

The transition of proteins targeted for autophagic degradation from the soluble to the insoluble phase is regulated in an ATG1-independent mechanism by TORC1. This process is likely a critical mechanism for maintaining protein homeostasis when challenged with proteomic stress.
Abstract
Accumulation of insoluble protein in cells is associated with aging and aging-related diseases; however, the roles of insoluble protein in these processes are uncertain. The nature and impact of changes to protein solubility during normal aging are less well understood. Using quantitative mass spectrometry, we identify 480 proteins that become insoluble during postmitotic aging in Saccharomyces cerevisiae and show that this ensemble of insoluble proteins is similar to those that accumulate in aging nematodes. SDS-insoluble protein is present exclusively in a nonquiescent subpopulation of postmitotic cells, indicating an asymmetrical distribution of this protein. In addition, we show that nitrogen starvation of young cells is sufficient to cause accumulation of a similar group of insoluble proteins. Although many of the insoluble proteins identified are known to be autophagic substrates, induction of macroautophagy is not required for insoluble protein formation. However, genetic or chemical inhibition of the Tor1 kinase is sufficient to promote accumulation of insoluble protein. We conclude that target of rapamycin complex 1 regulates accumulation of insoluble proteins via mechanisms acting upstream of macroautophagy. Our data indicate that the accumulation of proteins in an SDS-insoluble state in postmitotic cells represents a novel autophagic cargo preparation process that is regulated by the Tor1 kinase.
INTRODUCTION
Protein homeostasis is critical to cellular and organismal viability. Several key mechanisms have evolved to ensure protein homeostasis during normal cellular function and in the context of cellular and organismal stress. These processes include modulation of protein synthesis, increased chaperone activity, and enhanced protein degradation (Taylor and Dillin, 2011). Together these mechanisms work in concert to reestablish and maintain protein homeostasis during times of stress but can also trigger apoptosis (Rasheva and Domingos, 2009). In general, protein homeostasis is challenged with the increased levels of proteomic dysfunction associated with aging (Soskic et al., 2008). Age-related reduction in the cell's ability to clear damaged proteins contributes significantly to cellular dysfunction and associated decline in organismal integrity (Cuervo et al., 2005; Taylor and Dillin, 2011). Supporting this idea, several studies have shown that activation of processes known to promote protein homeostasis (e.g., chaperone function, macroautophagy, and translation inhibition) enhance longevity, whereas abrogation of these pathways generally leads to reduced lifespan (Juhasz et al., 2007; Pan et al., 2007; Hansen et al., 2008).
Much of our understanding about the relationship between compromised protein homeostasis and organismal function and viability has come from the study of late-onset neurodegenerative proteinopathies such as spongiform encephalopathy, amyotrophic lateral sclerosis, and Huntington, Parkinson, and Alzheimer diseases. The presence of specific insoluble protein aggregates in affected tissues is both a histopathological hallmark of these diseases and evidence of impaired protein homeostasis in these cells. In many cases, these disease-associated protein aggregates exist in conformations insoluble in the strong detergent SDS (Tanemura et al., 2006). Although microscopic protein inclusions were initially believed to cause cellular dysfunction in these disorders, in some cases their presence does not correlate positively with cell death (Tagawa et al., 2004). Moreover, some forms of large protein inclusions associated with neurodegenerative diseases are relatively benign or even protective as compared with more toxic small oligomeric aggregates (Ravikumar et al., 2002; Taylor et al., 2003; Arrasate et al., 2004; Selkoe, 2004).
One example of a protective regulated mechanism that promotes insoluble protein accumulation is the active sequestration of misfolded proteins into large inclusions called aggresomes. This microtubule-dependent process ultimately leads to lysosomal degradation of the misfolded proteins via macroautophagy (Kopito, 2000; Taylor et al., 2003). Aggresome formation promotes protein homeostasis by sequestering and ultimately degrading aberrant proteins, thus preventing toxic forms of these proteins from interfering with normal cellular processes (Riley et al., 2010). Whereas formation of SDS-insoluble protein aggregates has been primarily studied in the context of neurodegenerative diseases, a wide range of cellular proteins were recently shown to undergo a transition into SDS-insoluble conformations during aging in Caenorhabditis elegans (Reis-Rodrigues et al., 2012). This suggests that this phenomenon likely plays a role in protein homeostasis during healthy aging, as well as in response to proteinopathic diseases.
The target of rapamycin (TOR) complex plays a central role in protein homeostasis. The TOR complex, specifically TOR complex 1 (TORC1), has been well characterized as a fundamental regulator that couples cell growth and proliferation to growth factor and/or nutrient signaling cues. Although active in nutrient-rich conditions, nutrient limitation inactivates TORC1, resulting in decreased protein production, mobilization of nutrient stores, degradation of proteins, activation of the stress-response mechanisms, and suppression of growth and proliferation (Dann and Thomas, 2006). Although this has placed TORC1 at the nexus of nutrient signaling and growth, TORC1 also has a critical role in regulating protein homeostasis. TORC1 activity is down-regulated in response to several challenges to protein homeostasis, including oxidative stress, heat shock, osmotic stress, and aging (Reiling and Sabatini, 2006). Furthermore, in models of neurodegenerative diseases TORC1 inhibition has been shown to reduce levels of toxic protein species through autophagy-dependent mechanisms and ameliorate phenotypes associated with expression of the disease proteins (Ravikumar et al., 2004; Tsvetkov et al., 2010; Barnett and Brewer, 2011).
Here we test the idea that insoluble protein accumulates with age in Saccharomyces cerevisiae and that this event is a regulated cellular process. We go on to characterize different stimuli that promote insoluble protein accumulation and hypothesize that this process is critical in the maintenance of protein homeostasis. Using quantitative mass spectrometry, we identify and compare insoluble proteins that accumulate in response to aging and nutrient signaling. Our results indicate that the TORC1 complex regulates protein homeostasis in two distinct ways: first, by sequestering proteins from the soluble fraction into SDS-insoluble inclusions, and second, by activating the autophagic pathway to promote protein degradation. We suggest that the transition of proteins from the soluble to the insoluble phase may be a process for preparing autophagic cargo for loading into forming autophagosomes and subsequent degradation via macroautophagy. Understanding the mechanisms involved in the accumulation of SDS-insoluble protein will illuminate the pathways involved in maintaining cellular homeostasis while providing novel insights into mechanisms underlying neurodegenerative and other protein-folding diseases.
RESULTS
Age-dependent accumulation of insoluble proteins is conserved across taxa
Although the accumulation of insoluble protein has been extensively studied in the context of late-onset neurodegenerative diseases, this phenomenon and the cellular processes that regulate it during normal aging are less well characterized. We set out to identify insoluble proteins that normally accumulate in eukaryotic cells using a chronologic aging model in the yeast S. cerevisiae. Using an insoluble protein extraction protocol and quantitative mass spectrometry, we compared the insoluble protein content of postmitotic young and aged cells (Figure 1A).
