

Semliki Forest virus nsP3 sequesters G3BP to inhibit stress granule formation on viral mRNAs. Furthermore, the efficient translation of viral mRNAs containing a translation enhancer element assists disruption of SGs in infected cells. This work thus describes a novel mechanism for SG disruption.
Abstract
Dynamic, mRNA-containing stress granules (SGs) form in the cytoplasm of cells under environmental stresses, including viral infection. Many viruses appear to employ mechanisms to disrupt the formation of SGs on their mRNAs, suggesting that they represent a cellular defense against infection. Here, we report that early in Semliki Forest virus infection, the C-terminal domain of the viral nonstructural protein 3 (nsP3) forms a complex with Ras-GAP SH3-domain–binding protein (G3BP) and sequesters it into viral RNA replication complexes in a manner that inhibits the formation of SGs on viral mRNAs. A viral mutant carrying a C-terminal truncation of nsP3 induces more persistent SGs and is attenuated for propagation in cell culture. Of importance, we also show that the efficient translation of viral mRNAs containing a translation enhancer sequence also contributes to the disassembly of SGs in infected cells. Furthermore, we show that the nsP3/G3BP interaction also blocks SGs induced by other stresses than virus infection. This is one of few described viral mechanisms for SG disruption and underlines the role of SGs in antiviral defense.
INTRODUCTION
Stress granules (SGs) are dynamic sites of mRNA storage, which are formed in the cytoplasm of cells under many types of environmental stress. The sequestration of mRNAs into these translationally silent cytoplasmic foci contributes to the rapid redirection of translation from housekeeping proteins to heat-shock proteins and other stress response factors (Anderson and Kedersha, 2009). The mechanism of SG assembly has been studied in considerable molecular detail. Eukaryotic translation initiation factor 2α (eIF2α) becomes phosphorylated by one of its kinases, and abortive translation initiation complexes form around mRNA molecules. The RNA-binding proteins TIA-1 and TIAR bind to these abortive complexes and sequester them into SGs. Assembly of the granules is also dependent on the RNA-binding ability of the Ras-GAP SH3 domain–binding protein (G3BP; Tourriere et al., 2003). G3BP is a multifunctional RNA-binding protein that is present in two forms, G3BP-1 and -2 (here collectively referred to as G3BP). In a process referred to as mRNA triage, the mRNA is stored pending either degradation or resumption of normal translation in the absence of stress. SGs are dynamic, and all components rapidly shuttle into and out of the complexes, so that changing stress conditions are dealt with rapidly. Previously, we reported that early events in Semliki Forest virus (SFV) infection induce the formation of SGs in the cytoplasm of infected cells but that the SGs are rapidly disassembled in the vicinity of newly formed viral RNA replication complexes as the infection progresses (McInerney et al., 2005). Since the publication of that report, SGs have been shown to be induced by a number of viral infections and have been implicated in cellular defense against infection (Beckham and Parker, 2008; Lloyd, 2012; White and Lloyd, 2012). In many cases, viral gene products were shown to inhibit SG assembly such that their formation is transient or undetectable in wild-type virus infections (Beckham and Parker, 2008; Lloyd, 2012; White and Lloyd, 2012). In general, SGs appear to have an antiviral role in many infections, with a prevailing feature being that their formation is counteracted by the virus to allow robust viral gene expression and replication.
