sheet-rich fibrils (Chiti and Dobson 2006). These fibrils share a characteristic cross-β spine in which the β strands run perpendicular to the fibril axis (Toyama and Weissman 2011). Among protein aggregates, amyloid fibrils are unusually stable, typically exhibiting SDS resistance. A hallmark of amyloid aggregation is that it proceeds via a self-seeding mechanism with a characteristic lag phase that can be eliminated by the addition of preformed fibrils (Chiti and Dobson 2006).
Among those proteins that are known to form amyloid aggregates under physiological conditions are the culprits in various devastating neurodegenerative diseases, including Alzheimer's, Parkinson's, Huntington's, and the transmissible spongiform encephalopathies (TSEs) (Chiti and Dobson 2006). In addition to these disease-associated proteins that have a propensity to misfold as amyloid, mammalian proteins that assemble into amyloid aggregates to perform normal biological functions have been described. For example, various endocrine hormones are stored as amyloid aggregates in secretory granules (Maji et al. 2009).
Fungal prion proteins make up a particularly intriguing class of amyloidogenic proteins (Wickner et al. 2007; Tuite and Serio 2010; Liebman and Chernoff 2012). In general, fungal prion proteins have the potential to adopt alternative stable conformations, a so-called native fold and a self-propagating amyloid fold, which is the basis for prion formation. Often, but not in all cases, conversion to the prion form phenocopies a partial or full loss-of-function mutation. Although conversion to the prion form is typically a rare event, once formed, prions are stably transmitted from generation to generation and “infectious” when transferred to naive strains. Thus, fungal prions act as non-Mendelian protein-based hereditary elements that can confer new phenotypic traits on the cells that harbor them.
In bacteria, all known amyloid-forming proteins aggregate extracellularly, in most cases forming surface-attached amyloid fibers (Blanco et al. 2011). In Escherichia coli, these fibers, known as curli fibers, are composed of two proteins, CsgA and CsgB, that are directed to the outside of the cell by a dedicated export system (Blanco et al. 2011). Here we demonstrate that the curli export apparatus can be appropriated for the production of extracellular amyloid fibers composed of heterologous amyloidogenic proteins derived from yeast and humans. Our findings indicate that protein secretion through the curli export pathway facilitates acquisition of the amyloid fold specifically for proteins that have an inherent amyloid-forming propensity. The bacteria-based system that we describe thus provides a simple and efficient means to distinguish amyloidogenic proteins from those that do not readily undergo conversion to an amyloid state.
Results
Experimental plan
Our goal was to establish a generalizable cell-based system that would recapitulate aspects of widely used in vitro assays for studying amyloid aggregation. Such in vitro assays involve purifying the protein of interest and subsequently monitoring its aggregation from a soluble and fully or partially unfolded state (for example, see Wang et al. 2007). We reasoned that a cell-based secretion system could similarly enable the separation of the protein of interest from the bulk of cellular protein in a fully or partially unfolded state that might facilitate acquisition of the amyloid fold. We therefore sought to determine whether heterologous amyloid-forming proteins could be directed for export via the E. coli curli system and, if so, whether they would form extracellular amyloid fibrils.
Curli fibers are composed of two related amyloidogenic proteins: the major subunit CsgA and the minor subunit CsgB, which remains anchored in the outer membrane, where it nucleates the polymerization of the fully secreted CsgA subunits (Chapman et al. 2002; Hammer et al. 2007; Blanco et al. 2011). Both CsgA and CsgB are translocated across the inner membrane into the periplasm by the general Sec translocon system; subsequently, they are directed through a curli-specific pore-like structure in the outer membrane that is formed by the CsgG protein (Robinson et al. 2006). The specificity of this outer membrane secretion process depends on a 22-amino-acid signal sequence at the N terminus of the mature CsgA and CsgB proteins (Robinson et al. 2006). Under native conditions, curli biogenesis also depends on several accessory proteins (Blanco et al. 2011). Nevertheless, despite the complex requirements for curli biogenesis, previous work indicates that CsgG overproduction in the absence of all other curli factors enables the efficient secretion of CsgA, which does not assemble into amyloid fibrils, however, due to the absence of the nucleator CsgB (Chapman et al. 2002; Robinson et al. 2006). Thus, our strategy for assessing the fate of heterologous amyloid-forming proteins directed to the curli export channel was to fuse the CsgA signal sequence to a set of target amyloidogenic proteins and overproduce these fusion proteins along with CsgG in a strain lacking CsgA and CsgB.
The prion-forming domain of yeast Sup35 protein forms amyloid-like material when exported from E. coli using the curli system
We initially tested the well-characterized yeast prion protein Sup35 in this system. An essential translation release factor, Sup35 has a modular structure with an N-terminal region (N) that contains the critical prion-forming determinants, a highly charged middle region (M), and a C-terminal domain (C) that carries out the translation release function (Glover et al. 1997; Liebman and Chernoff 2012). Together, the N and M regions can function as a separable prion-forming module that is transferable to heterologous proteins (Li and Lindquist 2000). Accordingly, we constructed a plasmid vector designed to direct the arabinose-inducible synthesis of Sup35 NM (hereafter NM) fused to the bipartite CsgA signal sequence (CsgAss), consisting of a SecA-dependent secretion signal (which is cleaved after passage through the Sec translocon) and the CsgG targeting sequence (which is retained at the N terminus of the mature protein). As a control, we constructed an otherwise identical plasmid directing the synthesis of the M domain (which lacks the essential prion-forming determinants and does not undergo conversion to an amyloid conformation) (Glover et al. 1997) fused to the CsgAss.
We introduced each of these plasmids into a ΔcsgBAC strain of E. coli containing a second plasmid that directs the IPTG-inducible overproduction of CsgG. We first plated cells containing either the NM plasmid or the M plasmid onto inducing (i.e., arabinose + IPTG) medium containing Congo red (CR), an amyloid-binding dye that can be used to detect the presence of curli fibers on E. coli cells (Hammar et al. 1995; Chapman et al. 2002). Like curli-positive cells of wild-type E. coli, cells producing CsgAss-NM formed colonies that stained bright red on this medium, whereas cells producing CsgAss-M formed pale colonies (Supplemental Fig. S1). Furthermore, samples of the CsgAss-NM cells that had been plated on inducing medium revealed an abundance of fibrillar aggregates when examined by transmission electron microscopy (EM), and the protein content of these fibrils was confirmed by immuno-gold labeling (Fig. 1A). In contrast, no such aggregates were observed in the case of the CsgAss-M cells. A control experiment indicated that the CsgAss directed the export of CsgAss-M and that the absence of CsgAss-M aggregates was not due to lower levels of secreted protein (Supplemental Fig. S2). We also used bright-field microscopy to show directly that the CsgAss-NM fibrils bind CR and manifest “apple-green” birefringence when examined between crossed polarizers (Fig. 1B), a property that is diagnostic of amyloid material (Teng and Eisenberg 2009).
