

Arachidonic acid (AA) is remarkably enriched in phosphatidylinositol (PI). Studies using knockout mice of lysophosphatidylinositol acyltransferase 1, which selectively incorporates AA into PI, reveal that AA-containing PI plays a crucial role in cortical lamination and neuronal migration during brain development.
Abstract
Dietary arachidonic acid (AA) has roles in growth, neuronal development, and cognitive function in infants. AA is remarkably enriched in phosphatidylinositol (PI), an important constituent of biological membranes in mammals; however, the physiological significance of AA-containing PI remains unknown. In an RNA interference–based genetic screen using Caenorhabditis elegans, we recently cloned mboa-7 as an acyltransferase that selectively incorporates AA into PI. Here we show that lysophosphatidylinositol acyltransferase 1 (LPIAT1, also known as MBOAT7), the closest mammalian homologue, plays a crucial role in brain development in mice. Lpiat1−/− mice show almost no LPIAT activity with arachidonoyl-CoA as an acyl donor and show reduced AA contents in PI and PI phosphates. Lpiat1−/− mice die within a month and show atrophy of the cerebral cortex and hippocampus. Immunohistochemical analysis reveals disordered cortical lamination and delayed neuronal migration in the cortex of E18.5 Lpiat1−/− mice. LPIAT1 deficiency also causes disordered neuronal processes in the cortex and reduced neurite outgrowth in vitro. Taken together, these results demonstrate that AA-containing PI/PI phosphates play an important role in normal cortical lamination during brain development in mice.
INTRODUCTION
Polyunsaturated fatty acids (PUFAs) in membrane phospholipids play critical roles in regulating the structure, dynamics, and permeability of membranes. In mammals, the PUFA composition affects many cellular processes, including modulation of ion channels (Chyb et al., 1999; Xiao et al., 2001) and activities of membrane-associated enzymes that are sensitive to the biophysical properties of lipid membrane (Goldberg and Zidovetzki, 1997). Postnatal PUFA depletion causes various abnormalities, including sterility, ulceration, and dermatitis, and PUFA supplementation can alleviate most of those symptoms (Stoffel et al., 2008; Stroud et al., 2009; Roqueta-Rivera et al., 2010; Williard et al., 2001). Arachidonic acid (AA; 20:4n-6) is the most enriched n-6 PUFA in the brain and is involved in multiple aspects of neuronal development and function, including neurite outgrowth, signal transduction, and membrane fluidity. The prenatal and postnatal status of AA is associated with early postnatal neurological function (Dijck-Brouwer et al., 2005; Zhao et al., 2009). There are several lines of evidence suggesting that AA has more beneficial effects than other n-6 fatty acids for perinatal infants. Human infants, as well as neonatal animals, have a reduced ability to elongate/desaturate fatty acids to an appropriate degree, and supplementation with linoleic acid and linolenic acid is not sufficient to overcome a PUFA-deficiency state (Abad-Jorge, 2008). AA is preferentially transferred across the placenta compared with other n-6 fatty acids (Davis-Bruno and Tassinari, 2011). Moreover, a neurological optimality score that quantifies the quality of neonatal neurological functioning is positively related with AA but not with other n-6 fatty acids (Dijck-Brouwer et al., 2005). Nutritional studies also suggest that AA supplementation ameliorates cognitive function in aged animals (Kotani et al., 2003; Okaichi et al., 2005) and helps to protect against a variety of mental disorders (Maekawa et al., 2009).
Among membrane phospholipids, phosphatidylinositol (PI) is unique in its fatty acid composition, that is, most of the fatty acid attached to the sn-2 position of PI is AA (20:4n-6), whereas other major membrane phospholipids such as phosphatidylcholine (PC) and phosphatidylethanolamine (PE) contain various PUFAs, including docosahexaenoic acid (DHA; 22:6n-3; Jungalwala et al., 1984; Patton et al., 1982; Nakagawa et al., 1985; Tanaka et al., 2003). However, the physiological significance of the enrichment of AA in PI is unclear. AA-containing PI is formed by fatty acid remodeling after the de novo synthesis of PI through sequential deacylation and reacylation reactions (Holub and Kuksis, 1971; Baker and Thompson, 1973; Luthra and Sheltawy, 1976). Although high levels of lysophosphatidylinositol acyltransferase (LPIAT) activities were detected from various mammalian tissues by in vitro assays (Baker and Thompson, 1973; Inoue et al., 1984; Sanjanwala et al., 1989; Yashiro et al., 1995), the gene responsible for the activity was not identified until recently. In an RNA interference–based genetic screen using Caenorhabditis elegans, we identified a gene encoding an acyltransferase that selectively incorporates AA into PI and named it mboa-7 (Lee et al., 2008). We also showed that a human orthologue of mboa-7 exhibited arachidonoyl-CoA (AA-CoA):LPIAT activity and that small interfering RNA–mediated knockdown selectively reduced the AA-CoA:LPIAT activity and [14C]AA incorporation into the PI fraction in HeLa cells. Voelker's group named a mammalian orthologue of mboa-7 “MBOAT7” and showed that MBOAT7 is an LPIAT with remarkable specificity for AA-CoA by using mass spectrometry–based enzyme assays (Gijón et al., 2008). Drosophila orthologue also prefers AA-CoA as an acyl donor (Steinhauer et al., 2009). Shindou and Shimizu (2009) renamed MBOAT7 as LPIAT1 based on its substrate specificity and by the order of its publication in their review. In the present study, we generated LPIAT1-knockout mice to elucidate the physiological significance of AA in PI.
RESULTS
Targeted deletion of the Lpiat1 gene in the mouse
To determine the physiological role of LPIAT1 in mammals, we generated LPIAT1-deficient mice (Lpiat1−/− mice) by gene targeting (Figure 1, A and B). A targeting vector substituted a neomycin-resistant gene for exons 2–4 of the Lpiat1 gene, deleting the initiation codon. Western blot analysis of wild-type mice revealed high expression of LPIAT1 in the brain among the tissues tested (Figure 1C). In the fetal brain, expression of LPIAT1 was observed in the cerebral cortex (Figure 1D), the CA regions and the dentate gyrus of the hippocampus (Figure 1E), the external plexiform layer and the mitral cell layer of the olfactory bulb (Figure 1F), and the granular cell layer of the cerebellum (Figure 1G). In contrast, LPIAT1 protein was not detected in tissues from Lpiat1−/− mice (Figure 1, C–G). LPIAT activity with AA-CoA as an acyl donor was almost absent in the membranes of the brain (Figure 1H), but acyltransferase activities toward other lysophospholipids were not changed (Figure 1I). AA-CoA:LPIAT activity was also undetectable in the membranes of the liver, kidney, and testis from Lpiat1−/− mice (Figure 1H). These data indicate that LPIAT1 is the predominant enzyme that catalyzes the incorporation of AA into lysoPI in mice.
FIGURE 1:

Lpiat1−/− mice lack AA-CoA:LPIAT activity. (A) Diagram of the Lpiat1 genomic locus and the targeting vector. The positions of the PCR primers (P1, P2, and P3) are indicated. All three primers were used in the same PCR. (B) PCR analysis of genomic DNAs from Lpiat1+/+, Lpiat1+/−, and Lpiat1−/− mice. (C) Immunoblot analysis of LPIAT1. Each tissue was prepared from 2-wk-old Lpiat1+/+ and Lpiat1−/− mice. Control GAPDH was run simultaneously in a different gel. The same amount of protein was loaded in each lane. (D–G) LPIAT1 expression in the brain. Sagittal sections of the brains from E18.5 Lpiat1+/+ and Lpiat1−/− littermates were stained with antibodies against mouse LPIAT1. Neocortex (D), hippocampus (E), olfactory bulb (F), and cerebellum (G) are shown. LPIAT1 was also detected in granule cells in accessory olfactory bulb. Scale bar, 100 μm. (H) AA-CoA:LPIAT activity in the membrane fractions of each tissue from Lpiat1+/+, Lpiat1+/−, and Lpiat1−/− mice at 2 wk of age. [14C]AA-CoA, lysoPI, and 5 μg of protein were used. Data are means ± SD (n = 3). (I) AA-CoA:lysophospholipid acyltransferase activity of the Lpiat1+/+, Lpiat1+/−, and Lpiat1−/− brains using the indicated lysophospholipids as acyl acceptors. PS, phosphatidylserine; PA, phosphatidic acid. Protein at 20 μg was used. Data are means ± SD (n = 3).