FIGURE 1:
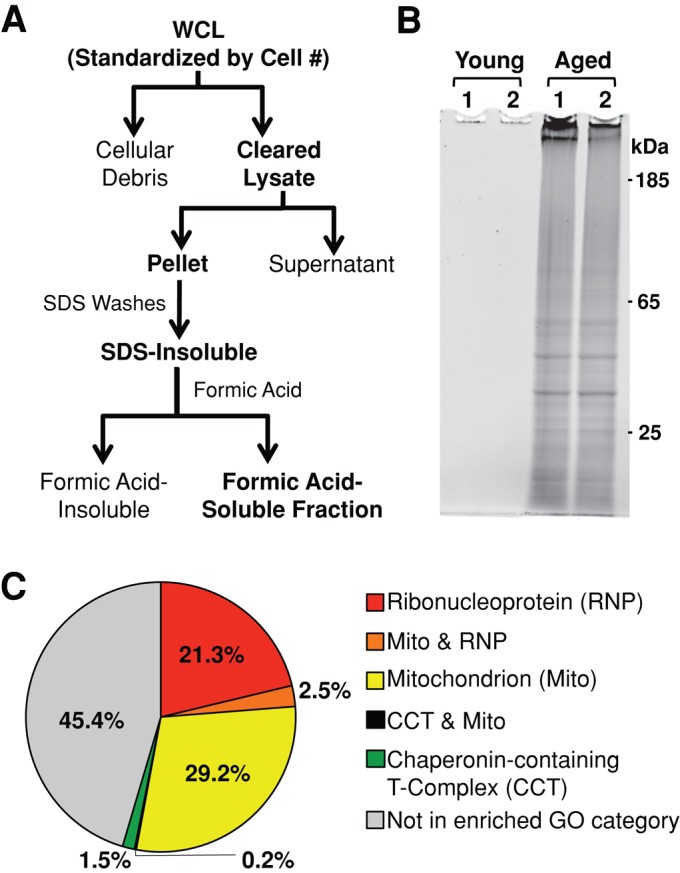
Insoluble proteins accumulate in chronologically aged S. cerevisiae. (A) Whole-cell lysates (WCL) were cleared of cellular debris via a low-speed centrifugation. Insoluble protein was pelleted from cleared lysates via high-speed centrifugation, washed with 1% SDS, and resolubilized in 70% formic acid. Formic acid was removed via desiccation, and remaining protein was resuspended in buffers compatible with downstream proteomic analysis. (B) Cells that had just entered the postmitotic state were harvested (young) or aged for 48 h postmitotically (aged) before preparation of insoluble protein. Formic acid–soluble protein was resuspended in loading buffer, run on polyacrylamide gels, and stained for total protein. Two biological replicates of each time point are shown (1, 2). SDS-insoluble protein from 8 × 107 cells was loaded in each lane. (C) GO analysis of the 480 SDS-insoluble proteins identified in aged cells revealed three significantly enriched cellular component GO categories: mitochondrion (GO:0005739), ribonucleoprotein complex (GO:0030529), and chaperonin-containing T-complex (GO:0005832).
Yeast were grown in synthetic complete media and harvested soon after the diauxic shift (young) and 48 h after entrance into this postmitotic state (aged; Supplemental Figure S1A). At both time points, cultures had similar viability, allowing us to exclude the effect of cell death on insoluble protein accumulation (Supplemental Figure S1B). SDS-insoluble protein was subsequently extracted from cell lysates, resolubilized in formic acid, and analyzed by PAGE. Total protein staining was used to show the overt differences in the amount of insoluble protein extracted from young and aged cells (Figure 1B). We observed that a significant amount of SDS-insoluble protein accumulated in aged yeast, but no SDS-insoluble protein was detected in young cells on polyacrylamide gels. To determine the effective range of detection using this method, serial dilutions of aged samples showed young samples had at least 200-fold lower levels of SDS-insoluble protein as compared with that present in aged cells (Supplemental Figure S1C).
Isobaric tag for relative and absolute quantitation (iTRAQ) mass spectrometry was used to identify insoluble proteins and determine variation between biological replicates (Supplemental Figure S2). This technique allowed us to directly compare pairs of protein preparations from biological replicates of young and aged samples with a high degree of accuracy and sensitivity. Overall we identified 480 unique proteins from 6819 distinct peptides at a 95% confidence interval (Supplemental Table S1). Nearly all detected peptides were derived from aged samples, as we found essentially no iTRAQ reporter ions corresponding to peptides originating from the young samples. Because this multiplexed iTRAQ tagging approach was normalized to yeast cell counts for all samples, the inability to detect iTRAQ-specific reporter ions for peptides originating from either of the young biological replicates led us to conclude that very little insoluble protein was extracted from young cells. In contrast, aged biological replicates had strong yet remarkably similar protein levels between the biological replicates. Of the 480 proteins identified, we accurately measured the relative quantitation of 434 proteins between biological replicates, only six of which showed significantly different levels between replicates (Supplemental Table S1). The plasma membrane ATPase1 (encoded by the PMA1 gene) had the largest difference (2.3-fold) between replicates and yet was orders of magnitude less than the total variation between young and old samples (Supplemental Figure S1C). We therefore concluded that all 480 identified proteins were enriched in the aged sample and thus had undergone a chronological aging–dependent transition from the soluble to the insoluble fraction.
We next compared the age-dependent insoluble proteins identified in yeast to insoluble proteins that accumulate with age in a metazoan aging model. Using a similar method, our group recently identified SDS-insoluble proteins that accumulate in aging C. elegans (Reis-Rodrigues et al., 2012). This study found 203 proteins enriched in the SDS-insoluble fraction of aged nematodes. When we compared the age-dependent SDS-insoluble proteins detected in yeast to those identified in nematode, we found a high degree of overlap. Of the 480 insoluble yeast proteins, 246 have direct homologues in C. elegans. Of these 246 genes, 30.5% (75 of 246 genes) encode proteins that were identified in the insoluble fraction in aged C. elegans (Supplemental Table S2). Overall this represents 15.6% (75 of 480) of the proteins identified in our yeast study having a direct homolog present in the insoluble fraction in aged C. elegans. This represents a 5.7-fold enrichment over the chance expectation (p < 0.001). This significant overlap indicates that many of the proteins that become insoluble with age are conserved between these two species.
We used Gene Ontology (GO) analysis to identify shared characteristics that might contribute to an age-dependent change in solubility of identified yeast insoluble proteins. We searched our list of age-dependent insoluble proteins against the yeast genome to determine enriched GO categories using the DAVID Bioinformatics Databases Functional Annotation Tool. We found that three cellular complexes were significantly enriched (false discovery rate [FDR] < 0.05) in the age-dependent insoluble fraction: ribonucleoprotein complex (RNP; FDR = 1.1E-8), mitochondrion (1.3E-15), and chaperonin-containing T complex (9.8E-3; Figure 1C and Supplemental Table S2). More than 53% of the proteins identified in this study had either the RNP or mitochondrion GO annotations (Figure 1C and Supplemental Table S2). Of note, GO analysis of the C. elegans age-dependent insoluble protein using identical parameters revealed enrichment in both mitochondrion and ribonucleoprotein categories as well (Reis-Rodrigues et al., 2012). Because both mitochondrial and ribosomal proteins are enriched in insoluble fractions extracted from the aged population of these divergent organisms, we suggest that these highly conserved organelles undergo an aging-dependent change in solubility through a conserved mechanism.
Among the ribosomal proteins present in the insoluble fraction of postmitotic yeast, we identified 93 and 82% of the structural proteins making up the 40S and 60S ribosomal subunits, respectively (Supplemental Figure S3A). This indicates that ribosomes are partitioned into the insoluble fraction as an intact protein complex. We also noted that the mitochondrial proteins identified in the insoluble fraction come from all major suborganelle regions within the mitochondria (Supplemental Figure S3B), suggesting similarly that the entire organelle undergoes a transition into the insoluble phase with age.
Insoluble proteins accumulate in the nonquiescent subpopulation of postmitotic yeast
On entrance into stationary phase, yeast cultures partition into two physiologically distinct subpopulations: a quiescent population made up of daughter cells of the final mitotic division, and a nonquiescent population comprising the corresponding mother cells (Allen et al., 2006). Differences in buoyant density between the denser quiescent cells and less dense nonquiescent cells allow these two populations to be separated using gradient centrifugation. Several features have been described that distinguish these populations. Nonquiescent cells have several characteristics indicative of proteomic stress. These include higher levels of reactive oxygen species, increased sensitivity to heat stress, lower levels of glycogen accumulation, and reduced ability to reenter the cell cycle as compared with quiescent cells (Allen et al., 2006). Given the evidence for impaired protein homeostasis in nonquiescent cells, we reasoned that insoluble protein accumulated during chronologic aging might be asymmetrically distributed between quiescent and nonquiescent cells.
To test this idea, we separated quiescent and nonquiescent cells in young and aged cultures and assayed each population for insoluble protein accumulation. Gradient centrifugation of young and aged cultures separated distinct populations that varied in density. As expected, young cells migrated primarily to the denser quiescent fraction, indicating that the two subpopulations had yet to be established. However, after 48 h of postmitotic aging, both quiescent and nonquiescent populations had been established and cells were distributed approximately equally between the two populations (Figure 2A). Because the young cells partitioned nearly entirely (>95%) to the denser fraction, we analyzed the insoluble protein content of three different populations: the young population, the aged quiescent population, and the aged nonquiescent population. Consistent with our previous observations, no insoluble protein was found in cells from the young population. The aged culture, however, showed a marked difference in insoluble protein content between the quiescent and nonquiescent populations. Whereas aged quiescent cells lacked insoluble protein, aged nonquiescent cells contained significant insoluble protein (Figure 2B), indicating that only nonquiescent cells accumulate significant levels of insoluble protein during postmitotic aging. Alternatively, these data would indicate that SDS-insoluble proteins are preferentially retained in the mother cell during the final cell division events that establish the quiescent and nonquiescent populations (Liu et al., 2010).