SFV is a member of the alphavirus genus in the family Togaviridae. The alphavirus genome is single-stranded RNA of positive polarity containing two open reading frames, coding for the early nonstructural (ns) polyprotein and the later-expressed structural proteins. The ns polyprotein is composed of four proteins, nsP1 to nsP4, which interact with each other and with host proteins to form the viral RNA replication complexes (RCs). nsP1 is the capping enzyme for viral mRNAs (Ahola et al., 1999) and is the main determinant for the membrane association of the RCs (Salonen et al., 2003). nsP2 is the protease that initially processes the polyprotein to the mature nsPs but also has other roles in altering the cellular environment to facilitate viral replication (Garmashova et al., 2006; Breakwell et al., 2007; Akhrymuk et al., 2012). The viral RNA-dependent RNA polymerase activity resides in the nsP4 protein (Rubach et al., 2009). Functions for nsP3, however, have been more difficult to define. The N-terminal region contains a macrodomain that binds poly(ADP-ribose) (PAR; Neuvonen and Ahola, 2009) and PAR polymerase-1 (Park and Griffin, 2009) and also has a role in the viral ns polyprotein processing (Lulla et al., 2012). The C-terminal one-third of the protein is largely unconserved, apart from two conserved seven–amino acid (aa) repeat sequence elements L/ITFGDFD located close to the terminus and a degradation signal in the terminal 10 amino acids (Varjak et al., 2010). Recently nsP3 was shown to interact with amphiphysin-1 and -2 in infected cells via a conserved proline-rich, SH3-domain–binding motif, and, although this interaction was shown to promote viral replication, the mechanism of this is not known (Neuvonen et al., 2011). Several reports have shown colocalization and coprecipitation of G3BP proteins with alphavirus nsP3 (Cristea et al., 2006; Frolova et al., 2006; Gorchakov et al., 2008; Fros et al., 2012). However, because G3BP is also reported to interact with nsP2 (Atasheva et al., 2007) and nsP4 (Cristea et al., 2010), it is not clear which of the components of the RNA RCs is the direct interacting partner of G3BP. The functional consequence of G3BP interaction with alphavirus nsPs is not well described, but the G3BP proteins do not seem to be important for RNA replication (Cristea et al., 2010).
Because G3BP-1 is necessary for SG assembly (Tourriere et al., 2003) and is cleaved by poliovirus 3Cpro to inhibit formation of SGs on viral RNAs (White et al., 2007), we wanted to determine whether G3BP is targeted by SFV in a manner that explains the inhibition of SGs on its own RNAs. We found that soon after infection with SFV, G3BP is recruited to viral RNA RCs. This recruitment is dependent on a seven-aa repeat domain in the C-terminus of nsP3. Infection with a viral mutant that lacks this domain induces a more robust SG response than wild-type (wt) SFV infection. Of importance, cells infected with this mutant were also able to mount a stress response to secondary stress with pateamine A (Pat A), whereas wt SFV–infected cells did not. This work thus describes the mechanism for the inhibition of SGs in SFV-infected cells and provides a previously unknown function for the alphaviral nsP3 protein.
RESULTS
SFV nsP3 forms a complex with G3BP and sequesters it into viral RNA RCs
To determine whether SFV nsP3 bound to G3BP-1 in infected cells, we infected baby hamster kidney (BHK) cells with wt SFV at a multiplicity of infection (MOI) of 10. Lysates were prepared 8 h postinfection (hpi) and immunoprecipitated with nsP3 antisera or nonimmune sera. Immunoprecipitates were then immunoblotted with nsP3, G3BP-1, and actin antisera. G3BP-1 coprecipitated with nsP3 from infected cell lysates but not with control serum (Figure 1A, top). When the same lysates were immunoprecipitated with G3BP-1 antisera and probed with nsP3, G3BP-1, and actin antisera, we found that nsP3 coprecipitated with G3BP-1 but not with control sera (Figure 1A, bottom). Similar results were obtained with G3BP-2 antisera (Supplemental Figure S1A). These results suggest that nsP3 and G3BP form a complex in SFV-infected cells. We next determined the localization of these proteins in infected cells. Mouse embryonic fibroblasts (MEFs) were infected with wt SFV at MOI 1. At 8 hpi, cells were fixed and stained for SFV nsP3 and G3BP-1. We observed that G3BP-1 strongly colocalized with nsP3 in SFV-infected cells (Figure 1B). In our hands, the G3BP-1 antisera used in this experiment did not efficiently recognize the protein when its localization was diffuse. We also observed that the G3BP-1 puncta colocalized with double-stranded (ds) RNA (Figure 1C) and with nsP1 (Figure 1D), suggesting that the nsP3 puncta most likely represent SFV RNA RCs. To further confirm that G3BP-1 colocalizes with SFV RCs, we infected BHK–enhanced green fluorescent protein (EGFP)–G3BP-1 cells, stably expressing an EGFP–G3BP-1 fusion reporter, with wt SFV at MOI 1 and determined the localization of the reporter protein. We found that EGFP–G3BP-1 colocalized with SFV nsP3 (Figure 1E). Taken together, the data in Figure 1 show that SFV nsP3 complexes with G3BP-1 and sequesters it into viral RNA RCs.