Figure 1.

Amyloid-like aggregates formed by secreted CsgAss-NM. (A) Electron micrographs of immunolabeled CsgAss-NM (left) and CsgAss-M (right) scraped cell samples. Fibrils were detected only with the CsgAss-NM sample. (B) Micrographs of CsgAss-NM scraped cell sample harvested from CR-containing agar. Extracellular material binds CR (left) and displays apple-green birefringence when viewed between crossed polarizers (right). (C) CsgAss-NM, but not CsgAss-M, scraped cell samples contain SDS-resistant aggregates that are solubilized upon boiling, as detected by filter retention using an antibody that recognizes the M domain.
Another diagnostic characteristic of amyloid aggregates is their resistance to denaturation in the presence of SDS (Bagriantsev et al. 2006). To determine whether the aggregates produced by CsgAss-NM cells were SDS-resistant, we scraped colonies (together with the fibrillar aggregates) off inducing medium (without CR), resuspended the material in 2% SDS, and used a filter retention assay (Alberti et al. 2009) to test for the presence of SDS-stable NM aggregates (detectable with an anti-NM antibody). This analysis revealed an abundance of SDS-resistant aggregated material specifically with the CsgAss-NM cells that was solubilized when the samples were boiled (Fig. 1C).
An additional key feature of amyloid is that aggregation proceeds via a self-seeding mechanism with a characteristic lag phase that can be eliminated by the addition of preformed fibrils (Chiti and Dobson 2006). We therefore tested the ability of the scraped cell suspensions to seed the conversion of soluble NM protein to the amyloid-aggregated (SDS-stable) form. To carry out this test, we diluted the cell suspensions into extracts prepared from E. coli cells containing soluble NM-GFP fusion protein and used the filter retention assay to monitor the appearance of SDS-stable NM aggregates over time. We showed previously that these E. coli cell extracts support the slow, spontaneous conversion of NM-GFP to the amyloid form and that this conversion reaction is accelerated in the presence of preassembled seed particles (Garrity et al. 2010). As expected based on these prior observations, we detected the accumulation of a relatively small amount of SDS-stable NM aggregates when we used the fibril-free CsgAss-M cell suspension as seed (Fig. 2A). However, the conversion reaction was significantly accelerated when we used the CsgAss-NM suspension as seed (Fig. 2A). As positive and negative controls, respectively, we used [PSI+] and [psi−] yeast extracts as seed (i.e., extracts prepared from yeast cells containing Sup35 in the prion and nonprion forms, respectively). A scraped cell suspension prepared from cells exporting native CsgA, which also forms amyloid fibrils under our experimental conditions (see below), served as an additional negative control because cross-seeding is not expected to occur. Another pair of control reactions indicated that no SDS-stable NM aggregates accumulated when the seeding-competent samples were diluted into extract prepared from E. coli cells containing unfused GFP (empty extract).
Figure 2.

Amyloid-like aggregates of CsgAss-NM are seeding-competent and infectious. (A) E. coli cell extracts containing SDS-soluble NM-GFP were seeded with scraped cell samples (*) (CsgAss-NM, CsgAss-M, or CsgA) or with yeast extracts (**) prepared from a [PSI+] or [psi−] strain. Seed-only control samples consisted of the CsgAss-NM scraped cell sample or the [PSI+] yeast extract diluted into E. coli cell extracts containing overproduced GFP only. Cartoon depicts experimental protocol. Samples from seeded reactions were removed at the indicated time points and treated with 2% SDS, and the presence of SDS-stable aggregates was assayed by filter retention. SDS-stable aggregates that were retained were probed with an anti-GFP antibody. (B) Infection of [pin−][psi−] yeast spheroplasts with NM-GFP aggregates isolated by centrifugation from the seeding reactions at the 30-min time point and either sonicated (post-sonication) or not (presonication). Cartoon depicts experimental protocol.
Material derived from extracellular NM aggregates produced by E. coli can induce prion formation when introduced into yeast cells
Having determined that cells exporting CsgAss-NM produce material with all the hallmarks of amyloid, we sought to find out whether this amyloid-like material was infectious when introduced into yeast cells. To do this, we took advantage of a well-established protocol for introducing exogenous prion aggregates into yeast cells and monitoring the conversion of Sup35 from the nonprion [psi−] form to the prion [PSI+] form (Tanaka and Weissman 2006). Because [PSI+] cells are deficient in translation termination and manifest a heritable nonsense suppression phenotype, they can readily be distinguished from [psi−] cells on appropriate indicator medium. Importantly, the spontaneous conversion of Sup35 to the prion form in yeast cells is strictly dependent on the presence of a so-called [PSI+] inducibility (PIN) factor, which is itself a prion (Derkatch et al. 1997, 2001; Osherovich and Weissman 2001). Thus, the use of a [pin−] strain ensures that only seeded conversion events are detected. Accordingly, we tested scraped cell suspensions of our E. coli strains (supplemented with plasmid DNA encoding a yeast-selectable marker) for infectivity by using them to transform yeast spheroplasts prepared from a suitably marked [pin−] [psi−] yeast strain. [PSI+] transformants were obtained when we used the CsgAss-NM cells (at a frequency of 0.4%) but not when we used the CsgAss-M cells (at a frequency of <0.05%) (Supplemental Table S1).