Lpiat1−/− mice show reduced AA content in PI and PI phosphates
PI participates in various types of signal transduction through distinct phosphorylated derivatives of the inositol head group (Di Paolo and De Camilli, 2006; Sasaki et al., 2009). Therefore we analyzed the fatty acyl species of PI and PI phosphates in the Lpiat1−/− brain. The amount of AA in total PI fatty acids in the Lpiat1−/− brain (28%) was significantly less than that in the Lpiat1+/+ brain (43%; Figure 2A). Conversely, other fatty acids, such as palmitic acid (16:0) and DHA in PI, were increased in the Lpiat1−/− brain. Consistent with this observation, liquid chromatography–electrospray ionization mass spectrometry (LC/ESI-MS) analysis revealed that the amount of 18:0/20:4 PI (38:4 PI) was significantly reduced in the Lpiat1−/− brain (Figure 2, B and C). The molecular species compositions of PC and PE were not significantly affected (Figure 2, D and E). PI content was slightly decreased in the Lpiat1−/− brain (Figure 2F). In addition to the reduced content of AA in PI, the fatty acyl species of PI monophosphate (PIP; primarily phosphatidylinositol 4-phosphate [PI4P]) and PI bisphosphate (PIP2; primarily phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2]) were affected in a manner similar to that of PI (Figure 2, G and H). The contents of PI phosphates in the Lpiat1+/+ and Lpiat1−/− brains could not be measured because the values fluctuated from experiment to experiment.
FIGURE 2:

Lipid analysis of Lpiat1−/− mice. (A) Analysis of PI fatty acid composition of the brains by gas chromatography. C16:0, palmitic acid; C18:0, stearic acid; C18:1n-9, oleic acid; C18:1n-7, vaccenic acid; C20:4n-6, arachidonic acid; C22:4n-6, docosatetraenoic acid; C22:6n-3, docosahexaenoic acid. Data are means ± SEM (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001. The unpaired, two-tailed t test was used. (B) LC-MS/MS analysis of PI. Negative-ionization LC-MS spectra of PI molecular species of the Lpiat1+/+ and Lpiat1−/− brains. 34:1, 16:0/18:1; 36:1, 18:0/18:1; 36:2, 18:1/18:1; 36:4, 16:0/20:4; 38:4, 18:0/20:4; 38:5, 18:1/20:4; 40:6, 18:0/22:6; 40:7, 18:1/22:6. Data are means ± SEM (n = 5). *p < 0.05; **p < 0.01; ***p < 0.001. The unpaired, two-tailed t test was used. (C) Negative-ionization LC-MS spectra of PI molecular species of the Lpiat1+/+ (top) and Lpiat1−/− (bottom) brains. 36:4, 16:0/20:4; 38:4, 18:0/20:4; 40:6, 18:0/22:6. (D) Positive-ionization LC-MS spectra of PC molecular species. 32:1, mainly 16:0/16:1; 32:0, 16:0/16:0; 34:1, 16:0/18:1; 36:4, mainly 16:0/20:4; 36:1, 18:0/18:1; 38:6, 16:0/22:6; 38:4, mainly 18:0/20:4; 40:6, 18:0/22:6. (E) Positive-ionization LC-MS spectra of PE molecular species. 34:1, 16:0/18:1; 38:6 alkenyl, 16:0 alkenyl/22:6; 38:6, mainly 16:0/22:6; 38:4, mainly 18:0/20:4; 40:6 alkenyl, 18:0 alkenyl/22:6; 40:6, 18:0/22:6. (F) The content of individual phospholipid of the brains. Data are means ± SEM (n = 4-5). *p < 0.05. The unpaired, two-tailed t test was used. (G, H) LC/MS analysis of PIP (primarily PI4P; G) and PIP2 (primarily PI(4,5)P2; H) of Lpiat1+/+ and Lpiat1−/− mouse hippocampus. Data are means ± SEM (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001. Unpaired, two-tailed t test was used.
As shown previously (Palmer, 1986), acyltransferase activity with AA-CoA as an acyl donor toward lysoPI4P or lysoPI(4,5)P2 was almost undetectable in the membranes of the brain from wild-type mice compared with the activity toward lysoPI (Figure 3A). Overexpression of LPIAT1 did not increase the acyltransferase activity toward lysoPI4P or lysoPI(4,5)P2 (Figure 3B). These data indicate that AA-containing PI phosphates are not formed by incorporation of AA into lysoPI phosphates.
FIGURE 3:

LPIAT1 does not use lysoPI4P or lysoPI(4,5)P2 as acyl acceptors. (A) AA-CoA:acyltransferase activity toward lysoPI, lysoPI4P, or lysoPI(4,5)P2 in the membrane fractions of the Lpiat1+/+ and Lpiat1−/− brains at P0. Protein at 5 μg was used. ND, not detected or present only in trace amounts. Data are means ± SD (n = 3). (B) AA-CoA:acyltransferase activity toward lysoPI, lysoPI4P, or lysoPI(4,5)P2 in the membrane fractions of HEK 293A cells transfected with vector control or Lpiat1 expression plasmid. ND, not detected or present only in trace amounts. Protein at 1 μg was used. Data are means ± SD (n = 3).
AA metabolites in Lpiat1−/− mice
AA cleaved from phospholipids is converted to bioactive mediators such as prostaglandins and leukotrienes. An LC-MS/MS analysis showed that the amounts of free AA and its metabolites such as prostaglandin D2 (PGD2) and 5-hydroxyeicosatetraenoic acid (5-HETE) were slightly but not significantly reduced in the Lpiat1−/− mouse brain compared with the Lpiat1+/+ mouse brain (Table 1). Leukotrienes were not detectable under the present conditions.
TABLE 1:
AA metabolites of Lpiat1+/+ and Lpiat1−/− brains.
| AA metabolite | Lpiat1+/+ | Lpiat1−/− |
|---|---|---|
| AA | 1620 ± 307 | 971 ± 120 |
| PGE2 | 0.581 ± 0.113 | 0.482 ± 0.121 |
| PGD2 | 1.95 ± 0.64 | 0.873 ± 0.236 |
| PGF2α | 0.447 ± 0.094 | 0.412 ± 0.080 |
| 6-keto-PGF1α | 1.02 ± 0.14 | 0.809 ± 0.204 |
| TXB2 | 3.84 ± 0.94 | 4.74 ± 0.98 |
| 5-HETE | 1.15 ± 0.30 | 0.424 ± 0.070 |
| 12-HETE | 0.165 ± 0.043 | 0.069 ± 0.030 |
| 15-HETE | 0.201 ± 0.052 | 0.168 ± 0.042 |
P0 brains were harvested, weighed, and frozen immediately in liquid nitrogen. Data are pg/mg wet weight expressed as means ± SEM (n = 6). TXB2, thromboxane B2.
Lpiat1−/− mice show abnormal brain morphology
Matings between Lpiat1 heterozygous mice yielded significantly fewer P0 Lpiat1−/− progeny than expected according to Mendelian inheritance (18.0%; p = 0.0027, chi-square test), whereas the genotypes of embryonic day 16.5 (E16.5) and E18.5 progeny followed the expected Mendelian frequencies (Table 2). The number of surviving Lpiat1−/− mice diminished markedly in the 4 wk after birth (Figure 4A). Lpiat1−/− mice were significantly smaller than Lpiat1+/− and Lpiat1+/+ littermates (Figure 4, B and C).
TABLE 2:
Genotypes of litters from Lpiat1+/− intercrosses.
| Lpiat1 Genotype | |||||
|---|---|---|---|---|---|
| Stage | +/+ | +/− | −/− | Total | |
| E16.5 | Number of animals | 28 | 52 | 28 | 108 |
| Percentage (%) | 26.0 | 48.1 | 26.0 | 100 | |
| E18.5 | Number of animals | 27 | 43 | 23 | 93 |
| Percentage (%) | 29.0 | 46.2 | 24.7 | 100 | |
| Birth (P0) | Number of animals | 126 | 247 | 82 | 455 |
| Percentage (%) | 27.7 | 54.3 | 18.0* | 100 | |
Neonates and embryos were harvested at the times indicated. Genomic DNA was extracted from the tail of each pup and subjected to PCR analysis to determine the genotype. Only live-born animals are counted at birth (P0). *p = 0.0027. The p values were calculated using the chi-square test.
FIGURE 4:

Abnormal morphology of the Lpiat1−/− brain. (A) Juvenile lethality of Lpiat1−/− mice (n = 14). (B) Representative photograph of Lpiat1−/− mouse compared with a wild-type littermate at 3 wk of age. Scale bar, 1 cm. (C) Body weights of Lpiat1+/+, Lpiat1+/−, and Lpiat1−/− mice at different points in time. Littermates of all genotypes (+/+, n = 29; +/−, n = 41; −/−, n = 14) were weighed at 1 and 2 wk of age. Newborn littermates (+/+, n = 6; +/−, n = 12; −/−, n = 4) were weighed before killing for primary hippocampal culture (see Figure 9). NB, newborn. Data are means ± SEM *p < 0.05; **p < 0.01. The analysis of variance with Tukey-Kramer post hoc test was used. (D) Brains from Lpiat1+/+ and Lpiat1−/− littermates at E18.5. cx, cortex; hb, hindbrain; mb, midbrain; ob, olfactory bulb. Scale bar, 2.5 mm. (E, F) Sagittal sections of the brains from E18.5 Lpiat1+/+ (E) and Lpiat1−/− (F) littermates were stained with hematoxylin and eosin (H&E). cb, cerebellum; cx, cortex; hi, hippocampus; mb, midbrain; ob, olfactory bulb. The red asterisk indicates the atrophic cortex, and the yellow star indicates the atrophic hippocampus. Scale bar, 500 μm.