FIGURE 1:

SFV nsP3 forms a complex with G3BP and sequesters it into viral RNA RCs. (A) BHK cells were infected at MOI 10 with SFV. At 8 hpi, cell lysates were prepared and immunoprecipitated with nsP3 antisera (top), G3BP-1 antisera (bottom), or control sera and separated by SDS–PAGE. Lysates and IPs were probed for nsP3, G3BP-1, and actin. Results are representative of more than three similar experiments. The position of immunoglobulin heavy chain (HC) is indicated. (B–D) MEFs were infected at MOI 1 with SFV or mock infected. Cells were fixed at 8 hpi and stained with nsP3 and mouse G3BP-1 antisera (B), dsRNA and rabbit G3BP-1 antisera (C), or nsP1 and rabbit G3BP-1 (D) and analyzed by confocal microscopy. Bar, 10 μm. (E) BHK-EGFP-G3BP-1 cells were infected with SFV (MOI 1), fixed, and stained at 8 hpi for nsP3 and analyzed by confocal microscopy. Bar, 10 μm.
Sequestration of G3BP-1 is dependent on the C-terminal repeat domains of SFV nsP3
Because the viral RCs are multicomponent protein complexes with all four nsPs, as well as numerous host proteins, the coimmunoprecipitation of G3BP-1 and nsP3 could be the result of binding to any other protein(s) in these complexes or to viral RNA. To determine whether sequestration of G3BP-1 was dependent on nsP3 alone, we analyzed the binding of G3BP-1 to nsP3 in HEK293 cells directing the inducible expression of nsP3 in the absence of other viral proteins. In these experiments we also included cell lines expressing variants of nsP3 carrying C-terminal deletion mutations. The C-terminus of nsP3 contains two seven-aa repeat sequence elements L/ITFGDFD, conserved in the Old World alphaviruses, terminating 10 amino acids from the C-terminus and separated by a nonconserved stretch of 10 amino acids (Figure 2A). These repeat motifs have unknown but important function, since their deletion attenuated the virus (Varjak et al., 2010). Cell lines inducibly expressing wt nsP3 (T-REx-nsP3), a mutant lacking the C-terminal 10 amino acids but retaining the conserved repeats (T-REx-nsP3∆10) or a mutant lacking the C-terminal 30 amino acids retaining only four amino acids of one repeat (T-REx-nsP3∆30) were described previously (Varjak et al., 2010). With the use of these cell lines, nsP3 expression was induced and lysates were taken and analyzed by SDS–PAGE and immunoblotting. Expression levels of nsP3∆10 and nsP3∆30 were considerably higher than for wt nsP3 (Figure 2B) due to the ablation of a degradation signal normally present in the extreme C-terminal 10 amino acids of nsP3 (Varjak et al., 2010). When lysates were analyzed by immunoprecipitation with G3BP-1 antisera, we observed that wt nsP3 and nsP3∆10 coprecipitated with G3BP-1 at ratios correlating with their expression level. Neither protein immunoprecipitated with control sera. Despite high relative expression of nsP3∆30, it did not coprecipitate with G3BP-1 to any detectable level, suggesting that formation of the nsP3/G3BP-1 complex is independent of the C-terminal degradation signal but dependent on the presence of the 20 residues encompassing half of one repeat, the spacer region, and the second repeat motif. To determine whether the complex required only the C-terminal domain and no other sequence of nsP3, we fused 31 amino acids of the C-terminus of nsP3 with EGFP (Figure 2C) and determined the ability of this reporter to form a complex with G3BP-1. BHK cells were transfected with vectors encoding wt EGFP or EGFP-31. Lysates were immunoprecipitated with EGFP antisera and immunoblotted with sera to G3BP-1 (Figure 2D). G3BP-1 coprecipitated with EGFP from lysates of cells expressing EGFP-31 but not wt EGFP, indicating that only this region is required and suggesting that G3BP-1 binds directly to the C-terminal domain of nsP3. Similar results were obtained with G3BP-2 antisera (Supplemental Figure S1B). The results in Figure 2 show that formation of a complex with G3BP requires only the C-terminal repeat motifs of nsP3.