FIGURE 2:
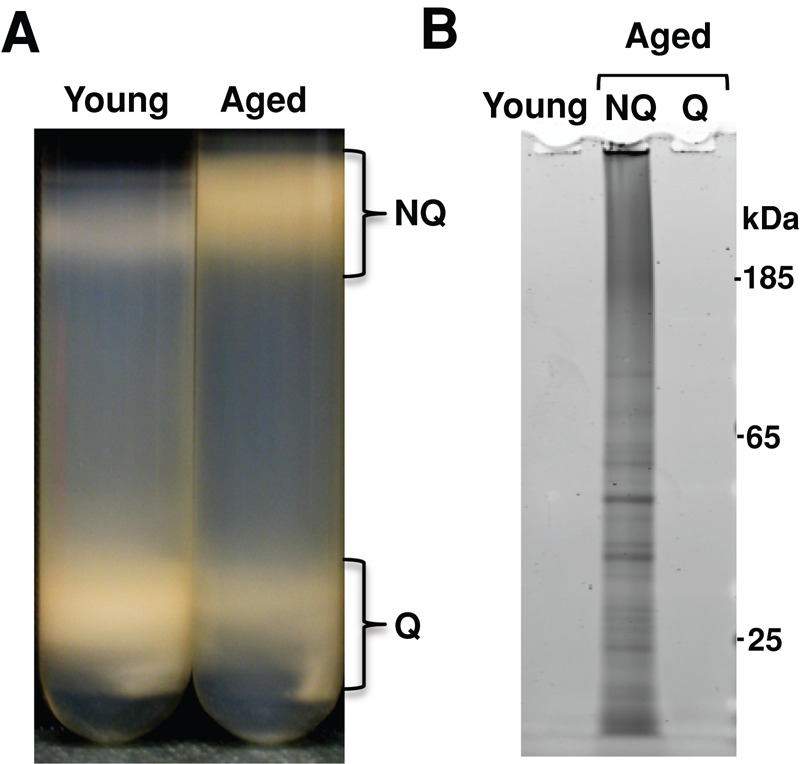
Insoluble proteins accumulate in the nonquiescent population of postmitotic yeast. (A) Postmitotic cultures that had just entered stationary phase (young) or chronologically aged in stationary phase for 48 h (aged) were separated by Percoll gradient centrifugation into quiescent (Q) and nonquiescent (NQ) populations. (B) Young cells, aged Q, and aged NQ populations were harvested and insoluble protein extracted from a standard number of cells. SDS-insoluble protein from 8 × 107 cells was loaded in each lane.
Insoluble protein formation correlates with presence of autophagic bodies in postmitotic cells
One adaptive cellular response to proteomic stress is increased protein turnover via autophagy (Taylor and Dillin, 2011). Highly specialized forms of autophagy, termed mitophagy and ribophagy, are responsible for sequestering and degrading mitochondria and ribosomes (Kraft et al., 2008; Kanki et al., 2009; Okamoto et al., 2009). Because we found the insoluble fraction to be enriched for proteins from ribosomes and mitochondria, we hypothesized that autophagic processes might influence their accumulation into this form. Consistent with this idea, we observed an accumulation of autophagic bodies in the vacuole of aged cells (Figure 3A). Autophagic bodies (ABs) are substrates of autophagy bound by a single membrane vesicle in the lumen of the vacuole. These vesicles are distinguishable by light microscopy because they exhibit extensive Brownian motion within the vacuole (Takeshige et al., 1992). Although inhibition of vacuolar proteases is generally needed to detect ABs during nitrogen starvation of logarithmically growing cells, we found this treatment unnecessary to detect ABs in postmitotically aged yeast. This indicates that postmitotic cells deliver autophagic substrates to the vacuole more rapidly than the proteases degrade them, resulting in higher steady-state levels of ABs in this compartment. Studies describing ABs showed that they contain both mitochondrial and ribosomal proteins (Takeshige et al., 1992). We found the vast majority of young postmitotic cells had enlarged but empty vacuoles. Those with ABs had only one to two discrete particles per vacuole. Conversely, aged cells had vacuoles that had many (more than three) particles per vacuole (Figure 3A). We concluded that the accumulation of ABs in postmitotic cells correlates with the appearance of SDS-insoluble protein that we detected biochemically.
FIGURE 3:

Accumulation of SDS-insoluble protein correlates with the presence of autophagic bodies. (A) Young and aged cells were harvested as described, and vacuolar morphology was evaluated for the presence of ABs (arrows) by light microscopy. (B) After 48 h of postmitotic growth in synthetic complete media (SC), wild-type (TN121) or atg1Δ (TN123) cells carrying the Pho8Δ60 reporter were starved for nitrogen (SC-N) for 8 h, and alkaline phosphatase (ALP) activity was measured (n = 4). (C–E) Wild-type cells were grown into the postmitotic state and harvested (young) or aged in SC or SC-N media for 48 h. Cells were harvested, and SDS-insoluble protein extracted from 4 × 107 cells was analyzed by SDS–PAGE (C) or the percentage of cells with ABs quantitated (D, E). More than 200 cells were quantitated for each replicate (n = 3). White bars, 10 μm. *p < 0.01.
The coincident appearance and similar protein content of the SDS-insoluble protein fraction and ABs suggested that insoluble protein detected biochemically represents AB cargo. We therefore tested whether induction of autophagy through nitrogen starvation would clear both insoluble protein and ABs from the cell. Nitrogen starvation is known to induce autophagy during logarithmic growth. We first confirmed that nitrogen starvation activated autophagy in postmitotic cells using the Pho8Δ60 alkaline phosphatase assay (Takeshige et al., 1992). Alkaline phosphatase (ALP) activity was compared between cells that were postmitotically aged in synthetic complete (SC) media for 56 h to those postmitotically aged for 48 h and nitrogen starved (SC-N) for 8 h. Although there was low ALP activity in both wild-type and autophagy-null (atg1Δ) cells aged in complete media, those cells that were nitrogen starved showed a fivefold increase in ALP activity compared with similarly treated atg1Δ cells (Figure 3B). This confirmed that nitrogen starvation activates autophagy in postmitotic cells.
To test the effect of autophagic activation on protein solubility, we compared the presence of ABs and insoluble protein accumulation between cells that had just entered stationary phase to those that were aged in SC or SC-N for 48 h. We again found that young cells grown in SC had no detectable insoluble protein, whereas aging promoted accumulation of SDS-insoluble protein (Figure 3C). Surprisingly, cells aged in SC-N media accumulated considerably more insoluble protein than those aged in SC even though autophagy is induced by nitrogen starvation (Figure 3, B and C). Again, we found that the SDS-insoluble protein load correlated with the percentage of cells containing ABs under these circumstances. Only 2.3% of young cultures had ABs, and these cells had only one to two ABs per vacuole. Conversely, 92.2% of the cells in cultures aged in SC had ABs (Figure 3, D and E). Nitrogen starvation exacerbated this phenotype. Although only slightly more cells in the SC-N cultures contained ABs (92.2% vs. 97.7%), nitrogen-starved cells had more ABs per vacuole in the cultures aged in SC-N. Overall these results demonstrate that the accumulation of insoluble protein is correlated with presence of ABs and suggest that both phenotypes are similarly modulated by nutrient signaling pathways.
Nitrogen starvation is sufficient to induce accumulation of insoluble protein
Levels of ABs and the biochemically defined SDS-insoluble protein fraction are similarly affected by aging and nitrogen starvation in the postmitotic state. Furthermore, they contain common protein complexes (Figure 1C and Table S2; Baba et al., 1994b). We therefore reasoned that SDS-insoluble protein could be an intermediate substrate of an autophagic process. If this were the case, starvation of logarithmically growing cells lacking vacuolar proteolytic function would promote accumulation of insoluble protein to levels similar to that observed in aged postmitotic cells.