At E18.5, the forebrain of Lpiat1−/− mice was smaller in size, whereas the midbrain appeared to be normal in size (Figure 4D). Histological analysis revealed that the sizes of the cerebral cortex and hippocampus were reduced in the Lpiat1−/− brain (Figure 4, E and F). The laminar structures of the cerebral cortex and hippocampus were disarranged in the Lpiat1−/− brain. These results indicate that LPIAT1 deficiency causes atrophy of the cerebral cortex and hippocampus and disordered lamination in the cortical layer.
The mammalian neocortex has a characteristic laminar structure composed of six layers of different cells. The cortical layer formation begins around E10–12 (Gleeson and Walsh, 2000). First, the earliest-born cells from cortical ventricular zone migrate toward the pial surface to form the preplate. Subsequently generated neurons arise from the ventricular zone and migrate into the preplate to form the cortical plate (layer 6), splitting the preplate into the marginal zone (layer 1) and the subplate below. Thereafter, newly born neurons migrate past the subplate and older cortical plate neurons to form more superficial layer, thereby making layers 2–6 in an inside-out pattern by E18.5 (Rakic, 1988; Tissir and Goffinet, 2003). We analyzed the layer formation in Lpiat1−/− mice with specific markers. Tbr1 is a marker of early-born neurons. At E18.5, Tbr1-positive neurons were located mainly in the lower cortical plate (layer 6; Hevner et al., 2001) in wild-type mice (Figure 5A), whereas they were broadly scattered throughout the cortical plate in Lpiat1−/− mice (Figure 5B). Brn1 is a marker of the late precursor cells of the ventricular and subventricular zones and the migrating neurons. By E18.5, Brn1-positive neurons in wild-type mice had migrated into the cortical plate (Figure 5C), as shown previously (McEvilly et al., 2002), whereas most of the Brn1-positive neurons were stacked in the intermediate zone in Lpiat1−/− mice (Figure 5D). Cells positive for Reelin, a marker of Cajal–Retzius neurons (Alcántara et al., 1998; D'Arcangelo et al., 1995, 1997; Ogawa et al., 1995), were normally distributed in the marginal zone of the Lpiat1−/− cortex (Figure 5, A and B). In the wild-type cortex, MAP2-positive neuronal processes were arranged radially and formed a tight, palisade-like structure (Figure 5E). In contrast, in Lpiat1−/− mice, the palisade-like neuronal processes were disordered in the cortex, and the subplate neurons, which are also stained by MAP2 (Luskin and Shatz, 1985), were dispersed (Figure 5F). Neuronal differentiation during early corticogenesis at E14.5 appeared normal in Lpiat1−/− mice as assessed by the expressions of βIII-tubulin, Tbr1, and MAP2 (Figure 6). These data indicate that LPIAT1 is required for normal cortical lamination.
FIGURE 5:
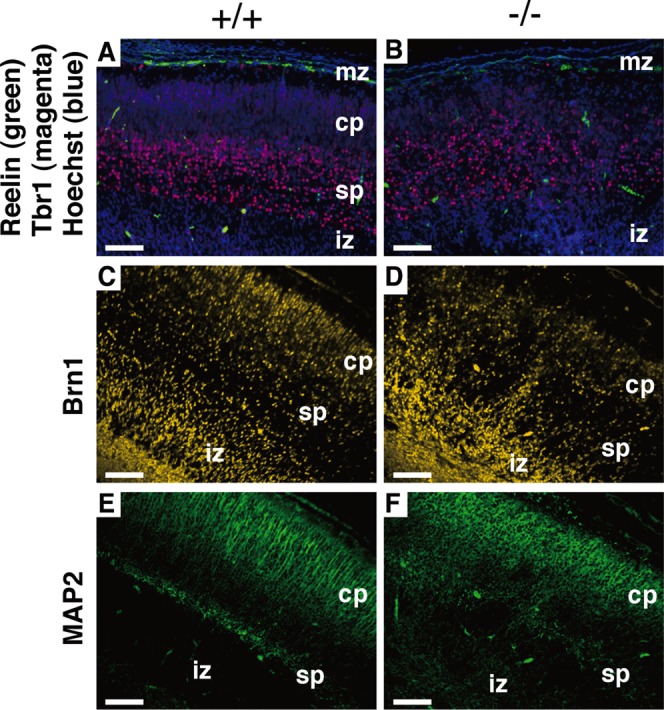
Abnormal cortical lamination in Lpiat1−/− mice. Sagittal sections of neocortices from E18.5 Lpiat1+/+ (A, C, E) and Lpiat1−/− (B, D, F) littermates were stained with antibodies against Reelin and Tbr1, and DNA dye Hoechst 33342 (green, magenta, and blue, respectively; A, B), Brn1 (yellow; C, D), and MAP2 (green; E, F). cp, cortical plate; iz, intermediate zone; mz, maginal zone; sp, subplate. Scale bar, 100 μm.
FIGURE 6:
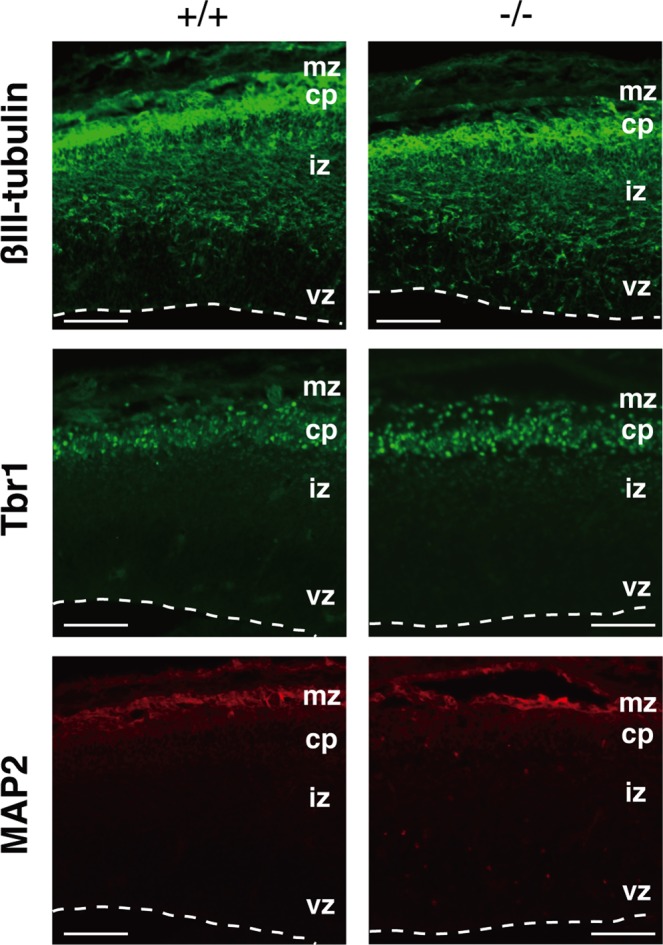
Loss of LPIAT1 does not affect neuronal differentiation. Coronal sections of the E14.5 neocortices were stained with antibodies against βIII-tubulin, Tbr1, and MAP2. cp, cortical plate; iz, intermediate zone; mz, maginal zone; vz, ventricular zone. Scale bar, 50 μm.
Neuronal migration is delayed in Lpiat1−/− mice
The disturbed laminar organization of the Lpiat1−/− cortex suggested that the migrations of cortical cells were abnormal. We injected pregnant females with 5-bromodeoxyuridine (BrdU) at E15.5, when neurogenesis of the middle and superficial layers is at a peak, and examined the localization of BrdU-positive cortical neurons at E18.5. Most of the BrdU-positive cells were superficially distributed in the Lpiat1+/+ cortex (Figure 7A). In the Lpiat1−/− cortex, the number of BrdU-positive cells was comparable to that of the Lpiat1+/+ cortex, suggesting that neurogenesis is not affected in Lpiat1−/− mice at E15.5. However, the proportion of cells in the superficial cortex was significantly reduced in the Lpiat1−/− cortex (Figure 7B). These results suggest that loss of LPIAT1 leads to a significant delay in neuronal migration. We also found that the number of apoptotic cells increased in the Lpiat1−/− cortex as judged by terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assays (Figure 8), suggesting that atrophy of the cerebral cortex in Lpiat1−/− mice is caused by increased apoptosis of neuronal cells.