FIGURE 2:
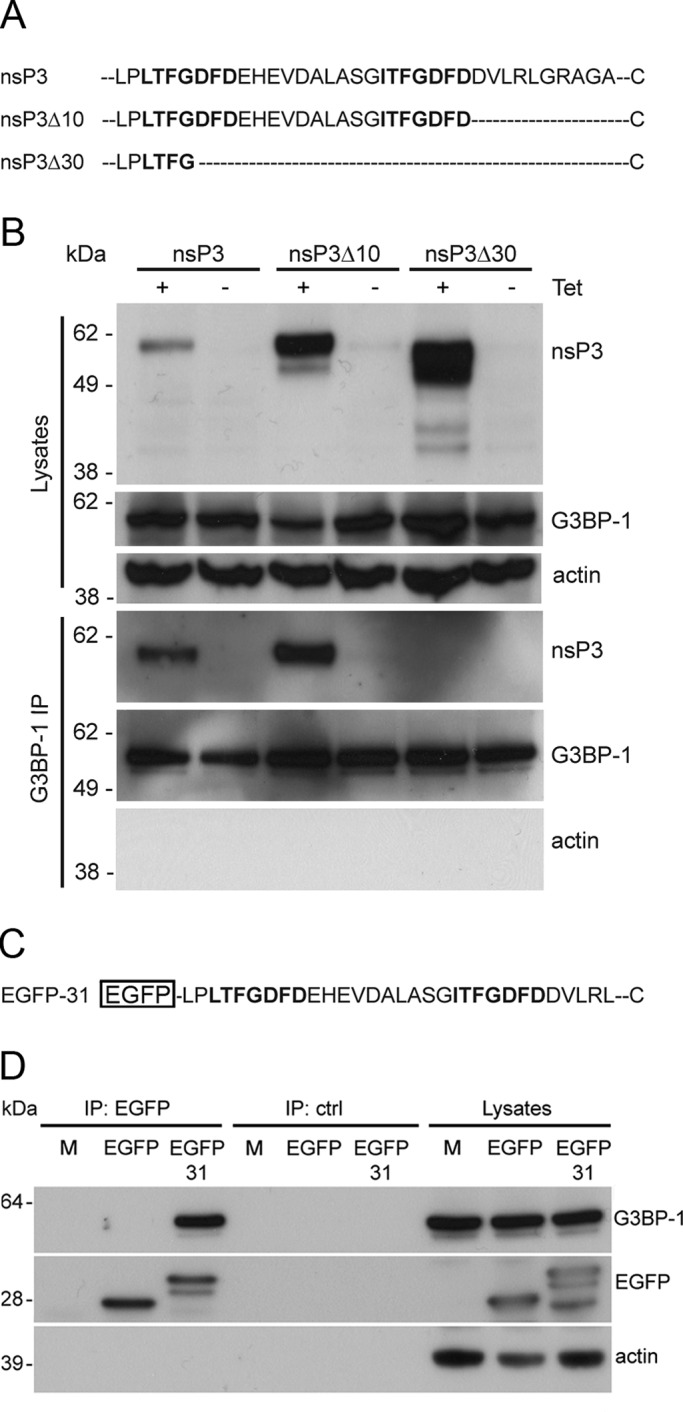
Formation of a complex with G3BP-1 is dependent on the C-terminal repeat domains of SFV nsP3. (A) Extreme C-terminal sequences of wt nsP3, nsP3Δ10, and nsP3Δ30. Two highly similar repeat motifs are indicated in boldface. (B) T-REx-nsP3, T-REx-nsP3∆10, and T-REx-nsP3∆30 cells were induced with tet for 16 h. Cell lysates were prepared and immunoprecipitated with G3BP-1 or control antisera and separated by SDS–PAGE. Lysates and IPs were probed for nsP3, G3BP-1, and actin. Results are representative of more than three similar experiments. (C) C-terminal sequence of EGFP-31. (D) BHK cells were transfected with pBK-CMV-EGFP or pEGFP-31. Cell lysates were prepared 16 h after transfection and immunoprecipitated with EGFP or control antisera and separated by SDS–PAGE. Lysates and IPs were probed for G3BP-1, EGFP, and actin. Results are representative of three independent experiments.