We suspected that this relatively low [psi−]-to-[PSI+] conversion frequency was attributable to the fact that the fibrillar material, which appeared as a dense meshwork of long fibers (Supplemental Fig. S3), likely was inefficiently taken up by the yeast cells and, once internalized, may have provided relatively few free ends to nucleate the polymerization of soluble Sup35. In fact, in vitro generated Sup35 aggregates are typically fragmented by sonication to increase their infectivity (King and Diaz-Avalos 2004; Tanaka and Weissman 2006). However, because sonication can also stimulate the assembly of soluble NM into amyloid aggregates, we could not readily test whether sonication increased the infectivity of the fibrillar material in the scraped cell suspensions due to the presence of intact cells that could potentially release soluble CsgAss-NM. As an alternative strategy, we assessed the infectivity of the material generated in the seeding reactions of Figure 2A. To do this, we isolated the high-molecular-weight aggregates from the seeding reactions (at the 30-min time point) by centrifugation and tested them for infectivity before and after sonication (Fig. 2B); that is, we used the isolated aggregates to transform [pin−] [psi−] yeast spheroplasts, as described above. In accord with our previous observations using E. coli cell extracts containing soluble NM-GFP as substrate for the conversion reaction (Garrity et al. 2010), sonication dramatically increased the infectivity of the aggregates. Furthermore, the seeded reactions exhibited a marked increase in infectivity as compared with the mock-seeded reactions. Thus, whereas the mock-seeded reactions (containing the CsgAss-M suspension, the CsgA suspension, or the [psi−] yeast extract as seed) resulted in conversion frequencies of 9%–10%, the reactions seeded with the CsgAss-NM suspension and the [PSI+] extract resulted in conversion frequencies of 39% and 27%, respectively. We conclude that E. coli cells exporting CsgAss-NM produce amyloid-like material that is capable of seeding the conversion of soluble NM to an infectious, prion conformation.
The curli-based export system provides a general method for detecting amyloid-forming potential
To evaluate the generality of our findings with CsgAss-NM, we tested three other yeast prion proteins (Rnq1, Cyc8, and New1) and four additional yeast proteins that were identified bioinformatically as having candidate prion domains (PrDs) (Alberti et al. 2009). The candidate PrDs of two of these four proteins (Mss11 and Pub1) were shown to form amyloid aggregates in vitro and to behave as prions in vivo, whereas the candidate PrDs of the other two (Snf2 and Med2) did not form amyloid aggregates in vitro or behave as prions in vivo (Alberti et al. 2009); thus, Snf2 and Med2 served as negative controls. For each of the seven proteins, we fused the PrD (or candidate PrD in the case of the negative controls) to the CsgAss and provided a His6 tag at the C terminus of the fusion protein. As for CsgAss-NM, transmission EM of cell samples scraped from inducing medium revealed extracellular fibrillar aggregates for each of the confirmed PrDs (Fig. 3A) but not for the negative controls. Furthermore, the aggregates (detected with an antibody to the C-terminal His tag) were SDS-resistant, as determined by the filter retention assay, and were solubilized when the material was boiled (Fig. 3B); as expected, the filter retention assay revealed no SDS-resistant material in the case of the negative control samples (Supplemental Fig. S4). We also tested CsgA itself (with a C-terminal His tag) and similarly observed fibril formation and the presence of SDS-resistant aggregates (Supplemental Fig. S5), indicating that under our experimental conditions, the local concentration of exported CsgA is sufficiently high to allow polymerization in the absence of the CsgB nucleator (Hammer et al. 2007).
Figure 3.
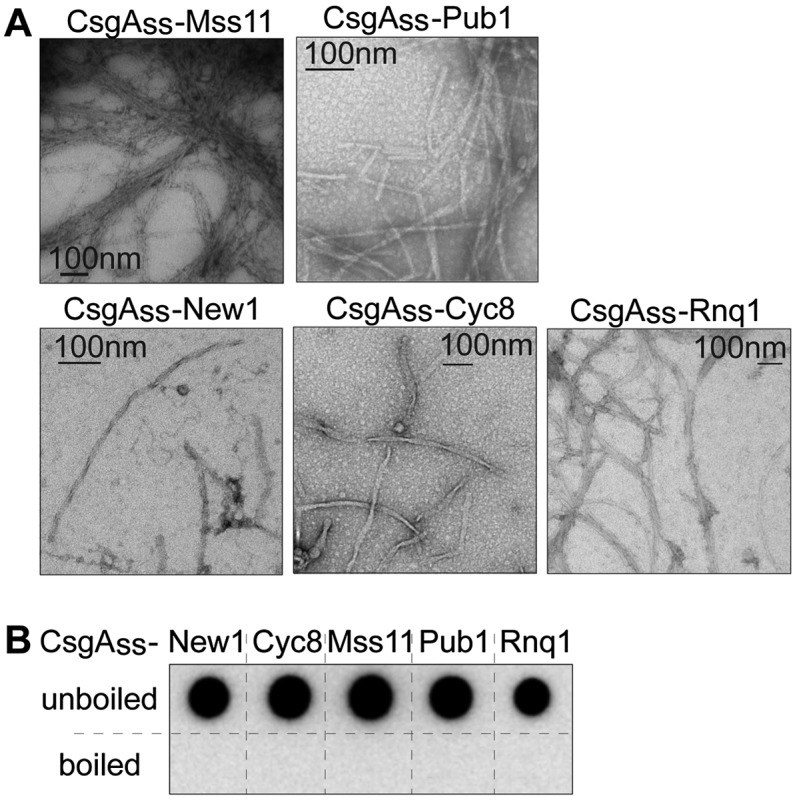
Amyloid-like aggregates formed by other secreted yeast prion proteins. (A) Electron micrographs of scraped cell samples containing various yeast prion proteins as CsgAss fusions. Fibrillar aggregates were detected for all samples. (B) All samples contain SDS-resistant aggregates (as detected by filter retention), which were solubilized upon boiling.