FIGURE 7:
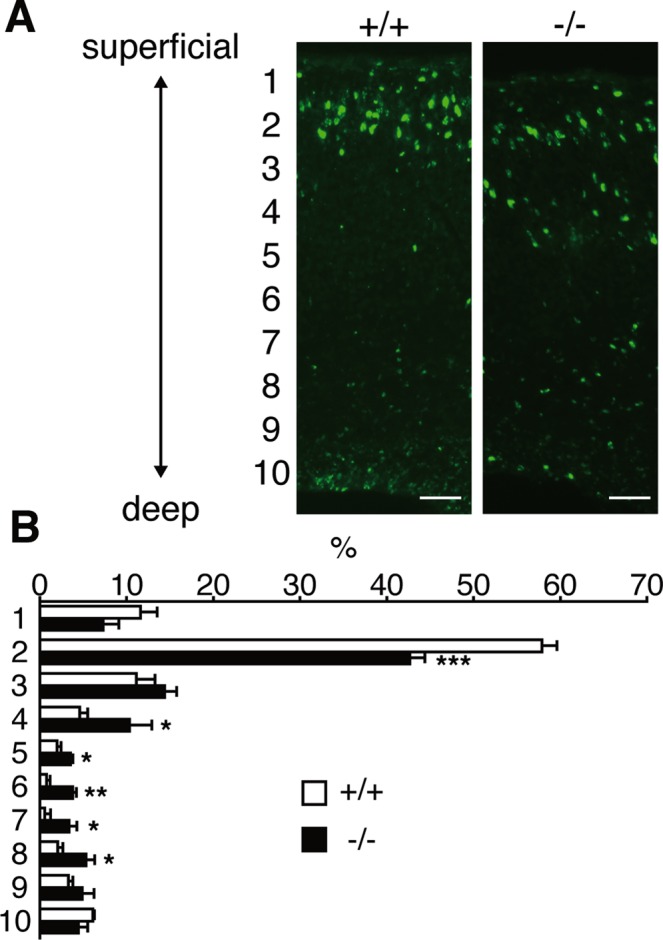
Neuronal migration is delayed in Lpiat1−/− mice. (A) Coronal sections of neocortices from E18.5 Lpiat1+/+ and Lpiat1−/− littermates labeled with BrdU at E15.5. Scale bar, 50 μm. (B) The radial distribution of BrdU-labeled cells. Data are means ± SEM (n = 4). *p < 0.05; **p < 0.01; ***p < 0.001. Unpaired, two-tailed t test was used.
FIGURE 8:
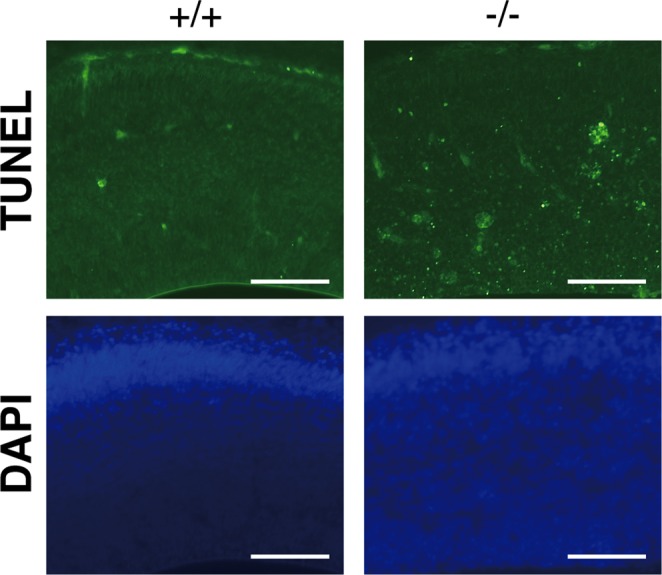
Loss of LPIAT1 causes increased apoptosis in early corticogenesis. Apoptotic cell death in coronal sections was examined by TUNEL staining at E14.5. Scale bar, 100 μm.
Neurite outgrowth is reduced in Lpiat1−/− hippocampal neurons
We then examined the role of LPIAT1 in neurite outgrowth of primary hippocampal neurons. Hippocampal neurons obtained from P0 brains were cultured for 72 h and immunostained with a βIII-tubulin antibody to visualize neuronal morphology. Hippocampal neurons from Lpiat1−/− mice had significantly fewer cells with neurites than did those from Lpiat1+/+ mice (Figure 9, A and B). Moreover, the total neuritic length per cell and the length of the longest neurite in each cell were significantly reduced in Lpiat1−/− cells (Figure 9, C and D). Taken together, these results indicate that LPIAT1 deficiency causes defects in neuronal migration and neurite outgrowth, leading to cortical and hippocampal malformation.
FIGURE 9:

LPIAT1 deficiency causes defects in neurite elongation. (A) Cultured hippocampal neurons from P0 brains were stained with neuronal marker anti–βIII-tubulin antibody (green) and 4′,6-diamidino-2-phenylindole (blue) at DIV3. Scale bar, 50 μm. Percentage of cells with neurites (B), total neurite length (C), and longest neurite length (D) of cultured hippocampal neurons from Lpiat1+/+ and Lpiat1−/− mice. At least 50 neurons were measured for each group. Data are means ± SEM (n = 4). *p < 0.05; **p < 0.01. Unpaired, two-tailed t test was used.
DISCUSSION
In this article, we showed that Lpiat1−/− mice have reduced content of AA in PI and have defects in cortical lamination during brain development. Intensive efforts have been made to understand the physiological roles of AA-derived metabolites such as prostaglandins and leukotrienes. However, little is known about the function of AA when attached to phospholipids other than serving as a source of free AA. Metabolites of AA have been reported to be involved in neuronal functions (Williams and Bliss, 1988; Chen et al., 2002; Shaw et al., 2003; Besana et al., 2005). As far as we know, mice with knockout of any AA-metabolizing enzyme, such as cPLA2α, MAGL, and 5-LOX, or knockout of the receptors for prostaglandins and leukotrienes, such as PGE2 receptors, do not show phenotypes similar to those of Lpiat1−/− mice (Chen et al., 1994; Bonventre et al., 1997; Uozumi et al., 1997; Ushikubi et al., 1998). Moreover, the levels of major AA metabolites were not significantly changed in the Lpiat1−/− brain. Thus the defects in brain development seen in Lpiat1−/− mice are unlikely caused by the reduction or the impairment of the signaling pathways regulated by AA-derived metabolites.
In addition to PI, AA contents of PIP and PIP2 were also reduced in the Lpiat1−/− brain. Previous in vitro studies demonstrated that whereas lysoPI was acylated with [3H]AA by the microsomes from rat liver and brain in the presence of ATP, CoA, and Mg2+, neither lysoPI4P nor lysoPI(4,5)P2 was acylated with [3H]AA under the same assay conditions (Palmer, 1986). In the present study, we confirmed this observation and also showed that overexpression of LPIAT1 in cultured cells did not increase acyltransferase activity toward lysoPI4P or lysoPI(4,5)P2. These results indicate that LPIAT1 does not use lysoPI4P or lysoPI(4,5)P2 as an acyl acceptor. Nevertheless, the AA contents of PI phosphates and PI showed similar decreases in the Lpiat1−/− brain, suggesting that the reduction in the AA content in PI phosphates may result from the reduction of AA content in PI, a precursor of PI phosphates.
PI phosphates are synthesized by PI kinases and phosphatases and play crucial roles in the regulation of a wide variety of cellular processes via specific interactions of PIP-binding proteins (Di Paolo and De Camilli, 2006; Sasaki et al., 2009). Among PI phosphates, PI 3-phosphate (PI3P) regulates a variety of vesicular trafficking pathways, including endocytosis, endosome-to-Golgi retrograde transport, autophagy, and the target of rapamycin (TOR) signaling pathway (Backer, 2008). PI3P is synthesized from PI by a class III PI 3-kinase, Vps34 (Backer, 2008). Recently we showed that knockdown of vps-34, a C. elegans orthologue of Vps34, causes severe growth defects in mboa-7 mutants (Lee et al., 2012). Moreover, PI3P signaling pathways such as autophagy and endosome morphology were impaired in mboa-7 mutants. Of interest, Zhou (2010) recently showed that neuron-specific Vps34 conditional knockout mice exhibit defects in cortical lamination, which is very similar to that of Lpiat1−/− mice. On the basis of these observations, we hypothesize that reduction of AA content in PI3P impaired PI3P function(s), which leads to cortical lamination defects in Lpiat1−/− mice. The observation that the fatty acid compositions of PIP and PIP2 were changed similarly to that of PI in Lpiat1−/− mice suggests that the enzymes involved in the synthesis of PIP and PIP2 do not prefer AA-containing PI as a substrate. Change in the fatty acid composition in PI phosphates may affect the interaction with PI phosphate–binding proteins and/or the intramembrane localization of PI phosphates in subcellular organelles. We are testing this hypothesis.