We then tested whether mutation of this domain affected G3BP-1 recruitment to viral RCs in infected cells. Mutants were constructed to ablate one or both of these repeat domains (Figure 3A). In all cases the multifunctional C-terminal 10-aa sequence was retained to ensure correct processing of the polyprotein. SFV-∆8 lacks eight of the 10 residues in the nonconserved spacer region between the two repeat motifs; SFV-∆78 lacks the same eight residues, as well as the first of the two repeat motifs; and SFV-∆789 lacks both repeats and the spacer region. BHK cells were infected at MOI 10 with these viruses, and lysates were taken at 8 hpi. Immunoblotting revealed that the nsP3 variants were expressed at levels equivalent to wt and that SFV-∆789 had a slight delay in polyprotein processing, as evidenced by the presence of increased amounts of P34 processing intermediate (Figure 3B). Immunoprecipitation with anti–G3BP-1 sera efficiently coprecipitated wt nsP3. nsP3 from SFV-∆8 also coprecipitated with G3BP-1, although at slightly lower levels than the wt protein, whereas the coimmunoprecipitation of nsP3 from SFV-∆78 with G3BP-1 was drastically reduced. nsP3 from SFV-∆789, which lacks both repeat motifs, did not coprecipitate with G3BP-1. To confirm these findings, we determined the localization of G3BP-1 with viral RCs in MEFs infected at MOI 1 for 8 h with each of the viruses used in Figure 3B. Infected cells were fixed and stained for SFV nsP3 and G3BP-1. RCs of wt SFV and SFV-∆8 both contained G3BP-1, whereas only very few RCs in SFV-∆78– and none in SFV-∆789–infected cells stained positive for G3BP-1 (Figure 3C). Similar results were obtained for G3BP-2 (Supplemental Figure S1C). To determine whether the abrogation of G3BP recruitment negatively affected viral replication, we performed single-step and multistep growth curves. These revealed that SFV-∆789 propagated to titers of between one and two orders of magnitude lower that wt SFV in MEFs (Figure 3D) and in BHK cells (Supplemental Figure S2). Although it cannot be excluded that this replication defect may have partially resulted from the slight delay in polyprotein processing, it was apparent that removal of the C-terminal repeat motifs led to a slow-propagation phenotype in all conditions tested. Taken together, the results in Figure 3 show that G3BP-1 is recruited to SFV RCs in a manner dependent on one or both of the conserved seven-aa repeat motifs near the C-terminus of nsP3 and that removal of these motifs limits viral propagation.