To test this idea, we first measured protease activity in cells lacking the major vacuolar aspartyl protease Pep4 after 8 h of nitrogen starvation using a green fluorescent protein (GFP)–Atg8 processing assay. This assay measures the vacuolar cleavage of a GFP-Atg8 fusion protein as detected through Western blotting (Klionsky et al., 2007). The release of free GFP is indicative of delivery of autophagosomes to the vacuole and activation of vacuolar proteases. We found that 8 h of nitrogen starvation promotes the production of the free GFP in wild-type cells but not in pep4Δ cells (Figure 4A). When we compared the level of insoluble protein that accumulates in wild-type and pep4Δ cells, we surprisingly found that both strains accumulate insoluble protein in response to nitrogen starvation (Figure 4B). This indicates that nitrogen starvation is itself sufficient to induce the accumulation of insoluble protein.
FIGURE 4:

Nitrogen starvation is sufficient to induce the accumulation of a similar set of SDS-insoluble proteins. (A) Wild-type and pep4Δ cells expressing a GFP-Atg8 fusion protein were harvested after 8 h of nitrogen starvation. Protein extracts from 1 × 107 cells were subjected to SDS–PAGE and blotted for GFP. GFP-Atg8 and free GFP are indicated. The amount of free GFP as a percentage of the total GFP is shown for each sample. (B) Insoluble protein from wild-type and pep4Δ cells during logarithmic growth and after 8 h of nitrogen starvation were extracted and compared as previously described. Material from 8 × 107 cells was loaded into each lane. (C) Insoluble proteins were extracted as previously described and compared between cells growing logarithmically (SC), cells nitrogen starved for 8 h (SC-N), postmitotic cells (young), and cells aged postmitotically for 48 h (aged). Two biological replicates (1, 2) of each treatment are shown. Material from 8 × 107 cells was loaded from each sample.
We next wanted to compare the identity of proteins that accumulate in response to nitrogen starvation to those that accumulate during postmitotic aging. To do this, we extracted insoluble protein from logarithmically growing cells before (SC) and after (SC-N) 8 h of nitrogen starvation. We also extracted insoluble proteins from postmitotic cells before (young) and after (aged) 48 h of aging as described previously. Total protein staining after gel electrophoresis showed that 8 h of nitrogen starvation was sufficient to induce the accumulation of SDS-insoluble protein to levels similar to those observed in postmitotically aged cells (Figure 4C). This indicates that nutrient signaling pathways are capable of regulating insoluble protein accumulation in a manner that is independent of postmitotic aging.
Overall, insoluble proteins from nitrogen-starved cells and postmitotically aged cells had a similar banding pattern and abundances as indicated by total protein staining on a polyacrylamide gel (Figure 4C). To quantitatively compare the levels and identity of insoluble proteins that accumulated in response to these two conditions, we used quantitative iTRAQ labeling mass spectrometry (Supplemental Figure S2). We found that the majority of the insoluble proteins identified in this study (70.2%, or 337 of 480 proteins) were present at similar levels in the age-induced and starvation-induced insoluble fractions (Supplemental Table S3). Of note, independent GO analysis of proteins found at similar levels between these insoluble fractions indicated significant enrichment in the mitochondrion and ribonucleoprotein GO categories (unpublished data). Thus we conclude that nitrogen starvation and postmitotic chronological aging induce similar cellular processes that result in the transition of a core ensemble of proteins from the soluble to an SDS-insoluble phase.
Insoluble protein accumulates independently of autophagic pathway activation
More than half of the proteins that we identified in the insoluble fraction of either nitrogen-starved or postmitotically aged cells are part of protein complexes or organelles that are degraded by specialized forms of autophagy (Figure 1C). Furthermore, these same proteins make up autophagic cargoes that are present in ABs after nitrogen starvation (Baba et al., 1994a). Given these similarities, we hypothesized that the biochemically identified insoluble protein fraction either represent proteins that are sequestered for subsequent autophagic degradation or are autophagic substrates that are in the process of being transported to the vacuole for degradation. To examine this, we tested whether the canonical autophagic pathway was required for the accumulation of insoluble protein during nitrogen starvation by comparing insoluble protein accumulation in wild-type and two autophagy-null strains, atg1Δ and atg7Δ. Atg1p kinase is responsible for activating the canonical autophagic pathway, whereas Atg7p is critical to the formation of much of the machinery responsible for assembly of the autophagosome. Loss of either protein leads to the inability to form autophagosomes and thus inability to degrade proteins via macroautophagy (Abeliovich and Klionsky, 2001). We found that nitrogen starvation for 8 h induced similar levels of insoluble proteins in wild-type, atg1Δ, and atg7Δ strains (Figure 5). This demonstrates that the canonical autophagic pathway is not required for the transition of proteins into the insoluble phase and that a nutrient-responsive mechanism acting upstream of autophagy (i.e., ATG1) can control the accumulation of SDS-insoluble protein.
FIGURE 5:
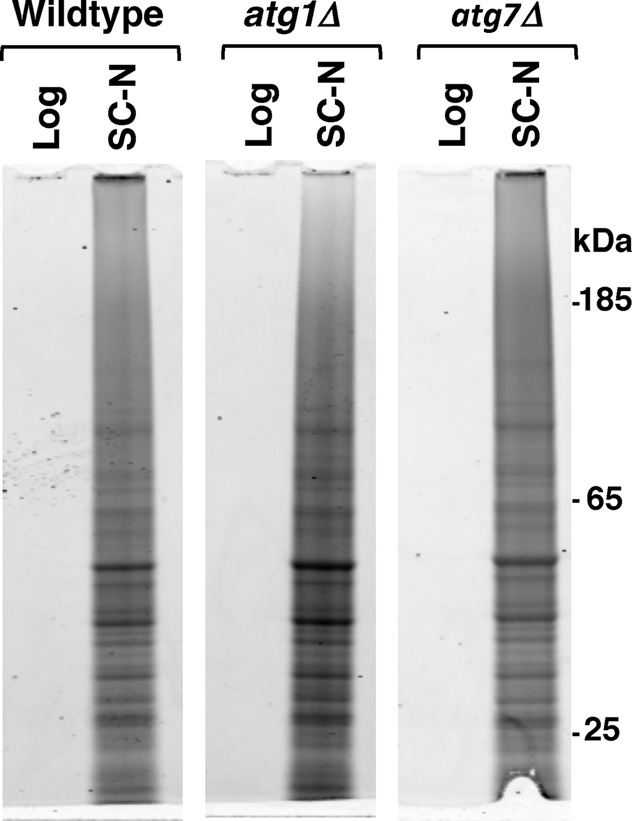
SDS-insoluble proteins accumulate in autophagy-deficient cells. Total protein staining of SDS-insoluble protein extracted from wild-type, atg1Δ, or atg7Δ cells growing logarithmically in synthetic complete media (Log) or after 8 h of nitrogen starvation (SC-N). Each lane contains extract from 8 × 107 cells.
Inhibition of TORC1 signaling promotes accumulation of insoluble protein
Upon nutrient depletion, TORC1-dependent phosphorylation of Atg1 and Atg13 is reduced. This promotes formation of the Atg1/13 complex and subsequent activation of autophagy (Dann and Thomas, 2006). Given that accumulation of insoluble protein occurs in response to nitrogen starvation via an ATG1-independent mechanism, we tested whether TORC1 might be involved in this process.
We first compared insoluble protein accumulation in wild-type and tor1Δ cells during logarithmic growth, under conditions of nitrogen starvation, and during postmitotic aging. Both strains accumulated SDS-insoluble protein to similar levels upon nitrogen starvation and did not accumulate insoluble protein during logarithmic growth in synthetic complete media (unpublished data). Similarly, neither strain contained insoluble protein immediately after the shift to the postmitotic state (Figure 6A). However, when aged in the postmitotic state, tor1Δ cells contained significantly more SDS-insoluble protein than aged wild-type cells (Figure 6A). This enhanced level of SDS-insoluble protein accumulation is similar to that observed in aged wild-type cells upon nitrogen starvation (Figure 3C). This demonstrates that loss of the TOR1 enhances insoluble protein accumulation during postmitotic aging.