∆6-Fatty acid desaturase (FADS2) converts linoleic acid (18:2n-6) to γ-linolenic (18:3n-6), and ∆5-fatty acid desaturase (FADS1) converts dihomo-γ-linolenic acid (20:3n-6) to AA. Fads2−/− mice show reduced content of PUFAs, including AA in tissues (Stoffel et al., 2008; Stroud et al., 2009). Fads2−/− mice exhibit a variety of symptoms, including sterility, ulceration, and dermatitis. PUFA supplementation restores the symptoms in Fads2−/− mice and in patients with ∆6-desaturase deficiency (Stoffel et al., 2008; Stroud et al., 2009; Roqueta-Rivera et al., 2010; Williard et al., 2001). Fads1−/− mice began to die gradually starting at 5–6 wk of age, with no survivors past 12 wk of age, although no overt physical differences between Fads1−/− and wild-type mice were observed (Fan et al., 2012). Thus Fads1−/− and Fads2−/− mice did not show any phenotypes similar to those of Lpiat1−/− mice. Although Fads1−/− and Fads2−/− mice are ideal for studying the function of AA and other PUFAs in vivo, PUFAs are supplied from the heterozygous mother through placental transfer in the homozygous embryos. Moreover, the content of PUFAs in the brain tends to remain stable even under PUFA-depleted conditions (García-Calatayud et al., 2002; Stroud et al., 2009). Lpiat1−/− embryos have a defect in the synthesis of AA-containing PI using AA derived from their heterozygous mother. Thus it is plausible that Fads2−/− mice and Lpiat1−/− mice show distinct brain phenotypes in the embryonic stage.
The fact that a considerable amount of AA remained in PI of Lpiat1−/− mice suggests that there are other pathways to enrich AA in PI. Lysophosphatidic acid acyltransferase (LPAAT) may catalyze the incorporation of PUFAs into the sn-2 position of lysophosphatidic acid (lysoPA) during de novo phospholipid synthesis. Mammalian LPAATs show relatively broad substrate specificity for acyl-CoAs and incorporate AA into lysoPA (Stamps et al., 1997; Eberhardt et al., 1997). Cytidine 5′-diphosphate-diacylglycerol synthase (CDS) catalyzes the biosynthesis of CDP-diacylglycerol, a direct precursor of PI, from phosphatidic acid (PA). CDS1, an isoform of CDS that is highly expressed in the brain, prefers AA-containing PA as a substrate (Saito et al., 1997). Therefore it is possible that PI-containing AA can be synthesized once AA is incorporated into PA via the de novo pathway.
It has been recognized for decades that AA-containing PI is a major molecular species in PI in mammals. By knocking out LPIAT1—the enzyme responsible for incorporation of AA into PI—we showed for the first time that AA-containing PI is essential for brain development in mammals.
MATERIALS AND METHODS
Materials
[1-14C]-labeled fatty acids and fatty acyl-CoAs were purchased from American Radiolabeled Chemicals (St. Louis, MO). Arachidonoyl-CoA was obtained from Sigma-Aldrich (St. Louis, MO). LysoPI from bovine liver, lysophosphatidylserine from porcine brain, lysoPE from porcine liver, lysoPC from egg yolk, sn-1-oleoyl-lysoPA, PI4P from porcine brain, and PI(4,5)P2 from porcine brain were purchased from Avanti Polar Lipids (Alabaster, AL). Dipalmitoyl PI was purchased from Serdary Research Laboratories (London, Canada). PLA2 from honey bee venom was purchased from Sigma-Aldrich. AA-d8, 15-HETE-d8, LTB4-d4, and PGE2-d4 were purchased from Cayman Chemical (Ann Arbor, MI).
Animals
Specific pathogen-free female C57BL/6 mice were obtained from CLEA Japan (Tokyo, Japan). We used Lpiat1-deficient mice backcrossed eight times to C57BL/6 background. Mice were maintained in our animal facility and treated in accordance with the guidelines of the Institutional Animal Care Committee (Graduate School of Pharmaceutical Sciences, University of Tokyo, Tokyo, Japan).
Generation of LPIAT1-deficient mice
E14K murine embryonic stem (ES) cells heterozygous for a deletion mutation of the Lpiat1 gene were generated. We replaced exons 2–4 of the murine Lpiat1 gene with a PGK-Neo cassette because the putative translation initiation codons were identified in exons 2–4 but not in exon 1. The targeting vector contained a 5.5-kb genomic murine (129/Ola) Lpiat1 fragment plus the PGK-Neo cassette inserted in antisense orientation to Lpiat1 transcription. The linearized construct was electroporated into 1 × 107 E14K ES cells. ES cell colonies resistant to G418 (0.3 mg/ml) were screened for homologous recombination by PCR. Four different targeted ES cells derived from two independent electroporation experiments were injected into C57BL/6 blastocysts. Chimeric male mice were crossed with C57BL/6 females to achieve germline transmission. After heterozygous matings, Lpiat1−/− mice were distinguished from Lpiat1+/− and Lpiat1+/+ mice by PCR. An oligo primer specific for the Lpiat1+ allele (P1, 5′-CACGCCCTTCACCAATGCTG-3′), a primer common to the Lpiat1+ and Lpiat1− alleles (P2, 5′-TGGAGGACGGTTTGCTACAGACTC-3′), and a primer specific for the Lpiat1− allele (P3, 5′-AGACTGCCTTGGGAAAAGCG-3′), were combined in the same PCR.
Generation of anti-mouse LPIAT1 antibody
A recombinant mouse LPIAT1 (amino acids 274–334 of mouse LPIAT1, GenBank accession no. NM_029934.3) that was expressed and purified by an Escherichia coli pCold TF expression system (TaKaRa, Otsu, Japan) was injected into the hind foot pads of WKY/Izm rat strain by using Freund's complete adjuvant. The enlarged medial iliac lymph nodes were used for cell fusion with mouse myeloma cells, PAI. In the present study, the established monoclonal antibody FT10 was used for immunoblotting and immunohistochemistry at dilutions of 1:1000 and 1:100, respectively.
Western blot analysis
Tissues from 2-wk-old mice were homogenized in 9× volume (wt/vol) of SET buffer (10 mM Tris-HCl, pH 7.4, 1 mM EDTA, 250 mM sucrose) with protease inhibitors (0.5 mM phenylmethylsulfonyl fluoride, 2 μg/ml pepstatin, 2 μg/ml leupeptin, 2 μg/ml aprotinin). After centrifugation at 1000 × g at 4°C, the supernatants were used as the total protein extracts. The protein concentrations of samples were determined by the bicinchoninic acid (BCA) assay (Pierce, Rockford, IL). Each total protein extract was separated by SDS–PAGE and transferred to either polyvinylidene difluoride (PVDF) or nitrocellulose membranes. The membranes were blocked with skim milk (Wako Pure Chemical Industries, Osaka, Japan) in TTBS buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.05% [wt/vol] Tween 20) and incubated with the specific monoclonal rat anti-mouse LPIAT1 antibody FT10 (see prior description). After incubation with horseradish peroxidase–conjugated goat anti–rat immunoglobulin G antibody (American Qualex, San Clemente, CA), LPIAT1 was detected by enhanced chemiluminescence (ECL Western blotting detection system; GE Healthcare, Piscataway, NJ). Anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 6C5) was purchased from Calbiochem (La Jolla, CA).
Acyltransferase assay
Murine tissues were pulverized under liquid nitrogen and homogenized in quadruple volumes (wt/vol) of SET buffer with protease inhibitors (0.5 mM phenylmethylsulfonyl fluoride, 2 μg/ml pepstatin, 2 μg/ml leupeptin, 2 μg/ml aprotinin). After centrifugation at 2330 × g for 20 min at 4°C, the resulting supernatant was further centrifuged at 105,000 × g for 60 min. The pellet was suspended in homogenizing buffer (without EDTA, phenylmethylsulfonyl fluoride, pepstatin, leupeptin, and aprotinin) and used for the enzyme assay. Acyl-CoA:1-acyl lysophospholipid acyltransferase assay was carried out as described before (Imae et al., 2010), except that the incubation temperature was 37°C and that 5 μg (Figure 1H) or 20 μg (Figure 1I) of microsomal protein was used. The protein concentrations of samples were determined by the BCA assay (Pierce).