FIGURE 3:

The C-terminal repeat domains of SFV nsP3 are required for sequestration of G3BP-1 into RCs. (A) Representation of C-terminal sequences of nsP3 from wt SFV, SFV-Δ8, SFV-Δ78, and SFV-Δ789. (B) BHK cells were infected with SFV, SFV-∆8, SFV-∆78, or SFV-∆789 (MOI 10). At 8 hpi, cell lysates were prepared, immunoprecipitated with G3BP-1 antisera, and separated by SDS–PAGE. Lysates and IPs were probed for nsP3, G3BP-1, and actin. Results are representative of three similar experiments. (C) MEFs were infected with SFV, SFV-∆8, SFV-∆78, or SFV-∆789 (MOI 1). Cells were fixed at 8 hpi and stained for nsP3 and G3BP-1 and analyzed by confocal microscopy. Results are representative of more than three similar experiments. Bar, 10 μm. (D) MEFs were infected with SFV or SFV∆789 at MOI 0.1 (top) or MOI 10 (bottom). At 4, 8, 12, or 24 hpi supernatants were collected, and SFV infectious units were quantified by plaque assay on BHK cells. Data are means of two independent experiments each titrated in duplicate. Error bars indicate SD.
G3BP-1 incorporation into RNA RCs correlates with disassembly of RNA stress granules
In a previous study, we showed that early in SFV infection, SGs are induced in a manner that correlates with the inhibition of host mRNA translation and that later, these SGs are disrupted and cannot be reinduced by further stress (McInerney et al., 2005). Because G3BP-1 is necessary for the assembly of SGs (Tourriere et al., 2003) and is a known target for viral modification of the SG machinery (White et al., 2007), we sought to determine whether the association of this host protein into viral RNA RCs correlates with the disassembly of SGs. Accordingly, we analyzed the localization of the SG marker TIA-1 together with G3BP-1 and SFV nsP3 in a time course of infection in MEFs (Figure 4). In line with our previous observations, TIA-1, present in the nucleus in noninfected cells, rapidly relocated to the cytoplasm in SFV-infected cells and at 4 hpi was observed in cytoplasmic puncta, which also contained G3BP-1. These puncta likely represented cellular SGs, induced by early events in virus replication and very rarely contained nsP3. At this time point, nsP3 was also detected in puncta, in this case representing viral RNA RCs, which often contained G3BP-1 but rarely contained TIA-1. As the infection progressed, TIA-1 staining remained cytoplasmic but became more diffuse as the SGs were disassembled, whereas G3BP-1 was found more in nsP3-positive puncta until the end of the experiment, when the majority of the cellular G3BP-1 appeared to be present in viral RCs. To confirm that the G3BP-1– and TIA-1–positive structures observed at 4 hpi contained stalled translation initiation complexes, we fixed cells after 4 h of SFV infection and stained for G3BP-1 and TIA-1/R together with eIF3 and eIF4E. We observed that G3BP-1 colocalized with eIF3 (Supplemental Figure S3A) and that TIA-1/R colocalized with eIF4E and eIF3 (Supplemental Figure S3, B and C) at this time point, and we concluded that these structures indeed represent true SGs, as defined as containing TIA-1 and stalled translation initiation complexes (Kedersha et al., 2002). The data in Figure 4 thus show that the disassembly of infection-induced SGs correlated with the sequestration of G3BP-1 into viral RNA RCs.
FIGURE 4:

G3BP incorporation into SFV RCs correlates with disassembly of SGs. MEFs were infected at MOI 1 with wt SFV and fixed and stained at the indicated time points for TIA-1, G3BP-1, and nsP3 and analyzed by confocal microscopy. Data are representative of more than three independent experiments. Cells containing predominantly SGs or RCs are indicated with yellow or cyan arrowheads, respectively. Bar, 10 μm.