FIGURE 6:
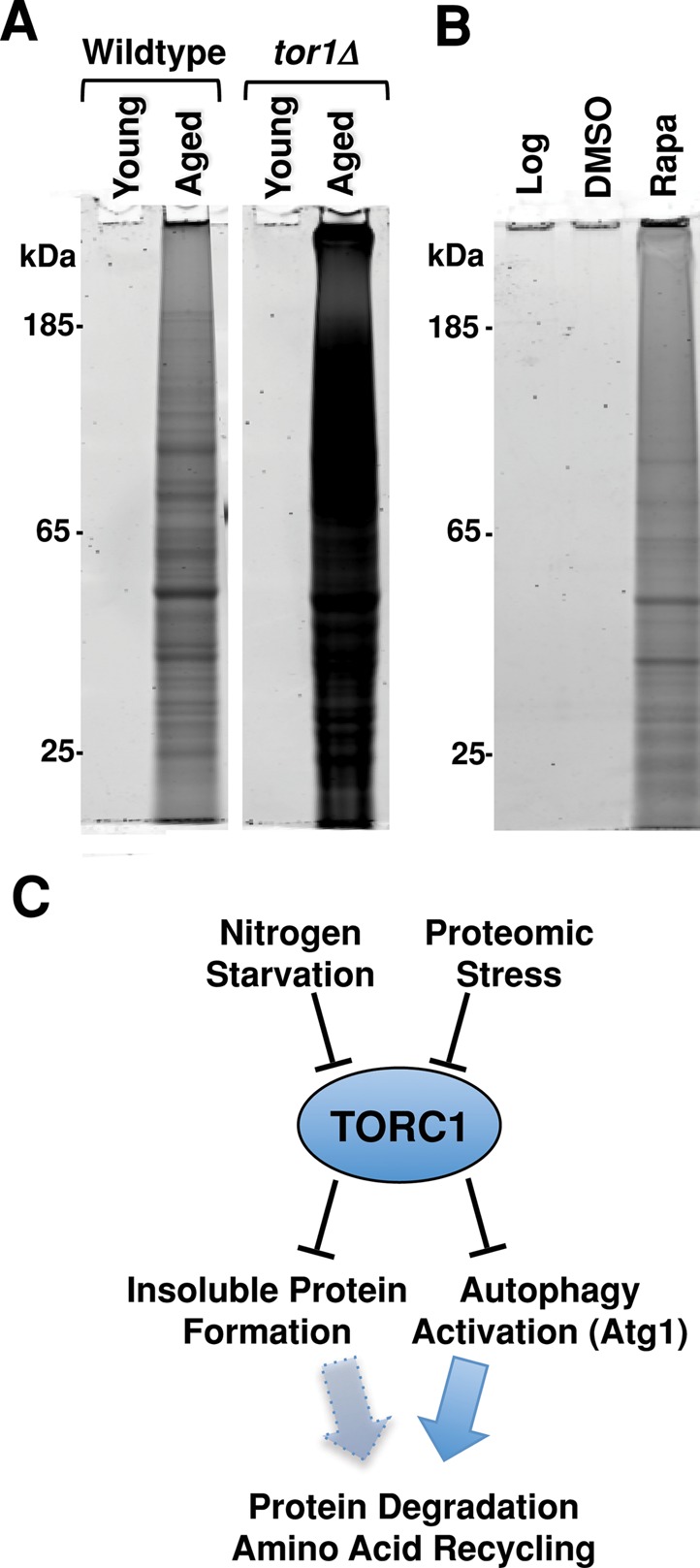
TORC1 inhibition promotes insoluble protein accumulation. (A) Total protein staining of SDS-insoluble protein extracted from 8 × 107 wild-type and tor1Δ cells that had just entered the postmitotic state (young) and those postmitotically aged for 48 h (aged). (B) Total protein staining of insoluble protein extracted from 8 × 107 wild-type cells growing logarithmically (Log), cells treated with either vehicle for 4 h (DMSO) or vehicle and rapamycin for 4 h (Rapa). (C) Proposed model of how the TOR pathway controls insoluble protein sequestration and autophagic pathway activation via distinct but parallel mechanisms.
We next tested whether acute inactivation of the TORC1 promotes formation of SDS-insoluble protein in logarithmically growing cultures. Wild-type cells in mid–log phase were treated with rapamycin (or dimethyl sulfoxide [DMSO] control) for 4 h before insoluble protein was extracted and analyzed. We found that rapamycin treatment was sufficient to induce the formation of SDS-insoluble protein in wild-type cells, indicating that acute TORC1 inactivation is sufficient to promote the accumulation of insoluble protein (Figure 6B). Taken together, these data suggest that inactivation of the TORC1 promotes the transition of soluble proteins to the insoluble phase in response to either nitrogen starvation or postmitotic aging. Given that accumulation of SDS-insoluble protein also occurs in response to nitrogen starvation in autophagy-null atg1Δ cells (Figure 5), the TORC1-sequestration of autophagic substrates into an insoluble state occurs through a mechanism distinct from autophagic activation. Thus the autophagy-independent regulation of protein solubility represents a novel role of TORC1 in maintenance of protein homeostasis (Figure 6C).
DISCUSSION
Here we report that a wide range of SDS-insoluble proteins accumulate in S. cerevisiae during postmitotic aging. These insoluble proteins are absent in logarithmically growing or young postmitotic cells. We find that the ensemble of proteins that become insoluble in postmitotic yeast is similar to that that accumulates during aging in adult C. elegans. This indicates that the types of proteins that undergo this conformation transition in response to age are conserved (Figure 1 and Supplemental Table S1). Furthermore, this shows that the causative factors and/or mechanisms governing age-dependent accumulation of insoluble proteins are conserved between fungi and nematodes and thus may be conserved in eukaryotes in general.
Recently an independent group identified proteins that became insoluble with age in C. elegans. Using techniques similar to those used in our yeast and nematode aging studies, David et al. (2010) identified 461 proteins that became insoluble with age in C. elegans. Although there is significant overlap with the proteins that we identified in aging C. elegans with those reported in the study by David and colleagues (Reis-Rodrigues et al., 2012), we note that there is also remarkable similarity with the age-dependent insoluble yeast proteins identified in this study. Of the 246 insoluble yeast proteins that have nematode homologues, 102 (41.5%) were found to accumulate in aged nematodes in the study carried out by David et al. (2010). Overall the similarity of the content of the age-dependent insoluble protein fraction found in distinct aging models in divergent species strongly supports the idea that this is a regulated and conserved phenomenon that is not unique to the yeast chronologic aging paradigm.
We show that insoluble protein only accumulates in the nonquiescent subpopulation of postmitotic yeast. This suggests that insoluble protein accumulation in the postmitotic cells is not merely due to nutrient depletion in stationary phase cultures. The quiescent subpopulation has no detectable insoluble protein, yet both subpopulations were subjected to the same nutrient conditions. Instead, we suggest that insoluble protein accumulation is related to the physiological state of the nonquiescent cells (Aragon et al., 2008). In this case, insoluble protein accumulation is another indication of impaired protein homeostasis in nonquiescent cells, yet the role of insoluble protein inclusions in this context remains unclear. Recently nonquiescent cells were shown to have lower levels of mitochondrial proteins as measured by fluorescence intensity in strains carrying single GFP fusions of mitochondrial-localized proteins (Davidson et al., 2011). The nonquiescent fraction was also shown to have lower levels of respiration, higher levels of reactive oxygen species, and higher rates of petite formation, all indicative of mitochondrial dysfunction (Davidson et al., 2011). Given that we find insoluble protein containing a significant amount of mitochondrial proteins in the same subpopulation of cells, we suggest that the lower levels of mitochondrial proteins in nonquiescent cells are due to their sequestration into the insoluble fraction. Thus, if insoluble protein accumulation is an active process that occurs in nonquiescent cells (discussed later), it may be a driving factor in decreasing mitochondrial function and the associated phenotypes of nonquiescent cells.
Asymmetrical inheritance of aggregated and or damaged proteins in yeast has been well documented and described as a form of “spatial protein quality control” (Nystrom, 2011). This phenomenon has been shown to involve a number of active processes dependent upon chaperones, deacetylases, and cytoskeletal proteins (Aguilaniu et al., 2003; Liu et al., 2010). We observed that SDS-insoluble protein is present in nonquiescent cells and absent in quiescent cells. The nonquiescent cell population comprises the mother cells from the last cell mitotic division upon entrance into stationary phase, whereas the quiescent cells are the daughters from this division (Allen et al., 2006). The difference in insoluble protein distribution may be a result of asymmetrical inheritance of this protein fraction during this final division and could contribute to the enhanced viability of the quiescent daughter cells in the postmitotic population.