To further evaluate the sensitivity of our curli-based export system, we took advantage of a previously characterized NM variant (NMRΔ) lacking four of the five oligopeptide repeat sequences that are essential for prion formation; previous analysis revealed that Sup35 NMRΔ is unable to undergo spontaneous conversion to the prion form in vivo and that purified NMRΔ assembles into amyloid aggregates much more slowly than NM in vitro (Liu and Lindquist 1999). We thus compared cells producing CsgAss-NM, CsgAss-NMRΔ, and CsgAss-M in order to test the potential of the export system to distinguish more or less amyloidogenic variants of a single protein. We found that cells exporting CsgAss-NMRΔ produced extracellular fibrils, but they were both thinner and far less abundant than those produced by the CsgAss-NM cells (Fig. 4A). Consistently, comparison of the scraped cell samples revealed that the CsgAss-NMRΔ sample contained much less SDS-stable NM material than the CsgAss-NM sample (Fig. 4B). These observations were paralleled by the colony colors as visualized on inducing medium containing CR, with the CsgAss-NMRΔ cells staining a pale shade of red (Fig. 4B) despite secreting an amount of fusion protein similar to the CsgAss-NM cells (Supplemental Fig. S2). While colony color can effectively be used to report on the amount of amyloid formed by a particular protein (e.g., NM) and its variants, we note that colony color cannot be used as a general surrogate for the presence of amyloid-like material because CR binding varies depending on the particular protein (Teng and Eisenberg 2009). In fact, when we examined cells producing each of the five other yeast prion proteins and CsgA, we observed a range of colony colors, with the CsgAss-Pub1 cells staining as red as the CsgAss-NM cells, and the CsgAss-Cyc8 cells staining only slightly darker than the negative control cells (Supplemental Fig. S1).
Figure 4.
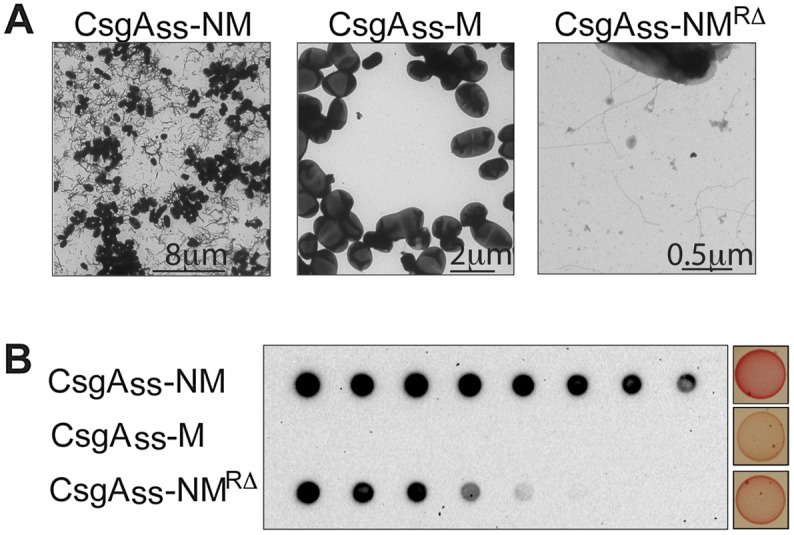
Aggregation propensity of CsgAss-NM variants. Fibril density within scraped cell samples (CsgAss-NM, CsgAss-M, or CsgAss-NMRΔ) as detected by EM (A) and the amount of SDS-resistant material as detected by filter retention (B) parallel the known amyloidogenicity of these protein variants, as does the colony color phenotype of the cells when plated on agar containing CR.
Having demonstrated that six different amyloidogenic yeast proteins form extracellular amyloid fibrils when directed to the curli export apparatus, we examined a disease-associated mammalian amyloid-forming protein, exon 1 of the human huntingtin protein (Htt). We took advantage of the fact that the amyloidogenicity of Htt depends on the number of glutamines within the so-called polyQ region (Scherzinger et al. 1997). Thus, we fused both a pathogenic polyQ expansion variant of Htt exon 1 (Htt72Q) and a nonpathogenic variant (Htt25Q) to the CsgAss and provided a His6 tag at the C terminus. Cells exporting CsgAss-Htt72Q produced an abundance of fibrils organized into fan-like structures (Fig. 5A), whereas no fibrils were observed with Htt25Q (although the two proteins were secreted at comparable levels) (Supplemental Fig. S6). This fibrillar material bound CR and exhibited apple-green birefringence when examined between crossed polarizers (Fig. 5B). Finally, the aggregates were SDS-stable; strikingly, they remained SDS-stable even when the samples were boiled, a property that is characteristic of Htt amyloid aggregates (Fig. 5C; Scherzinger et al. 1997).
Figure 5.
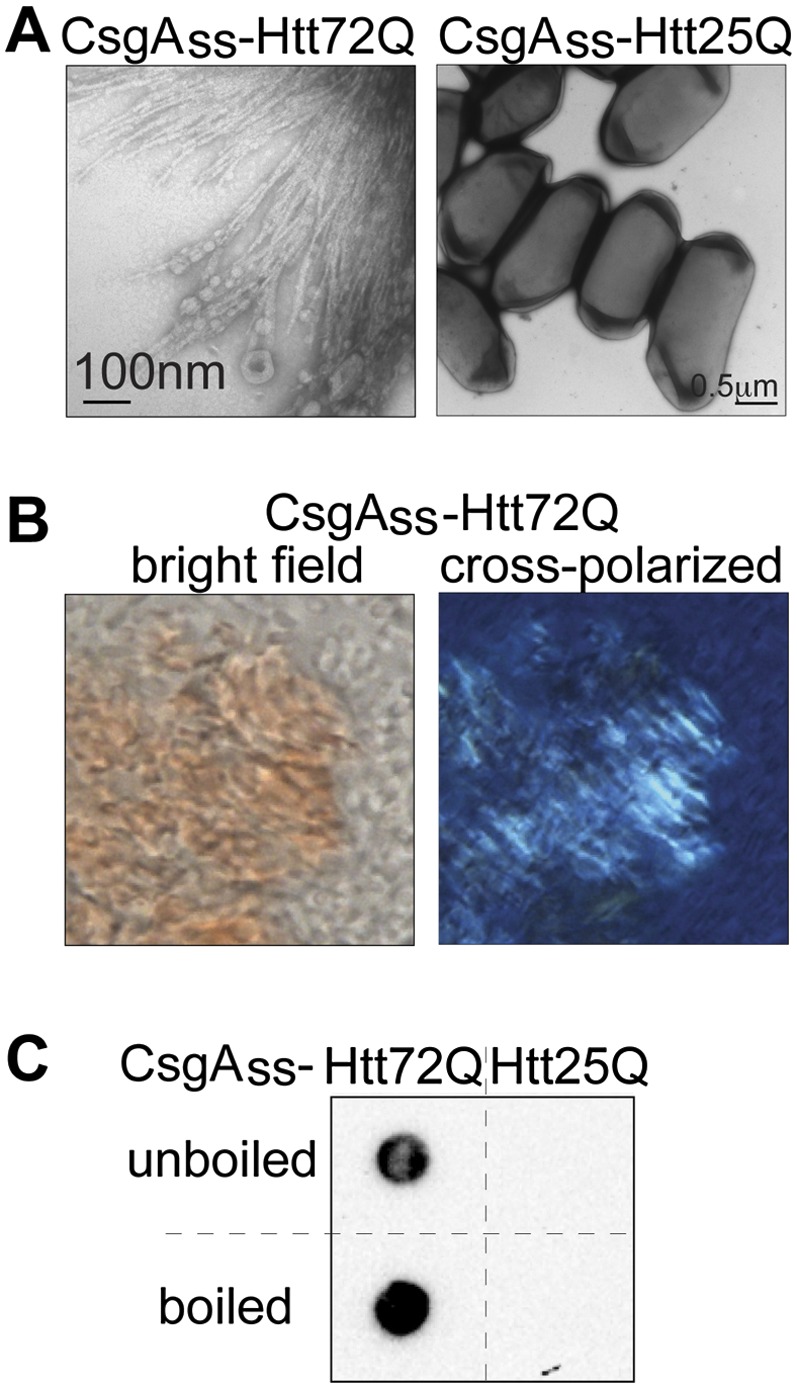
Amyloid aggregates formed by secreted CsgAss-Htt[exon1]. (A) Electron micrographs of CsgAss-Htt72Q (left) and CsgAss-Htt25Q (right) scraped cell samples. Fibrils were detected only with the CsgAss-Htt72Q sample. (B) Micrographs of CsgAss-Htt72Q scraped cell sample harvested from CR-containing agar. Extracellular material binds CR (left) and displays apple-green birefringence when viewed between crossed polarizers (right). (C) CsgAss-Htt72Q, but not CsgAss-Htt25Q, scraped cell samples contain SDS-resistant aggregates (as detected by filter retention), which are not solubilized by boiling.