G3BP incorporation into RNA RCs inhibits SG formation at early times after infection
To determine whether the sequestration of G3BP-1 into SFV RCs by nsP3 is responsible for the disassembly of virus-induced SGs, we quantified the extent of the SG response to wt SFV and SFV-∆789 infection. MEFs were infected with either virus at MOI 1, and coverslips were fixed at 1/2-h intervals and stained for TIA-1, G3BP-1, and nsP3. Infected cells at each time point (identified by nsP3 staining) were examined for the presence or absence of SGs by manual counting (Figure 5A). In cells infected with either virus, the appearance of SGs first occurred between 3 and 4 hpi. The proportion of SG-positive cells in wt SFV-infected cultures reached a maximum of 63% (±5%) at 4 hpi and dropped off rapidly thereafter. SFV-∆789 infected cultures, despite initiating an SG response to viral infection at the same time as wt, reached a peak of 71% (±2%) at 4.5 hpi and also declined at later times. Counting of SG-positive cells at later times in these experiments was complicated by the spreading infection and the induction of SG responses in newly infected cells. Nevertheless, these data suggest that the SG response to viral infection persists longer in cells infected with SFV-∆789 relative to wt SFV–infected cells. However, the observation that SGs are finally disassembled in SFV-∆789–infected cells suggested that there is an additional mechanism to disassemble SGs or to block their formation in SFV-infected cells that is not dependent on the nsP3/G3BP interaction. Because SG components are in dynamic equilibrium with polysomes (Kedersha et al., 2000), it was likely that the efficient translation of the viral mRNAs, most likely the 26S mRNA of either wt SFV or SFV-∆789, would lead to SG disassembly as SG-resident proteins and ribosomal subunits are recruited to polysomes for viral protein production. To test this, we infected cells with recombinant SFV vector particles carrying genomes that contain chicken egg ovalbumin (ova) as a reporter gene in place of the structural protein–coding region. These vectors lack a translational enhancer sequence known to confer to the viral mRNAs resistance to the high levels of phosphorylated eIF2α in the infected cell (Sjöberg et al., 1994; McInerney et al., 2005), and so the ova-expressing mRNAs are relatively inefficiently translated. MEF cells were infected with either SFV-ova or SFV-∆789-ova at MOI 0.2, and coverslips were fixed at 1-h intervals postinfection and stained for TIA-1, G3BP-1, and nsP3 (Figure 5B). Because the single-round vectors do not spread in the culture, we could expand the experiment to longer times postinfection. Again, both vectors led to the appearance of SGs in infected cells first at 3 hpi and, whereas the proportion of cells that were SG positive in cultures infected with the wt vector reached 51% (±4%) at 4 hpi, 80% (±2%) of SFV-∆789-ova–infected cells were SG positive at that time. As with the fully infectious viruses (Figure 5A), the proportion of cells that contained SGs diminished after reaching maximal values. However, the kinetics of this was considerably slower with the vectors lacking the translational enhancer. The SG-positive proportion remained at peak levels until 5 hpi in both cultures and diminished slowly in both infections, although it remained at significantly higher levels in SFV-∆789-ova–infected cells. We also quantified the number of SGs per cell in the different infections. In cells that were SG positive, the number of SGs/cell did not differ between cells infected with SFV-ova or SFV-∆789-ova at 4 hpi (Supplemental Figure S4A). However, this number was found to be significantly reduced in SFV-ova– relative to SFV-∆789-ova–infected cells at 8 hpi (Supplemental Figure S4B). Representative images from cells infected with these vectors or mock infected for 8 h are shown in Figure 5C. SFV-ova–infected cells contained strongly nsP3-positive foci, which were also G3BP-1 positive and represented RCs, whereas TIA-1 staining was diffuse (as previously observed in wt SFV infection; Figure 4). TIA-1–positive SGs were rare in SFV-ova–infected cells at this time postinfection. SFV-∆789-ova–infected cells also displayed strongly nsP3-positive foci, but these were G3BP-1 negative. In contrast to SFV-ova–infected cells, TIA-1/G3BP-1 double-positive SGs, although few, could be readily detected in SFV-∆789-ova–infected cells, often in the vicinity of viral RCs.