Postmitotic aging is not the only trigger for the formation of SDS-insoluble protein. We found that either nutrient limitation or TORC1 inactivation is sufficient to induce this process (Figures 3B and 6). Our observations indicate that TORC1 activity suppresses insoluble protein accumulation. Supporting this idea, we find that insoluble protein accumulation correlates with activation of the canonical autophagic pathway (Figures 3B and 4A), which is also negatively regulated by TORC1. However, insoluble protein accumulation does not require activation of autophagy (Figure 5). This indicates that TORC1 modulates insoluble protein accumulation and activation of autophagy through distinct mechanisms. Given that more than half of the insoluble proteins in yeast belong to complexes that have been observed in ABs and are known substrates of autophagy (Figure 2; Baba et al., 1994b; Kraft et al., 2008; Kanki et al., 2009; Okamoto et al., 2009), we suggest that insoluble protein accumulation plays a role in autophagic cargo preparation (Figure 6C). This idea is analogous to the sequestration of damaged proteins into aggresomes for degradation via autophagy (Taylor et al., 2003; Wang et al., 2009). Although aggresomes have been studied primarily in the context of misfolded proteins that accumulate in specific neurodegenerative diseases (i.e., expanded polyglutamine proteins), our work suggests that formation of SDS-insoluble proteins occurs during nutrient limitation, TORC1 inactivation, and postmitotic aging. Overall TORC1 regulation of cargo sequestration and autophagic degradation suggests that these mechanisms work in concert to promote protein degradation, amino acid recycling, and protein homeostasis.
The regulation of protein aggregation has been extensively studied in several models of aging and disease. A study in C. elegans reported that aggregation of the Aβ peptide into an insoluble form is promoted by a daf-16–dependent mechanism activated when proteasome capacity is overwhelmed (Cohen et al., 2006). This report suggests that active promotion of insoluble protein aggregation is an important mechanism facilitating protein homeostasis and is regulated in concert with disaggregase activity to promote protein degradation via the ubiquitin-proteasome pathway (Cohen et al., 2006). In studies using cell models of neurodegenerative diseases, the regulated formation of large insoluble protein aggregates has been correlated with enhanced survival (Arrasate et al., 2004; Cohen et al., 2006; Ben-Zvi et al., 2009). Overall these observations underscore the importance of regulating protein homeostasis through mechanisms that modulate protein conformation and solubility. The results presented in this study indicate that Tor1 plays a central role in regulation of protein solubility during aging and in response to nutrient stress. It is known that reduction of Tor1 activity by either genetic mutation or rapamycin treatment can increase longevity in yeast, fly, and mouse models (Kapahi et al., 2004; Kaeberlein et al., 2005; Powers et al., 2006; Harrison et al., 2009). Our results suggest that Tor1-dependent regulation of protein solubility and aggregation is likely to play a significant role in protein homeostasis, longevity, and disease.
MATERIALS AND METHODS
Yeast strains, media, and methods
Diploid yeasts used in this study were derived from S288C background (MATa/α; his3Δ1/his3Δ1; leu2Δ0/leu2Δ0; lys2Δ0/lys2Δ0; ura3Δ0/ura3Δ0, met15Δ 0/MET15; LYS2/lys2Δ 0). All deletion mutants were purchased from the Open Biosystems (Huntsville, AL) deletion collection (MATa his3Δ1/his3Δ1; leu2Δ0/leu2Δ0; lys2Δ0/lys2Δ0; ura3Δ0/ura3Δ0, met15Δ 0/MET15; LYS2/lys2Δ 0; XXX::kanMX4/XXX::kanMX4). TN121 (MATa; leu2-3, 112; trp1; ura3-52; pho8::pho8Δ60; pho13Δ::URA3) and TN123 (MATa; leu2-3, 112; trp1; ura3-52; pho8::pho8Δ60; pho13Δ::URA3; apg1Δ::LEU2) were kindly provided by Y. Ohsumi (Tokyo Institute of Technology). YTS187 (MATa; his3-Δ200; leu2-3112; lys2-801; trp1-Δ901; ura3-52; suc2-Δ9; GAL_URA3::GFP-ATG8) and YTS189 (MATa; his3-Δ200; leu2-3112; lys2-801; trp1-Δ901; ura3-52; suc2-Δ9; GAL_pep4Δ:: LEU2; GFP-ATG8::URA3) were kindly provided by D. Klionsky (University of Michigan). Ad libitum cultures were grown at 30°C in synthetic complete media (0.67% yeast nitrogen base with ammonium sulfate [Sigma-Aldrich, St. Louis, MO] and required amino acids; Sherman, 1991) supplemented with 2% glucose (SCD), and nitrogen-starved cells were grown in SC-N (0.17% yeast nitrogen base without amino acids or ammonium sulfate [BD-Difco, Sparks, MD]) supplemented with 2% glucose. Cells were treated with rapamycin at 2.0 μg/ml in 0.2% DMSO vehicle for 4 h. Postmitotic growth of cultures was determined by decreased budding index (<10%) and stabilization of culture density and determined empirically to be established ∼12 h after cultures reached OD of 1.0 for wild-type strains.
Viability analysis
Approximately 2 × 108 cells from all cultures and the same number of heat-killed cells were washed 2× in DPBS buffer (Cellgro, Manassas, VA) before being resuspended in 300 μl of DPBS buffer with 20 μM propidium iodide and incubated for 30 min 37°C. The percentage of stained cells was then determined using a BD LSR flow cytometer (BD, Franklin Lakes, NJ) and CellQuest 3.3 software measuring 500-nm emission from 10,000 cells excited at 488 nm. The percentage of unstained cells was reported after normalization of all samples to autofluorescence of unstained cells (>0.1% in all samples).
Insoluble protein preparation
A total of 2 × 109 cells harvested at each time point were washed 1× in RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 5 mM EDTA, 0.5% sodium deoxycholate, 0.1% SDS, 1.0% Nonidet P-4) and frozen. Defrosted samples were bead beat until 90% lysis was achieved in 200 μl of RIPA buffer containing complete protease inhibitor tablets (RIPAc; Roche, Indianapolis, IN). Lysate was diluted to 1 ml in RIPAc and centrifuged at 1000 × g. The resulting pellet was washed 2× with 1 ml of RIPAc. The final lysate was centrifuged at 20,000 × g for 20 min. Supernatant was removed and the pellet washed for 5 min 2× with wash buffer (150 mM NaCl, 50 mM Tris, pH 8.0), 2× with wash buffer containing 1% SDS, and 1× with water using the same centrifugation parameters. Resulting SDS-insoluble protein was agitated in 70% formic acid for 1 h and centrifuged at 20,000 × g for 20 min. Supernatant was aliquoted based on assay and SpeedVac desiccated. Resulting material was resuspended in 40 μl of pH 8.0, 10 mM Tris-ethyl ammonium bicarbonate buffer and 0.2% SDS (Applied Biosystems, Foster City, CA) for mass spectrometry analysis or empirically derived volumes of BSB buffer (1× LDS loading buffer [Invitrogen, Carlsbad, CA], 2% SDS) for SDS–PAGE analysis. Samples resuspended in BSB buffer were boiled at 100°C for 10 min, sonicated in a water bath for 5 min, and boiled at 100°C for 10 min before loading onto a gel. The relative amount of cells from the extract loaded onto each gel is noted. Polyacrylamide gels fixed with 10% methanol and 70% acetic acid were stained with Sypro Ruby (Bio-Rad, Hercules, CA) for 3 h, washed, and imaged at 532 nm with a Typhoon 8610 Variable Mode Imager and associated software (Molecular Dynamics, Sunnyvale, CA).
Mass spectrometry analysis
Trypsin digestion.
The pellet obtained form the dried formic acid–soluble protein fraction was resuspended in 30 μl of 100 mM triethylammonium bicarbonate, pH 8.0 (Sigma-Aldrich), 0.1% SDS, and 0.5% NP-40. Protein thiols were then reduced with 4.5 mM TCEP (Thermo, Rockford, IL) at 37°C for 1 h, alkylated with 10 mM iodoacetamide (Sigma-Aldrich; 30 min at room temperature), and incubated overnight at 37°C with 2 μg of sequencing-grade trypsin (Promega, Fitchburg, WI).
iTRAQ labeling and strong cation exchange chromatography.
Peptides were labeled per manufacturers instructions (ABSciex, Foster City, CA). Briefly, each iTRAQ reagent was individually reconstituted with 50 μl of isopropanol, combined with the digested peptide sample, and incubated at room temperature for 2 h.
Strong cation exchange chromatography.