Curli-based genetic screen enables identification of an amyloidogenic protein from E. coli
Our findings suggest that amyloidogenic proteins readily form extracellular amyloid fibrils when secreted via the curli export pathway. In principle, therefore, our curli-based export system should provide a convenient method for carrying out unbiased screens to identify amyloidogenic proteins from genomic or cDNA libraries. As a preliminary test of this possibility, we performed a pilot screen using a pool of ∼614 E. coli ORFs (approximately one-seventh of the complete ORF library) (Saka et al. 2005). We designed universal primers that enabled us to fuse the collection of ORFs to the CsgAss and to provide a His6 tag at the C terminus. We used the resulting library of plasmids directing the arabinose-inducible synthesis of these fusion proteins to transform ΔcsgBAC cells already containing the CsgG plasmid and plated the transformants on inducing medium supplemented with CR. We examined ∼10,000 transformants and identified two particularly bright-red colonies that resembled those formed by cells exporting CsgAss-NM. DNA sequence analysis revealed that both of these transformants contained the same plasmid encoding fliE. A component of the flagellar basal body, FliE is not known to form amyloid under physiological conditions; however, previous work indicates that FliE readily forms amyloid fibrils in vitro (Saijo-Hamano et al. 2004). Consistently, cells exporting the CsgAss-FliE fusion protein revealed an abundance of fibrillar aggregates when examined by transmission EM (Supplemental Fig. S7). We conclude that the curli-based export system can be exploited as a means to screen genomic libraries for amyloidogenic proteins (see the Discussion).
Discussion
Our results indicate that heterologous amyloid-forming proteins from yeast, humans, and bacteria readily adopt the amyloid fold when secreted via the E. coli curli export apparatus. We tested six yeast proteins that are capable of assembling as amyloid fibrils in vitro as well as two nonamyloidogenic controls (Alberti et al. 2009) and found that only the former generated extracellular amyloid fibrils upon export. Furthermore, in the case of the well-characterized yeast prion protein Sup35, we showed that the protein accesses an infectious prion conformation. In addition, we tested the human Htt protein and found that a pathogenic polyQ expansion variant (Htt72Q) formed amyloid fibrils when exported by the E. coli cells, whereas a nonpathogenic variant (Htt25Q) did not. Finally, a pilot screen performed with a partial E. coli ORF library establishes the feasibility of using this curli-based export system as a platform to screen for amyloidogenic proteins from genomic or cDNA libraries.
Secretion via the curli export pathway facilitates acquisition of the amyloid fold
Our findings suggest that passage of an amyloidogenic protein through the CsgG pore facilitates its assembly into amyloid fibrils in the extracellular milieu. We speculate that passage of substrate proteins through the export pore in a relatively unfolded conformation (Robinson et al. 2006; Nenninger et al. 2011) and their accumulation to a relatively high local concentration in the extracellular milieu facilitate their amyloid aggregation. Additionally, the lipid environment at the cell surface may be a contributing factor; we note that lipids have been shown previously to facilitate conversion of recombinant PrP to an infectious amyloid conformation (Wang et al. 2010).
Our results indicate further that the curli export process facilitates amyloid conversion for proteins that ordinarily access the amyloid conformation only under restrictive conditions and/or rarely. In particular, the spontaneous conversion of Sup35 NM to the prion form in yeast cells is strictly dependent on the presence of a PIN factor and occurs only rarely (Derkatch et al. 1997, 2001; Osherovich and Weissman 2001; Liebman and Chernoff 2012). In contrast, secretion through the curli export apparatus circumvents the requirement for a PIN factor.
A cell-based method for evaluating amyloidogenicity
Our findings indicate that the curli export system can serve as a general cell-based method for producing amyloid aggregates and distinguishing amyloidogenic proteins from those that do not readily undergo conversion to an amyloid state. This system, which we term C-DAG (curli-dependent amyloid generator), provides a convenient alternative to widely used in vitro assays for studying amyloid aggregation. In particular, C-DAG provides an efficient method for evaluating amyloid-forming potential without a need for protein purification.
In principle, C-DAG should facilitate the implementation of high-throughput screens for identifying amyloidogenic proteins and modulators of amyloid aggregation. The results of the pilot screen that we performed using a partial E. coli ORF library imply that plating the cells on solid medium containing CR can identify amyloidogenic proteins that bind CR efficiently. Furthermore, our findings with both strong and weak CR binders suggest that the use of the filter retention assay as a primary screening step should reliably identify a broader spectrum of amyloidogenic proteins.