Acyltransferase assay toward lysoPI phosphates
sn-1-acyl lysoPI4P and sn-1-acyl lysoPI(4,5)P2 were prepared as described previously (Palmer, 1986). P0 brains and HEK 293A cells were homogenized in phosphate buffer (pH 7.0) containing 0.15 M KCl, 0.25 M sucrose, 1 mM EDTA, 1 mM dithiothreitol, and 5 μg/ml pepstatin, leupeptin, and aprotinin (homogenizing buffer). After centrifugations at 1000 × g for 5 min and 10,000 × g for 30 min at 4°C, the supernatant was collected and centrifuged at 105,000 × g for 60 min at 4°C. The resulting pellet was resuspended in homogenizing buffer (without EDTA, dithiothreitol, and protease inhibitors) and used for the enzyme assay to be described. [14C]arachidonoyl-CoA (53 mCi/mmol) was diluted with the unlabeled arachidonoyl-CoA to 10 mCi/mmol before use. Reaction mixtures contained 10 μM of AA-CoA (10 mCi/mmol) and 10 μM of lysoPI(4,5)P2 and 5 μg (for brains) or 1 μg (for cells) of microsomal protein in a total volume of 0.2-ml assay buffer (0.15 M KCl, 0.25 M sucrose, 50 mM potassium phosphate buffer, pH 6.8). After incubation at 37°C for 5 min, reactions were stopped by the addition of 1 ml of methanol. The lipids were extracted as described previously (Serunian et al., 1991) and separated by one-dimensional TLC on silica gel 60 plates (Merck, Darmstadt, Germany) in chloro-form:methanol:concentrated NH4OH:water (45:45:4:11, vol/vol) (Tysnes et al., 1985). Acyltransferase activity toward lysoPI was also performed in the same assay condition to compare the acyl acceptor preference of LPIAT1.
Histological analysis
Brains were embedded in paraffin wax, and serial sagittal sections were mounted on slides and stained with hematoxylin and eosin. For immunohistochemistry, brain sections were blocked for 30 min with 2% bovine serum albumin (BSA; Sigma-Aldrich) diluted in phosphate-buffered saline (PBS) containing 0.1% Tween 20 at room temperature and then incubated in primary antibody diluted in 2% BSA-PBS for 2 h. Primary antibodies and their dilutions used in this study were as follows: Reelin (CR50; 1:1000; Ogawa et al., 1995), Tbr1 (AB9616; 1:500; Millipore, Billerica, MA), Brn1 (SC-6028; 1:100; Santa Cruz Biotechnology, Santa Cruz, CA), MAP2 (M1406; 1:1000; Sigma-Aldrich), βIII-tubulin (GT431195; 1:500; Genzyme-Techne, Minneapolis, MN). Sections were incubated with fluorescent secondary antibodies (Alexa series; Molecular Probes, Eugene, OR) diluted at 1:400 in 2% BSA-PBS for 1 h at room temperature. Slides were washed for several hours in PBS and coverslips applied with Dako fluorescent mounting medium (Dako, Glostrup, Denmark). TUNEL analysis was performed using the Apop Tag Plus fluorescein In Situ Apoptosis Detection Kit (Millipore) according to the manufacturer's instructions.
BrdU labeling and analysis
For BrdU experiments, pregnant mice were injected with BrdU (50 mg/g, intraperitoneally) at E15.5, and perinatal embryos were removed by cesarean section at E18.5. Embryo heads were fixed in 4% paraformaldehyde, and the brains were dissected out and soaked overnight in 20 and 30% sucrose and equilibrated and embedded in OCT on dry ice. Sections were stored frozen until use. After permeabilization with HCl, BrdU was detected by rat anti-BrdU antibodies (AB6326; 1:150; Abcam, Cambridge, MA). To determine the distribution of cells born at E15.5 in Lpiat1+/+ and Lpiat1−/− cortex, sections were divided into the horizontal bins from superficial to deep, and the percentage of cells in each bin was calculated.
Primary culture
The primary culture of neurons was prepared from hippocampi or cerebral cortices of E18.5 or P0 mice as described previously (Bannai et al., 2004). The dissociated cells were plated on poly-l-lysine (Sigma-Aldrich)–coated dishes or coverslips and cultured in Neurobasal Medium (for expression analysis of LPIAT1, E18.5) or Neurobasal-A Medium (for morphometric analysis, P0; Invitrogen, Carlsbad, CA) supplemented with 2.5 mM l-glutamine (Nacalai Tesque, Kyoto, Japan), 2.5% (vol/vol) B-27 (Invitrogen), and antibiotics (250 U/ml penicillin and 250 mg/ml streptomycin). As for morphometric analysis, neurons were cultured at a density of 2.5 × 104 cells/well in 24-well flat-bottom plates and fixed at 3 d in vitro (DIV) and then immunostained with the anti-TUJ1 antibody against the neuronal-specific βIII-tubulin (MAB1195; R&D Systems, Minneapolis, MN) at dilution of 1:5000. Cell images were captured by a fluorescence microscope (FSX100; Olympus, Tokyo, Japan), and then the length of neurites and the neurite number were determined.
Lipid analysis
Lipids of each tissue were extracted by the method of Bligh and Dyer (1959). The fatty acid composition was determined by gas chromatography as described previously (Lee et al., 2008), except that the solvent used in the one-dimensional TLC was chloroform:methyl acetate:1-propanol:methanol:0.25% KCl (25:25:25:10:9, vol/vol; Imae et al., 2010). LC/ESI-MS analysis was performed as described previously (Ogiso and Taguchi, 2008; Imae et al., 2010). Lipid phosphorus was determined by Bartlett's method (Bartlett, 1959).
Cell culture and transfection
Human embryonic kidney (HEK) 293A cells were maintained in DMEM supplemented with 10% fetal calf serum, penicillin (100 U/ml), streptomycin (100 mg/ml), and l-glutamine (2 mM). Transfection of the plasmid DNA into cells was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol.
Plasmid construction
Full-length mouse Lpiat1 was amplified by PCR from mouse cDNA with primers Lpiat1 forward (5′-ACGGTTGAATTCATGACACCCGAAGAATGGAC-3′) and reverse (5′-CAGCAGCTCGAGCTCTTCCCGGAGCTTTTCCT-3′) and was cloned into pCAGGS-myc vector (C-terminal Myc tag) at the EcoRI and XhoI sites.
Metabolite extraction and analysis
Arachidonic acid metabolites were extracted from P0 mouse brains by solid-phase extraction using Sep-Pak C18 cartridges (Waters, Milford, MA) with deuterium-labeled internal standards (AA-d8, 15-HETE-d8, LTB4-d4, and PGE2-d4). LC-MS/MS–based lipidomic analyses were performed using a HPLC system (Waters UPLC) with a linear ion trap quadrupole mass spectrometer (QTRAP 5500; AB Sciex, Foster City, CA) equipped with an Acquity UPLC BEH C18 column (1.0 mm × 150 mm × 1.7 μm; Waters). Samples were eluted with mobile phase composed of water/acetate (100:0.1, vol/vol) and acetonitrile/methanol (4:1, vol/vol) (73:27) for 5 min and ramped to 30:70 after 15 min, to 20:80 after 25 min and held for 8 min, ramped to 0:100 after 35 min, and held for 10 min with flow rates of 50 (0–30 min), 80 (30–33 min), and 100 μl/min (33–45 min). MS/MS analyses were conducted in negative-ion mode, and fatty acid metabolites were identified and quantified by multiple reaction monitoring. Quantitation was performed using calibration curves constructed for each compound, and recoveries were monitored using added deuterated internal standards.
Acknowledgments
We thank Tomohiko Taguchi for valuable discussion and Ryo Iwamoto for technical assistance in LC-MS/MS analysis. This work was supported by the Core Research for Evolutional Science and Technology, Japan Science and Technology Agency (to H.A.), the Program for Promotion of Basic and Applied Researches for Innovations in Bio-oriented Industry (to H.A.), grants-in-aid from the Japanese Ministry of Education, Culture, Sports, Science, and Technology (20370045 to H.A.), the Japanese Ministry of Health, Labor, and Welfare (to H. A.), and Research Fellowships of the Japan Society for the Promotion of Science for Young Scientists (to H.L. and S.M.).
Abbreviations used:
- AA
arachidonic acid
- AA-CoA
arachidonoyl-CoA
- FADS1
Δ5-fatty acid desaturase
- FADS2
Δ6-fatty acid desaturase
- LPIAT1
lysophosphatidylinositol acyltransferase 1
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PI
phosphatidylinositol
- PIP
PI monophosphate
- PI3P
PI 3-phosphate
- PIP2
PI bisphosphate
- PUFA
polyunsaturated fatty acid
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-09-0673) on October 24, 2012.
*Present address: Division of Cellular and Gene Therapy Products, National Institute of Health Sciences, Tokyo 158-8501, Japan.
Requests for the LPIAT1-knockout mice should be addressed to H.A. (harai@mol.f.u-tokyo.ac.jp) or T.S. (tsasaki@med.akita-u.ac.jp).