Strong cation exchange chromatography was performed using a Harvard Apparatus syringe pump (Holliston, MA) loaded with a polysulfoethyl A column (4.6 × 100 mm) packed with 5-μm, 200-Å beads (PolyLC, Columbia, MD). iTRAQ-labeled peptides were combined and diluted 10-fold with 10 mM KH2PO4/25% acetonitrile, pH 3.0, and loaded onto the column at 0.25 ml/min. The column was washed with 2 ml of buffer A before elution into four 1-ml fractions with 10 mM citric acid with increasing acidity of pH 4.5, 5.5, 7, and 8. Samples were concentrated to near dryness by vacuum centrifugation and resuspended in 0.1% formic acid and 1% acetonitrile.
Liquid chromatography–tandem mass spectrometry and mass spectrometry analysis.
Individual fractions were separated out and analyzed by reversed-phase nano–high-performance liquid chromatography (HPLC)–electrospray ionization tandem mass spectrometry (MS/MS) using an Eksigent nano-LC 2D HPLC system (Eksigent, Dublin, CA) connected to a QSTAR Elite (QqTOF) mass spectrometer (MDS SCIEX, Concorde, Canada). Settings for the 3-h gradient were as described previously (Drake et al., 2011) with minor modifications. Briefly, the fragment intensity multiplier was set to 6.0, with the maximum accumulation time set to 2.5 s and the enhanced reporter region adjusted for collision energy of the iTRAQ reagent in the data-dependent acquisition settings (Analyst QS 2.0). Two injection replicates were performed to maximize sampling efficiency.
Database searches.
Mass spectrometric data were analyzed using the database search engine ProteinPilot 4.0 (ABSciex) with revision #148085 using the Paragon algorithm (4.0.0.0, 148083). The following sample parameters were used: trypsin digestion, cysteine alkylation set to iodoacetamide, and species S. cerevisiae. Processing parameters were set to identify “Biological modifications” using a “thorough ID” search effort. All data files were searched using the SwissProt 2011_02 (8 Feb. 2011) protein database with 525,207 sequences. To determine FDR at the protein level we used the Proteomics System Performance Evaluation Pipeline tool in ProteinPilot 4.0 (Tang et al., 2008) to generate a global FDR at 1% for a peptide confidence score of 88.8%. Although a high threshold was set for peptide identifications in ProteinPilot, false-positive assignments are known to be higher for proteins identified by only one peptide. Therefore, to reduce false positives among this group, we manually examined each of the 94 MS/MS spectra in this category using a set of rigorous criteria we previously established for this purpose (Lee et al., 2010) and discarded 14 peptide spectra (14.9% attrition rate).
iTRAQ data analysis
Biological replicates.
Relative ratios of >1.5 with p > 0.05 were considered to be significantly different.
Nitrogen-starved versus postmitotically aged.
Biological replicates facilitated four distinct comparisons between treatments of each protein identified in this study. Statistical difference was determined to include those comparisons that had a relative ratios of >1.5 with p > 0.05 in at least two of the four comparisons and an average iTRAQ ratio that exceed biological variation.
Alkaline phosphatase assay
Approximately 1 × 107 cells carrying the Pho8Δ60 reporter gene were harvested during log phase and washed 3× in 1 ml of ALP lysis buffer (250 mM Tris-SO4, pH 9.4, 10 mM MgSO4, 10 M ZnSO4). The final cell pellet was resuspended in 250 μl of ALP lysis buffer, 50 μl was diluted to 1 ml and OD600 measured, and the remaining cells were lysed. The lysates were cleared, and 50 μl of cleared lysate was incubated with equal volume of 50 mM α-naphthyl phosphate disodium salt (Sigma-Aldrich) and 500 μl of ALP lysis buffer. After color development, 0.5 ml of 2 M glycine-NaOH, pH 11.0, was added and OD420 measured. Relative ALP activity was determined by the ratio OD420/OD600 and reported normalized to the corresponding atg1Δ samples.
GFP-Atg8 assay
The equivalent of 2 ml of cells at OD 1.0 was harvested from both wild-type (TN121) and pep4Δ (TN123) cultures. Cell lysates were created using trichloroacetic acetic acid precipitation and analyzed via PAGE and Western blot using an anti-GFP antibody (Cell Signaling Technology, Beverly, MA; Shintani and Klionsky, 2004). Signal intensity was measured using Image Quant TL (GE Healthcare Life Sciences, Piscataway, NJ).
Gene Ontology analysis
Age-dependent insoluble proteins were submitted to the DAVID Bioinformatics Databases Functional Annotation Tool v6.7 (National Institute of Allergy and Infectious Diseases, National Institutes of Health; http://david.abcc.ncifcrf.gov/). GO enrichment was determined using the yeast genome as background. Reported enriched cellular component categories have an FDR of <0.05 and are minimally redundant.
Homology analysis
UniProt accession numbers obtained from protein report of insoluble proteins identified in aged yeast were mapped to Entrez GeneIDs and queried for homology using the HomoloGene database (www.ncbi.nlm.nih.gov/homologene). Homologous C. elegans proteins were mapped to UniProt accession numbers and cross-referenced with the C. elegans age-dependent insoluble protein lists from two reports (David et al., 2010; Reis-Rodrigues et al., 2012). This process was repeated starting with nematode insoluble protein lists and mapping homologues found in the yeast insoluble protein list. Owing to multiple mapping discrepancies, these conversions found slightly different numbers of homologues (fewer than four) in either comparison. In both cases we report the more conservative estimate.
Postmitotic population separation
Percoll gradients were generated by centrifuging 10 ml of diluted Percoll (1 g/ml Percoll [GE Healthcare], 1.67 M NaCl) at 19,240 × g for 15 min. Cells were subsequently loaded onto gradient, and populations were separated via centrifugation at 400 × g for 60 min.
Microscopy
Nomarski differential interference contrast images of cells in SCD medium were taken at room temperature using an Eclipse E800 microscope with a 100×/1.40 oil objective, imaged using a DXM1200 camera (Nikon, Melville, NY), and processed with ACT-1v2.63 software.
Statistical analysis
All p values were generated using a Student's t test, except for the homologous insoluble protein study, which was generated using a permutation analysis. The permutation analysis was based on the 494 proteins that were identified at 95% confidence before the parsing of the 14 single-peptide false positives. In this analysis, 494 proteins were randomly selected from the yeast proteome and compared with the 192 nematode-to-yeast–converted insoluble proteins. This was repeated 100,000 times, and the average overlap between the permutations and nematode-converted insoluble protein list was 18 proteins.
Supplementary Material
Acknowledgments
We thank Yoshinori Ohsumi for kindly providing the Pho8Δ60 strains, Daniel Klionsky for kindly providing the GFP-ATG8 strains, Birgit Schilling for her expert spectral analysis of proteins identified from single peptides, and Maggie Werner-Washburne for helpful discussions. This work was supported by National Institutes of Health Grants RL1 GM084432 (R.E.H.) and 1R01 AG029631-01A1 (G.J.L.) and the Larry Hillblom Foundation (R.E.H. and G.J.L.). Mass spectrometry was supported by National Center for Research Resources Shared Instrumentation Grant S10 RR024615 (B.W.G.) and Geroscience Mass Spectrometry and Imaging Core Grant PL1 AG032118 (B.W.G.). T.W.P. and M.J.R. were supported by National Institutes of Health Training Grant T32 AG000266. G.C. and M.J.R. were supported by National Institutes of Health Grant PL1 AG032118 (BWG). U.S.E. was supported by National Institutes of Health Grant R01 LM009722NIH and the National Center for Biomedical Ontology (U54-HG004028 [PI: Musen]) to S.D.M.
Abbreviations used:
- AB
autophagic body
- ALP
alkaline phosphatase
- GFP
green fluorescent protein
- GO
Gene Ontology
- iTRAQ
isobaric tag for relative and absolute quantitation
- TOR
target of rapamycin
- TORC1
target of rapamycin complex 1
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-08-0620) on October 24, 2012.
T.W.P., G.J.L., B.W.G., and R.E.H. designed the experiments. T.W.P., M.J.R., G.C., and P.R.-R. carried out the experiments. T.W.P., M.J.R., G.C., U.S.E., S.D.M., B.W.G., and R.E.H. analyzed the data. T.W.P., B.W.G., and R.E.H. prepared the manuscript.