Several genome-wide screens have been carried out in yeast in order to identify new prion proteins and prion protein candidates. An essential Q/N-rich region found in the originally identified yeast prion proteins was exploited in developing algorithms to identify additional prion proteins (Michelitsch and Weissman 2000; Santoso et al. 2000; Sondheimer and Lindquist 2000). More recently, using a variant bioinformatic approach, Alberti et al. (2009) identified some 200 proteins in Saccharomyces cerevisiae with candidate Q/N-rich prion-forming domains; among the 100 that were examined experimentally, approximately one-fourth were found to contain a bona fide prion-forming domain. Taking a strictly genetic approach, Suzuki et al. (2012) performed a functional genome-wide screen by identifying yeast ORFs that could serve as PIN factors for a synthetic Sup35 variant; intriguingly, this screen uncovered a new prion that lacks the Q/N-rich signature region. As an unbiased screening platform, C-DAG may be particularly useful in uncovering novel classes of prion-like or other amyloidogenic proteins because the export process can enable bypass of restrictive conditions for amyloid conversion (see above).
In addition to identifying amyloidogenic proteins, C-DAG could facilitate the identification of modulators of amyloid aggregation. Thus, for any particular amyloid-forming protein that binds CR when assembled into amyloid fibrils, the use of CR-containing medium would facilitate the identification of mutations or small molecules that hinder or accelerate the conversion process, and the filter retention assay would provide a secondary screening step. A similar approach has enabled the genetic dissection of the curli system (Wang and Chapman 2008). Although most screens for small molecule modulators of amyloid aggregation depend on in vitro assembly reactions, cell-based systems have also been developed for the discovery of amyloid modulators, including a yeast-based assay that detects the loss of the [PSI+] phenotype (Bach et al. 2003) and two bacteria-based systems for studying Aβ aggregation (Kim et al. 2006; Lee et al. 2009).
A potential means to interrogate the amylome
Over the past decade, diverse computational approaches have been developed for predicting amyloid-forming propensity in order to define the amylome—the proteome subset capable of forming amyloid-like fibrils (Goldschmidt et al. 2010). These include both sequence-based approaches (Fernandez-Escamilla et al. 2004; Trovato et al. 2006; Tartaglia et al. 2008; Bryan et al. 2009; Maurer-Stroh et al. 2010; O'Donnell et al. 2011) and structure-based approaches (Thompson et al. 2006; Zhang et al. 2007; Goldschmidt et al. 2010), the latter of which are designed to identify short amyloidogenic motifs based on their steric zipper-forming potential (Sawaya et al. 2007). In particular, the Eisenberg group (Goldschmidt et al. 2010) has used three-dimensional profiling to evaluate short (six-residue) protein segments to identify high fibrillation propensity (HP) segments. Importantly, recent work indicates that despite the prevalence of HP segments (most proteins contain at least one), their ability to induce protein fibrillation is highly context-dependent (Goldschmidt et al. 2010). Specifically, such segments must be surface-exposed with sufficient conformational flexibility to drive fibrillation. Despite the progress that has been made in predicting amyloid-forming propensity, significant challenges remain, especially when no structural information is available. Additionally, different algorithms appear to be differentially suited for the identification of different classes of amyloid-forming proteins (Toombs et al. 2012). Furthermore, experimental methods that can be used for algorithm validation by rapidly assessing amyloidogenicity on a genome-wide scale are lacking. We suggest that C-DAG, which detects amyloid fibril formation under a uniform and physiologically relevant set of conditions, is particularly well suited for generating comprehensive data sets against which to test and refine computational models, thereby extending our understanding of the nature of amyloid formation.
Materials and methods
Strains, plasmids, and cell growth
A complete list of strains and plasmids is provided in Supplemental Table S2. E. coli strain VS16 was constructed by replacing the csgBAC genes of strain MC4100 with a kanamycin resistance gene using a previously described protocol (Datsenko and Wanner 2000). CsgG was produced under the control of the lacUV5 promoter on plasmid pVS76. Export-directed fusion proteins contained the first 42 residues of CsgA at the N terminus and a His6 tag at the C terminus and were produced under the control of the arabinose-inducible PBAD promoter. For fibril production, overnight cultures of VS16 transformed with compatible plasmids directing the synthesis of CsgG and an export-directed fusion protein were diluted to OD600 0.01 in LB supplemented with the appropriate antibiotics (100 μg/mL carbenicillin, 25 μg/mL chloramphenicol). After 30 min of growth at 37°C, 5 μL of the culture was spotted on LB agar plates supplemented with the appropriate inducers (0.2% [w/v] L-arabinose, 1 mM IPTG), antibiotics (100 μg/mL carbenicillin; 25 μg/mL chloramphenicol), and, where indicated, CR (5 μg/mL). Plates were then incubated for 120 h at room temperature.
Scraped cell suspension preparation
To prepare unlysed cell suspensions, cells that had been spotted on agar were scraped off the plates in PBS (phosphate-buffered saline) and normalized to OD600 1.0 in a volume of 100 μL. To prepare lysed cell suspensions, BugBuster protein extraction reagent (Novagen), rlysozyme (Novagen), and OmniCleave endonuclease (Epicentre) were added to the unlysed cell suspensions to final concentrations of 0.5×, 300 U/mL, and 10 U/mL, respectively, followed by incubation for 15 min at room temperature with gentle rocking. SDS was then added to 2% (v/w). Boiled samples were incubated for 20 min at 98°C after the addition of SDS.
Filter retention assay
The filter retention assay was performed as previously described (Garrity et al. 2010). Lysed cell suspensions were filtered through the membrane in a volume of 200 μL. The membrane was probed with either anti-Sup35 (yS-20, Santa Cruz Biotechnology) or anti-His6 (clone His-2, Roche) to detect immobilized protein. Note that the Sup35 antibody recognizes the M domain.
Extract seeding assay
The extract seeding assay was performed as previously described (Garrity et al. 2010) with the exception that samples used as seeds were unlysed cell suspensions (if bacterial) or cell extracts (if of yeast origin). Yeast extracts were prepared as previously described (Garrity et al. 2010).