REFERENCES
- Abad-Jorge A. The role of DHA and ARA in infant nutrition and neurodevelopmental outcomes. Today's Dietician. 2008;10:66. [Google Scholar]
- Alcántara S, Ruiz M, D'Arcangelo G, Ezan F, de Lecea L, Curran T, Sotelo C, Soriano E. Regional and cellular patterns of reelin mRNA expression in the forebrain of the developing and adult mouse. J Neurosci. 1998;18:7779–7799. doi: 10.1523/JNEUROSCI.18-19-07779.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backer JM. The regulation and function of class III PI3Ks: novel roles for Vps34. Biochem J. 2008;410:1–17. doi: 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]
- Baker RR, Thompson W. Selective acylation of 1-acylglycerophosphorylinositol by rat brain microsomes. Comparison with 1-acylglycerophosphorylcholine. J Biol Chem. 1973;248:7060–7065. [PubMed] [Google Scholar]
- Bannai H, Inoue T, Nakayama T, Hattori M, Mikoshiba K. Kinesin dependent, rapid, bi-directional transport of ER sub-compartment in dendrites of hippocampal neurons. J Cell Sci. 2004;117:163–175. doi: 10.1242/jcs.00854. [DOI] [PubMed] [Google Scholar]
- Bartlett G. Phosphorus assay in column chromatography. J Biol Chem. 1959;234:466–468. [PubMed] [Google Scholar]
- Besana A, Robinson R, Feinmark S. Lipids and two-pore domain K+ channels in excitable cells. Prostaglandins Other Lipid Mediat. 2005;77:103–110. doi: 10.1016/j.prostaglandins.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Bligh E, Dyer W. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Bonventre JV, Huang Z, Taheri MR, O'Leary E, Li E, Moskowitz MA, Sapirstein A. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature. 1997;390:622–625. doi: 10.1038/37635. [DOI] [PubMed] [Google Scholar]
- Chen C, Magee J, Bazan N. Cyclooxygenase-2 regulates prostaglandin E2 signaling in hippocampal long-term synaptic plasticity. J Neurophysiol. 2002;87:2851–2857. doi: 10.1152/jn.2002.87.6.2851. [DOI] [PubMed] [Google Scholar]
- Chen XS, Sheller JR, Johnson EN, Funk CD. Role of leukotrienes revealed by targeted disruption of the 5-lipoxygenase gene. Nature. 1994;372:179–182. doi: 10.1038/372179a0. [DOI] [PubMed] [Google Scholar]
- Chyb S, Raghu P, Hardie RC. Polyunsaturated fatty acids activate the Drosophila light-sensitive channels TRP and TRPL. Nature. 1999;397:255–259. doi: 10.1038/16703. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G, Nakajima K, Miyata T, Ogawa M, Mikoshiba K, Curran T. Reelin is a secreted glycoprotein recognized by the CR-50 monoclonal antibody. J Neurosci. 1997;17:23–31. doi: 10.1523/JNEUROSCI.17-01-00023.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis-Bruno K, Tassinari MS. Essential fatty acid supplementation of DHA and ARA and effects on neurodevelopment across animal species: a review of the literature. Birth Defects Res B Dev Reprod Toxicol. 2011;92:240–50. doi: 10.1002/bdrb.20311. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- Dijck-Brouwer D, Hadders-Algra M, Bouwstra H, Decsi T, Boehm G, Martini I, Boersma E, Muskiet F. Lower fetal status of docosahexaenoic acid, arachidonic acid and essential fatty acids is associated with less favorable neonatal neurological condition. Prostaglandins Leukot Essent Fatty Acids. 2005;72:21–28. doi: 10.1016/j.plefa.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Eberhardt C, Gray PW, Tjoelker LW. Human lysophosphatidic acid acyltransferase. cDNA cloning, expression, and localization to chromosome 9q34.3. J Biol Chem. 1997;272:20299–20305. doi: 10.1074/jbc.272.32.20299. [DOI] [PubMed] [Google Scholar]
- Fan YY, Monk JM, Hou TY, Callway E, Vincent L, Weeks B, Yang P, Chapkin RS. Characterization of an arachidonic acid-deficient (Fads1 knockout) mouse model. J Lipid Res. 2012;53:1287–1295. doi: 10.1194/jlr.M024216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Calatayud S, Ruiz JI, García-Fuentes M, Dierssen M, Flórez J, Crespo PS. Long-chain polyunsaturated fatty acids in rat maternal milk, offspring brain and peripheral tissues in essential fatty acid deficiency. Clin Chem Lab Med. 2002;40:278–284. doi: 10.1515/CCLM.2002.044. [DOI] [PubMed] [Google Scholar]
- Gijón MA, Riekhof WR, Zarini S, Murphy RC, Voelker DR. Lysophospholipid acyltransferases and arachidonate recycling in human neutrophils. J Biol Chem. 2008;283:30235–30245. doi: 10.1074/jbc.M806194200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson JG, Walsh CA. Neuronal migration disorders: from genetic diseases to developmental mechanisms. Trends Neurosci. 2000;23:352–359. doi: 10.1016/s0166-2236(00)01607-6. [DOI] [PubMed] [Google Scholar]
- Goldberg EM, Zidovetzki R. Effects of dipalmitoylglycerol and fatty acids on membrane structure and protein kinase C activity. Biophys J. 1997;73:2603–2614. doi: 10.1016/S0006-3495(97)78290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevner R, Shi L, Justice N, Hsueh Y, Sheng M, Smiga S, Bulfone A, Goffinet A, Campagnoni A, Rubenstein J. Tbr1 regulates differentiation of the preplate and layer 6. Neuron. 2001;29:353–366. doi: 10.1016/s0896-6273(01)00211-2. [DOI] [PubMed] [Google Scholar]
- Holub B, Kuksis A. Differential distribution of orthophosphate-32 P and glycerol-14 C among molecular species of phosphatidylinositols of rat liver in vivo. J Lipid Res. 1971;12:699–705. [PubMed] [Google Scholar]
- Imae R, Inoue T, Kimura M, Kanamori T, Tomioka N, Kage-Nakadai E, Mitani S, Arai H. Intracellular phospholipase A1 and acyltransferase, which are involved in Caenorhabditis elegans stem cell divisions, determine the sn-1 fatty acyl chain of phosphatidylinositol. Mol Biol Cell. 2010;21:3114–3124. doi: 10.1091/mbc.E10-03-0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Murase S, Okuyama H. Acyl coenzyme a:phospholipid acyltransferases in porcine platelets discriminate between omega-3 and omega-6 unsaturated fatty acids. Arch Biochem Biophys. 1984;231:29–37. doi: 10.1016/0003-9861(84)90359-x. [DOI] [PubMed] [Google Scholar]
- Jungalwala F, Evans J, McCluer R. Compositional and molecular species analysis of phospholipids by high performance liquid chromatography coupled with chemical ionization mass spectrometry. J Lipid Res. 1984;25:738–749. [PubMed] [Google Scholar]
- Kotani S, Nakazawa H, Tokimasa T, Akimoto K, Kawashima H, Toyoda-Ono Y, Kiso Y, Okaichi H, Sakakibara M. Synaptic plasticity preserved with arachidonic acid diet in aged rats. Neurosci Res. 2003;46:453–461. doi: 10.1016/s0168-0102(03)00123-8. [DOI] [PubMed] [Google Scholar]
- Lee H, Inoue T, Imae R, Kono N, Shirae S, Matsuda S, Gengyo-Ando K, Mitani S, Arai H. Caenorhabditis elegans mboa-7, a member of the MBOAT family, is required for selective incorporation of polyunsaturated fatty acids into phosphatidylinositol. Mol Biol Cell. 2008;19:1174–1184. doi: 10.1091/mbc.E07-09-0893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HC, Kubo T, Kono N, Kage-Nakadai E, Gengyo-Ando K, Mitani S, Inoue T, Arai H. Depletion of mboa-7, an enzyme that incorporates polyunsaturated fatty acids into phosphatidylinositol (PI), impairs PI 3-phosphate signaling in Caenorhabditis elegans. Genes Cells. 2012;17:748–757. doi: 10.1111/j.1365-2443.2012.01624.x. [DOI] [PubMed] [Google Scholar]
- Luskin M, Shatz C. Studies of the earliest generated cells of the cat's visual cortex: cogeneration of subplate and marginal zones. J Neurosci. 1985;5:1062–1075. doi: 10.1523/JNEUROSCI.05-04-01062.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthra M, Sheltawy A. The metabolic turnover of molecular species of phosphatidylinositol and its precursor phosphatidic acid in guinea-pig cerebral hemispheres. J Neurochem. 1976;27:1501–1511. doi: 10.1111/j.1471-4159.1976.tb02636.x. [DOI] [PubMed] [Google Scholar]