REFERENCES
- Abeliovich H, Klionsky DJ. Autophagy in yeast: mechanistic insights and physiological function. Microbiol Mol Biol Rev. 2001;65:463–479. doi: 10.1128/MMBR.65.3.463-479.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilaniu H, Gustafsson L, Rigoulet M, Nystrom T. Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science. 2003;299:1751–1753. doi: 10.1126/science.1080418. [DOI] [PubMed] [Google Scholar]
- Allen C, et al. Isolation of quiescent and nonquiescent cells from yeast stationary-phase cultures. J Cell Biol. 2006;174:89–100. doi: 10.1083/jcb.200604072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragon AD, Rodriguez AL, Meirelles O, Roy S, Davidson GS, Tapia PH, Allen C, Joe R, Benn D, Werner-Washburne M. Characterization of differentiated quiescent and nonquiescent cells in yeast stationary-phase cultures. Mol Biol Cell. 2008;19:1271–1280. doi: 10.1091/mbc.E07-07-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Baba M, Takeshige K, Baba N, Ohsumi Y. Ultrastructural analysis of the autophagic process in yeast: detection of autophagosomes and their characterization. J Cell Biol. 1994a;124:903–913. doi: 10.1083/jcb.124.6.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M, Takeshige K, Baba N, Ohsumi Y. Ultrastructural analysis of the autophagic process in yeast: detection of autophagosomes and their characterization. J Cell Biol. 1994b;124:903–913. doi: 10.1083/jcb.124.6.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett A, Brewer GJ. Autophagy in aging and Alzheimer's disease: pathologic or protective? J Alzheimers Dis. 2011;25:385–394. doi: 10.3233/JAD-2011-101989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Zvi A, Miller EA, Morimoto RI. Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc Natl Acad Sci USA. 2009;106:14914–14919. doi: 10.1073/pnas.0902882106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Bergamini E, Brunk UT, Droge W, Ffrench M, Terman A. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy. 2005;1:131–140. doi: 10.4161/auto.1.3.2017. [DOI] [PubMed] [Google Scholar]
- Dann SG, Thomas G. The amino acid sensitive TOR pathway from yeast to mammals. FEBS Lett. 2006;580:2821–2829. doi: 10.1016/j.febslet.2006.04.068. [DOI] [PubMed] [Google Scholar]
- David DC, Ollikainen N, Trinidad JC, Cary MP, Burlingame AL, Kenyon C. Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol. 2010;8:e1000450. doi: 10.1371/journal.pbio.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson GS, et al. The proteomics of quiescent and nonquiescent cell differentiation in yeast stationary-phase cultures. Mol Biol Cell. 2011;22:988–998. doi: 10.1091/mbc.E10-06-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake PM, et al. A lectin affinity workflow targeting glycosite-specific, cancer-related carbohydrate structures in trypsin-digested human plasma. Anal Biochem. 2011;408:71–85. doi: 10.1016/j.ab.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M, Chandra A, Mitic LL, Onken B, Driscoll M, Kenyon C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 2008;4:e24. doi: 10.1371/journal.pgen.0040024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DE, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhasz G, Erdi B, Sass M, Neufeld TP. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 2007;21:3061–3066. doi: 10.1101/gad.1600707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, Powers RW 3rd, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310:1193–1196. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell. 2009;17:98–109. doi: 10.1016/j.devcel.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14:885–890. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy. 2007;3:181–206. doi: 10.4161/auto.3678. [DOI] [PubMed] [Google Scholar]
- Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- Kraft C, Deplazes A, Sohrmann M, Peter M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat Cell Biol. 2008;10:602–610. doi: 10.1038/ncb1723. [DOI] [PubMed] [Google Scholar]
- Lee BY, Jethwaney D, Schilling B, Clemens DL, Gibson BW, Horwitz MA. The Mycobacterium bovis bacille Calmette-Guerin phagosome proteome. Mol Cell Proteomics. 2010;9:32–53. doi: 10.1074/mcp.M900396-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Larsson L, Caballero A, Hao X, Oling D, Grantham J, Nystrom T. The polarisome is required for segregation and retrograde transport of protein aggregates. Cell. 2010;140:257–267. doi: 10.1016/j.cell.2009.12.031. [DOI] [PubMed] [Google Scholar]
- Nystrom T. Spatial protein quality control and the evolution of lineage-specific ageing. Philos Trans R Soc Lond. B Biol Sci. 2011;366:71–75. doi: 10.1098/rstb.2010.0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell. 2009;17:87–97. doi: 10.1016/j.devcel.2009.06.013. [DOI] [PubMed] [Google Scholar]
- Pan KZ, Palter JE, Rogers AN, Olsen A, Chen D, Lithgow GJ, Kapahi P. Inhibition of mRNA translation extends lifespan in Caenorhabditis elegans. Aging Cell. 2007;6:111–119. doi: 10.1111/j.1474-9726.2006.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers RW 3rd, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006;20:174–184. doi: 10.1101/gad.1381406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasheva VI, Domingos PM. Cellular responses to endoplasmic reticulum stress and apoptosis. Apoptosis. 2009;14:996–1007. doi: 10.1007/s10495-009-0341-y. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–1117. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Reiling JH, Sabatini DM. Stress and mTORture signaling. Oncogene. 2006;25:6373–6383. doi: 10.1038/sj.onc.1209889. [DOI] [PubMed] [Google Scholar]
- Reis-Rodrigues P, et al. Proteomic analysis of age-dependent changes in protein solubility identifies genes that modulate lifespan. Aging Cell. 2012;11:120–127. doi: 10.1111/j.1474-9726.2011.00765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley BE, et al. Ubiquitin accumulation in autophagy-deficient mice is dependent on the Nrf2-mediated stress response pathway: a potential role for protein aggregation in autophagic substrate selection. J Cell Biol. 2010;191:537–552. doi: 10.1083/jcb.201005012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Cell biology of protein misfolding: the examples of Alzheimer's and Parkinson's diseases. Nat Cell Biol. 2004;6:1054–1061. doi: 10.1038/ncb1104-1054. [DOI] [PubMed] [Google Scholar]
- Sherman F. Getting started with yeast. Methods Enzymol. 1991;194:3–21. doi: 10.1016/0076-6879(91)94004-v. [DOI] [PubMed] [Google Scholar]
- Shintani T, Klionsky DJ. Cargo proteins facilitate the formation of transport vesicles in the cytoplasm to vacuole targeting pathway. J Biol Chem. 2004;279:29889–29894. doi: 10.1074/jbc.M404399200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soskic V, Groebe K, Schrattenholz A. Nonenzymatic posttranslational protein modifications in ageing. Exp Gerontol. 2008;43:247–257. doi: 10.1016/j.exger.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Tagawa K, Hoshino M, Okuda T, Ueda H, Hayashi H, Engemann S, Okado H, Ichikawa M, Wanker EE, Okazawa H. Distinct aggregation and cell death patterns among different types of primary neurons induced by mutant huntingtin protein. J Neurochem. 2004;89:974–987. doi: 10.1111/j.1471-4159.2004.02372.x. [DOI] [PubMed] [Google Scholar]
- Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J Cell Biol. 1992;119:301–311. doi: 10.1083/jcb.119.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanemura K, et al. Formation of tau inclusions in knock-in mice with familial Alzheimer disease (FAD) mutation of presenilin 1 (PS1) J Biol Chem. 2006;281:5037–5041. doi: 10.1074/jbc.M509145200. [DOI] [PubMed] [Google Scholar]
- Tang WH, Shilov IV, Seymour SL. Nonlinear fitting method for determining local false discovery rates from decoy database searches. J Proteome Res. 2008;7:3661–3667. doi: 10.1021/pr070492f. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Tanaka F, Robitschek J, Sandoval CM, Taye A, Markovic-Plese S, Fischbeck KH. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum Mol Genet. 2003;12:749–757. doi: 10.1093/hmg/ddg074. [DOI] [PubMed] [Google Scholar]
- Taylor RC, Dillin A. Aging as an event of proteostasis collapse. Cold Spring Harb Perspect Biol. 2011;3:a004440. doi: 10.1101/cshperspect.a004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetkov AS, Miller J, Arrasate M, Wong JS, Pleiss MA, Finkbeiner S. A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc Natl Acad Sci USA. 2010;107:16982–16987. doi: 10.1073/pnas.1004498107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Meriin AB, Zaarur N, Romanova NV, Chernoff YO, Costello CE, Sherman MY. Abnormal proteins can form aggresome in yeast: aggresome-targeting signals and components of the machinery. FASEB J. 2009;23:451–463. doi: 10.1096/fj.08-117614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.