Yeast transformations
Protein transformations were performed essentially as previously described (Garrity et al. 2010) with the following modifications. To test the infectivity of unlysed cell suspensions, tetracycline (10 μg/mL) was added to the selection plate to inhibit bacterial growth. To prepare transformation samples consisting of polymerized material from the seeding assay, aliquots of the various seeded reactions from the 30-min time point were centrifuged at 10,000g for 15 min at 4°C, washed in 500 μL of STC (1 M sorbitol, 10 mM Tris at pH 7.5, 10 mM CaCl2), centrifuged again at 10,000g for 15 min at 4°C, and resuspended in 500 μL of STC. Each resuspension was then split into two samples. One sample was subjected to sonication (Sonics Vibracell Microtip sonicator, 25% amplitude, pulsed 1 sec “on” and 3 sec “off” for a total of 10 sec of “on” time), and both samples were then used to transform [pin−][psi−] yeast cells.
EM and immunolabeling
Unlysed cell suspensions were adsorbed onto carbon or formvar/carbon-coated nickel grids in PBS, washed by floating the grid on 10 μL of distilled water, blotted dry, negatively stained with 1% uranyl acetate, blotted dry, and then viewed on a JEOL 1200EX microscope at an accelerating voltage of 80 kV. To immunolabel fibers (see Fig. 1A; Supplemental Fig. S5), sample-adsorbed nickel grids were floated on blocking buffer consisting of 1% BSA (bovine serum albumin) in PBS for 15 min. Samples containing CsgAss-NM and CsgAss-M were then incubated with anti-Sup35 antibody (diluted 1:20), whereas the sample containing CsgA was incubated with anti-His6 antibody (diluted 1:100) for 2 h in blocking buffer and rinsed in PBS. Samples exposed to anti-Sup35 were incubated with donkey anti-goat 12-nm gold secondary antibody (Jackson ImmunoResearch Laboratories) for 1 h before rinsing in PBS, whereas the sample exposed to anti-His6 was incubated with Protein-A gold 10 nm (CMC–UMC) for 1 h before rinsing in PBS. All grids were stained with 1% uranyl acetate. Images were taken with an AMT 2k CCD camera.
CR birefringence
Unlysed cell suspensions prepared using cells grown on CR-containing agar were spotted on a glass slide. Poly-L-lysine-coated coverslips were then placed on the samples, and the samples were viewed between cross polarizers on a Nikon 80i upright microscope with a Plan Apo 100× 1.4 NA objective. Images were acquired using a Nikon Digital Sight DS-Fi1 color camera and NIS-Elements acquisition software.
Library construction and screen
An expression library of csgAss fusions (with a C-terminal His6 tag) representing all of the ORFs from the E. coli ORF library (Saka at al. 2005) was constructed using a previously described method (Gibson et al. 2009). The library was constructed in a pooled format, with each pool representing approximately a seventh of the entire expression library. One pool from the plasmid library was then transformed into strain VS16 that already contained the csgG overexpression plasmid pVS76 and plated on LB agar supplemented with inducers (0.2% [w/v] L-arabinose, 1 mM IPTG), antibiotics (100 μg/mL carbenicillin, 25 μg/mL chloramphenicol), and CR (5 μg/mL). Plates were then incubated for 120 h at room temperature.
Acknowledgments
We thank S. Garrity, B. Nickels, and S. Dove for valuable discussion and comments on the manuscript; L. McPartland for expert technical assistance; M. Duennwald for generously providing the Htt constructs; M. Ericsson (HMS EM Facility) for help with electron microscopy; and the HMS Nikon Imaging Center. This work was supported by NIH grant DP1AI104284 (to A.H.) and NERCE post-doctoral fellowship U54AI057159 (to V.S.).
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.205310.112.
References
- Alberti S, Halfmann R, King O, Kapila A, Lindquist S 2009. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 137: 146–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach S, Talarek N, Andrieu T, Vierfond J, Mettey Y, Galons H, Dormont D, Meijer L, Cullin C, Blondel M 2003. Isolation of drugs active against mammalian prions using a yeast-based screening assay. Nat Biotechnol 21: 1075–1081 [DOI] [PubMed] [Google Scholar]
- Bagriantsev SN, Kushnirov VV, Liebman SW 2006. Analysis of amyloid aggregates using agarose gel electrophoresis. Methods Enzymol 412: 33–48 [DOI] [PubMed] [Google Scholar]
- Blanco LP, Evans ML, Smith DR, Badtke MP, Chapman MR 2011. Diversity, biogenesis and function of microbial amyloids. Trends Microbiol 20: 66–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan AW, Menke M, Cowen LJ, Lindquist SL, Berger B 2009. BETASCAN: Probably β-amyloids identified by pairwise probabilistic analysis. PLoS Comput Biol 5: e1000333 doi: 10.1371/journal.pcbi.1000333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, Hammar M, Normark S, Hultgren SJ 2002. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 295: 851–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F, Dobson CM 2006. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 75: 333–366 [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci 97: 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkatch IL, Bradley ME, Zhou P, Chernoff YO, Liebman SW 1997. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics 147: 507–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkatch IL, Bradley ME, Hong JY, Liebman SW 2001. Prions affect the appearance of other prions: The story of [PIN+]. Cell 106: 171–182 [DOI] [PubMed] [Google Scholar]
- Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L 2004. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol 22: 1302–1306 [DOI] [PubMed] [Google Scholar]
- Garrity SJ, Sivanathan V, Dong J, Lindquist S, Hochschild A 2010. Conversion of a yeast prion protein to an infectious form in bacteria. Proc Natl Acad Sci 107: 10596–10601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang R, Venter CJ, Hutchison CA, Hamilton SO 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6: 343–345 [DOI] [PubMed] [Google Scholar]
- Glover JR, Kowal AS, Schirmer EC, Patino MM, Liu JJ, Lindquist S 1997. Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell 89: 811–819 [DOI] [PubMed] [Google Scholar]
- Goldschmidt L, Teng PK, Riek R, Eisenberg D 2010. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc Natl Acad Sci 107: 3487–3492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammar M, Arnqvist A, Bian Z, Olsen A, Normark S 1995. Expression of two csg operons is required for production of fibronectin- and Congo red-binding curli polymers in Escherichia coli K-12. Mol Microbiol 18: 661–670 [DOI] [PubMed] [Google Scholar]