- Maekawa M, et al. Arachidonic acid drives postnatal neurogenesis and elicits a beneficial effect on prepulse inhibition, a biological trait of psychiatric illnesses. PLoS One. 2009;4:9. doi: 10.1371/journal.pone.0005085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEvilly R, de Diaz M, Schonemann M, Hooshmand F, Rosenfeld M. Transcriptional regulation of cortical neuron migration by POU domain factors. Science. 2002;295:1528–1532. doi: 10.1126/science.1067132. [DOI] [PubMed] [Google Scholar]
- Nakagawa Y, Sugiura T, Waku K. The molecular species composition of diacyl-, alkylacyl- and alkenylacylglycerophospholipids in rabbit alveolar macrophages. High amounts of 1-O-hexadecyl-2-arachidonyl molecular species in alkylacylglycerophosphocholine. Biochim Biophys Acta. 1985;833:323–329. [PubMed] [Google Scholar]
- Ogawa M, Miyata T, Nakajima K, Yagyu K, Seike M, Ikenaka K, Yamamoto H, Mikoshiba K. The reeler gene-associated antigen on Cajal-Retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron. 1995;14:899–912. doi: 10.1016/0896-6273(95)90329-1. [DOI] [PubMed] [Google Scholar]
- Ogiso H, Taguchi R. Reversed-phase LC/MS method for polyphosphoinositide analyses: changes in molecular species levels during epidermal growth factor activation in A431 cells. Anal Chem. 2008;80:9226–9232. doi: 10.1021/ac801451p. [DOI] [PubMed] [Google Scholar]
- Okaichi Y, Ishikura Y, Akimoto K, Kawashima H, Toyoda-Ono Y, Kiso Y, Okaichi H. Arachidonic acid improves aged rats’ spatial cognition. Physiol Behav. 2005;84:617–623. doi: 10.1016/j.physbeh.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Palmer FB. Metabolism of lysopolyphosphoinositides by rat brain and liver microsomes. Biochem Cell Biol. 1986;64:117–125. doi: 10.1139/o86-019. [DOI] [PubMed] [Google Scholar]
- Patton G, Fasulo J, Robins S. Separation of phospholipids and individual molecular species of phospholipids by high-performance liquid chromatography. J Lipid Res. 1982;23:190–196. [PubMed] [Google Scholar]
- Rakic P. Specification of cerebral cortical areas. Science. 1988;241:170–176. doi: 10.1126/science.3291116. [DOI] [PubMed] [Google Scholar]
- Roqueta-Rivera M, Stroud CK, Haschek WM, Akare SJ, Segre M, Brush RS, Agbaga MP, Anderson RE, Hess RA, Nakamura MT. Docosahexaenoic acid supplementation fully restores fertility and spermatogenesis in male delta-6 desaturase-null mice. J Lipid Res. 2010;51:360–367. doi: 10.1194/jlr.M001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito S, Goto K, Tonosaki A, Kondo H. Gene cloning and characterization of CDP-diacylglycerol synthase from rat brain. J Biol Chem. 1997;272:9503–9509. doi: 10.1074/jbc.272.14.9503. [DOI] [PubMed] [Google Scholar]
- Sanjanwala M, Sun GY, MacQuarrie RA. Purification and kinetic properties of lysophosphatidylinositol acyltransferase from bovine heart muscle microsomes and comparison with lysophosphatidylcholine acyltransferase. Arch Biochem Biophys. 1989;271:407–413. doi: 10.1016/0003-9861(89)90290-7. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Takasuga S, Sasaki J, Kofuji S, Eguchi S, Yamazaki M, Suzuki A. Mammalian phosphoinositide kinases and phosphatases. Prog Lipid Res. 2009;48:307–343. doi: 10.1016/j.plipres.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Serunian LA, Auger KR, Cantley LC. Identification and quantification of polyphosphoinositides produced in response to platelet-derived growth factor stimulation. Methods Enzymol. 1991;198:78–87. doi: 10.1016/0076-6879(91)98010-4. [DOI] [PubMed] [Google Scholar]
- Shaw K, Commins S, O'Mara S. Deficits in spatial learning and synaptic plasticity induced by the rapid and competitive broad-spectrum cyclooxygenase inhibitor ibuprofen are reversed by increasing endogenous brain-derived neurotrophic factor. Eur J Neurosci. 2003;17:2438–2446. doi: 10.1046/j.1460-9568.2003.02643.x. [DOI] [PubMed] [Google Scholar]
- Shindou H, Shimizu T. Acyl-CoA:lysophospholipid acyltransferases. J Biol Chem. 2009;284:1–5. doi: 10.1074/jbc.R800046200. [DOI] [PubMed] [Google Scholar]
- Stamps AC, Elmore MA, Hill ME, Kelly K, Makda AA, Finnen MJ. A human cDNA sequence with homology to non-mammalian lysophosphatidic acid acyltransferases. Biochem J. 1997;326:455–461. doi: 10.1042/bj3260455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhauer J, Gijón MA, Riekhof WR, Voelker DR, Murphy RC, Treisman JE. Drosophila lysophospholipid acyltransferases are specifically required for germ cell development. Mol Biol Cell. 2009;20:5224–5235. doi: 10.1091/mbc.E09-05-0382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoffel W, et al. Delta6-desaturase (FADS2) deficiency unveils the role of omega3- and omega6-polyunsaturated fatty acids. EMBO J. 2008;27:2281–2292. doi: 10.1038/emboj.2008.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud CK, et al. Disruption of FADS2 gene in mice impairs male reproduction and causes dermal and intestinal ulceration. J Lipid Res. 2009;50:1870–1880. doi: 10.1194/jlr.M900039-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Iwawaki D, Sakamoto M, Takai Y, Morishige J, Murakami K, Satouchi K. Mechanisms of accumulation of arachidonate in phosphatidylinositol in yellowtail. A comparative study of acylation systems of phospholipids in rat and the fish species Seriola quinqueradiata. Eur J Biochem. 2003;270:1466–1473. doi: 10.1046/j.1432-1033.2003.03512.x. [DOI] [PubMed] [Google Scholar]
- Tissir F, Goffinet A. Reelin and brain development. Nat Rev Neurosci. 2003;4:496–505. doi: 10.1038/nrn1113. [DOI] [PubMed] [Google Scholar]
- Tysnes OB, Aarbakke GM, Verhoeven AJ, Holmsen H. Thin-layer chromatography of polyphosphoinositides from platelet extracts: interference by an unknown phospholipid. Thromb Res. 1985;40:329–338. doi: 10.1016/0049-3848(85)90268-3. [DOI] [PubMed] [Google Scholar]
- Uozumi N, et al. Role of cytosolic phospholipase A2 in allergic response and parturition. Nature. 1997;390:618–622. doi: 10.1038/37622. [DOI] [PubMed] [Google Scholar]
- Ushikubi F, et al. Impaired febrile response in mice lacking the prostaglandin E receptor subtype EP3. Nature. 1998;395:281–284. doi: 10.1038/26233. [DOI] [PubMed] [Google Scholar]
- Williams J, Bliss T. Induction but not maintenance of calcium-induced long-term potentiation in dentate gyrus and area CA1 of the hippocampal slice is blocked by nordihydroguaiaretic acid. Neurosci Lett. 1988;88:81–85. doi: 10.1016/0304-3940(88)90319-9. [DOI] [PubMed] [Google Scholar]
- Williard DE, Nwankwo JO, Kaduce TL, Harmon SD, Irons M, Moser HW, Raymond GV, Spector AA. Identification of a fatty acid delta6-desaturase deficiency in human skin fibroblasts. J Lipid Res. 2001;42:501–508. [PubMed] [Google Scholar]
- Xiao YF, Ke Q, Wang SY, Auktor K, Yang Y, Wang GK, Morgan JP, Leaf A. Single point mutations affect fatty acid block of human myocardial sodium channel alpha subunit Na+ channels. Proc Natl Acad Sci USA. 2001;98:3606–3611. doi: 10.1073/pnas.061003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro K, Kameyama Y, Mizuno-Kamiya M, Shin SO, Fujita A. Substrate specificity of microsomal 1-acyl-sn-glycero-3-phosphoinositol acyltransferase in rat submandibular gland for polyunsaturated long-chain acyl-CoAs. Biochim Biophys Acta. 1995;1258:288–296. doi: 10.1016/0005-2760(95)00136-z. [DOI] [PubMed] [Google Scholar]
- Zhao J, Del Bigio M, Weiler H. Maternal arachidonic acid supplementation improves neurodevelopment of offspring from healthy and diabetic rats. Prostaglandins Leukot Essent Fatty Acids. 2009;81:349–356. doi: 10.1016/j.plefa.2009.09.005. [DOI] [PubMed] [Google Scholar]
- Zhou X. Function of Phosphatidylinositol 3-Kinase Class III in the Nervous System. Durham, NC: Duke University; 2010. PhD Thesis. [Google Scholar]