- Hammer ND, Schmidt JC, Chapman MR 2007. The curli nucleator protein, CsgB, contains an amyloidogenic domain that directs CsgA polymerization. Proc Natl Acad Sci 104: 12494–12499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Kim Y, Kim DJ, Chang Y-T, Hecht MH 2006. A high-throughput screen for compounds that inhibit aggregation of the Alzheimer's peptide. ACS Chem Biol 7: 461–469 [DOI] [PubMed] [Google Scholar]
- King C, Diaz-Avalos R 2004. Protein-only transmission of three yeast prion strains. Nature 428: 319–323 [DOI] [PubMed] [Google Scholar]
- Lee LL, Ha HH, Chang YT, DeLisa MP 2009. Discovery of amyloid-β aggregation inhibitors using an engineered assay for intracellular protein folding and solubility. Protein Sci 18: 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Lindquist S 2000. Creating a protein-based element of inheritance. Science 287: 661–664 [DOI] [PubMed] [Google Scholar]
- Liebman SW, Chernoff YO 2012. Prions in yeast. Genetics 191: 1041–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JJ, Lindquist S 1999. Oligopeptide-repeat expansions modulate ‘protein-only’ inheritance in yeast. Nature 400: 573–576 [DOI] [PubMed] [Google Scholar]
- Maji SK, Perrin MH, Sawaya MR, Jessberger S, Vadodaria K, Rissman RA, Singru PS, Nilsson KPR, Simon R, Schubert D, et al. 2009. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 325: 328–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer-Stroh S, Debulpaep M, Kuemmerer N, Lopez de la Paz M, Martins IC, Reumers J, Morris KL, Copland A, Serpell L, Serrano L, et al. 2010. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat Methods 7: 237–242 [DOI] [PubMed] [Google Scholar]
- Michelitsch MD, Weissman JS 2000. A census of glutamine/asparagine-rich regions: Implications for their conserved function and prediction of novel prions. Proc Natl Acad Sci 97: 11910–11915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nenninger AA, Robinson LS, Hammer ND, Epstein EA, Badtke MP, Hultgren SJ, Chapman MR 2011. CsgE is a curli secretion specificity factor that prevents amyloid fibre aggregation. Mol Microbiol 81: 486–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell CW, Waldispühl J, Lis M, Halfmann R, Devadas S, Lindquist S, Berger B 2011. A method for probing the mutational landscape of amyloid structure. Bioinformatics 27: i34–i42 doi: 10.1093/bioinformatics/btr238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osherovich LZ, Weissman JS 2001. Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI+] prion. Cell 106: 183–194 [DOI] [PubMed] [Google Scholar]
- Robinson LS, Ashman EM, Hultgren SJ, Chapman MR 2006. Secretion of curli fibre subunits is mediated by the outer membrane-localized CsgG protein. Mol Microbiol 59: 870–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo-Hamano Y, Uchida N, Namba K, Oosawa K 2004. In vitro characterization of FlgB, FlgC, FlgF, FlgG, and FlgE, flagellar basal body proteins of Salmonella. J Mol Biol 339: 423–435 [DOI] [PubMed] [Google Scholar]
- Saka K, Tadenuma M, Nakade S, Tanaka N, Sugawara H, Nishikawa K, Ichiyoshi N, Kitagawa M, Mori H, Ogasawara N, et al. 2005. A complete set of Escherichia coli open reading frames in mobile plasmids facilitating genetic studies. DNA Res 12: 63–68 [DOI] [PubMed] [Google Scholar]
- Santoso A, Chien P, Osherovich LZ, Weissman JS 2000. Molecular basis of a yeast prion species barrier. Cell 100: 277–288 [DOI] [PubMed] [Google Scholar]
- Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJW, McFarlane HT, et al. 2007. Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 447: 453–457 [DOI] [PubMed] [Google Scholar]
- Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE 1997. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90: 549–558 [DOI] [PubMed] [Google Scholar]
- Sondheimer N, Lindquist S 2000. Rnq1: An epigenetic modifier of protein function in yeast. Mol Cell 5: 163–172 [DOI] [PubMed] [Google Scholar]
- Suzuki G, Shimazu N, Tanaka M 2012. A yeast prion, Mod5, promotes acquired drug resistance and cell survival under environmental stress. Science 336: 355–359 [DOI] [PubMed] [Google Scholar]
- Tanaka M, Weissman JS 2006. An efficient protein transformation protocol for introducing prions into yeast. Methods Enzymol 412: 185–200 [DOI] [PubMed] [Google Scholar]
- Tartaglia GG, Pawar AP, Campioni S, Dobson CM, Chiti F, Vendruscolo M 2008. Prediction of aggregation-prone regions in structured proteins. J Mol Biol 380: 425–436 [DOI] [PubMed] [Google Scholar]
- Teng PK, Eisenberg D 2009. Short protein segments can drive a non-fibrillizing protein into the amyloid state. Protein Eng Des Sel 22: 531–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson MJ, Sievers SA, Karanicolas J, Ivanova MI, Baker D, Eisenberg D 2006. The 3D profile methods for identifying fibril-forming segments of proteins. Proc Natl Acad Sci 103: 4074–4078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toombs JA, Petri M, Paul KR, Kan G, Ben-Hur A, Ross ED 2012. De novo design of synthetic prion domains. Proc Natl Acad Sci 109: 6519–6524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama BH, Weissman JS 2011. Amyloid structure: Conformational diversity and consequences. Annu Rev Biochem 80: 557–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trovato A, Chiti F, Maritan A, Seno F 2006. Insight into the structure of amyloid fibrils from the analysis of globular proteins. PLoS Comput Biol 2: e170 doi: 10.1371/journal.pcbi.0020170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuite MF, Serio TR 2010. The prion hypothesis: From biological anomaly to basic regulatory mechanism. Nat Rev Mol Cell Biol 12: 823–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chapman MR 2008. Sequence determinants of bacterial amyloid formation. J Mol Biol 380: 570–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Smith DR, Jones JW, Chapman MR 2007. In vitro polymerization of a functional Escherichia coli amyloid protein. J Biol Chem 282: 3713–3719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Wang X, Yuan CG, Ma J 2010. Generating a prion with bacterially expressed recombinant prion protein. Science 327: 1132–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner RB, Edskes HK, Shewmaker F, Nakayashiki T 2007. Prions of fungi: Inherited structures and biological roles. Nat Rev Microbiol 5: 611–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Chen H, Lai L 2007. Identification of amyloid fibril-forming segments based on structure and residue-based statistical potential. Bioinformatics 23: 2218–2225 [DOI] [PubMed] [Google Scholar]