FIGURE 5:

G3BP-1 sequestration into SFV RCs inhibits virus-induced SGs at early times after infection. (A) MEFs were infected at MOI 1 with wt SFV or SFV-∆789. Cells were fixed at 30-min intervals and stained for G3BP-1, TIA-1, and nsP3. Fifty infected cells per time point were identified by nsP3 staining and scored as SG+ based on G3BP-1 and TIA-1 colocalization. Data are presented as mean +/– SD from three independent experiments. Student's t test, * = p < 0.05. (B) MEFs were infected at MOI 0.2 with SFV-ova or SFV-∆789-ova. Cells were fixed at 1 h intervals and stained for G3BP-1, TIA-1 and nsP3. Cells were counted as in (A). Data are presented as mean ± SD from three independent experiments. Student's t test, *p < 0.05, **p < 0.01, ***p < 0.001. (C) Representative images from MEFs infected for 8 h as in B. Bar, 10 μm.
G3BP incorporation into RNA RCs inhibits Pat A–induced SG formation at late times after infection
We previously showed that wt SFV–infected cells are unable to form SGs in response to sodium arsenite added at 8 hpi (McInerney et al., 2005). This led us to conclude that the formation of SGs is blocked in a manner that allows the SG components to be reassembled into polysomes on the 26S mRNA. Our present work suggested that the mechanism by which SFV achieves SG disassembly is initially by sequestration of G3BP-1 into viral RCs and that this is aided by the efficient translation of viral mRNAs. To further investigate the effect of the nsP3/G3BP interaction on the disassembly of SGs, we tested whether cells infected with SFV-ova or SFV-∆789-ova could mount a stress response to an independent stress inducer at 8 hpi. Because sodium arsenite stress is signaled via eIF2α phosphorylation, it may not in fact have any effect in an SFV-infected cell already containing high levels of phospho-eIF2α. Therefore we expanded this experiment to include Pat A, which induces phospho-eIF2α–independent SGs (Bordeleau et al., 2006; Dang et al., 2006). Wild-type MEFs were mock infected or infected with SFV-ova or with SFV-∆789-ova at MOI 0.2. At 7 hpi, cells were mock stressed or stressed with sodium arsenite or Pat A for 1 h before fixation and staining for TIA-1 to detect SGs and nsP3 to detect SFV infection (Figure 6A, left). Almost 100% of mock-infected cells responded to both sodium arsenite and Pat A by forming SGs, 21% of wt SFV-ova–infected cells had SGs, and this number was not significantly increased by either arsenite or Pat A treatment, confirming that wt SFV–infected cells cannot mount a stress response to secondary stress signals. As previously observed, SFV-∆789-ova–infected cultures had a higher number of SG-positive cells than wt SFV-ova–infected cultures (52% vs. 21%). Although this proportion was not increased by the addition of sodium arsenite, it was significantly increased by Pat A treatment, indicating that in the absence of nsP3/G3BP interaction, SGs can be formed in SFV-infected cells. Arsenite, however, does not induce the formation of additional SGs, most likely because the viral infection already induces high levels of phospho-eIF2α (McInerney et al., 2005). To determine the effect of Pat A on the induction of SGs in SFV-ova– and SFV-∆789-ova–infected cells in the absence of background virus-induced SGs, we performed these secondary stress experiments also in eIF2α-AA MEFs, which lack phosphorylatable eIF2α (Figure 6A, right). In these cells, the contribution of Pat A to SG induction is more easily appreciated. Although, as expected, sodium arsenite–treated eIF2α-AA MEFs did not induce SGs, Pat A still induced SGs in >98% of treated cells. SFV-ova infection of eIF2α-AA MEFs did not induce SGs, even in combination with sodium arsenite treatment, whereas Pat A treatment did induce an SG response in a very small number of SFV-ova–infected cells. Similar to the wt vector, SFV-∆789 ova infection, even in combination with sodium arsenite treatment, did not induce SGs. However, Pat A treatment of SFV-∆789-ova–infected cells induced a strong SG response: 74% of infected cells under these conditions had TIA-1– and G3BP-1-positive SGs in the cytoplasm. We also performed similar experiments comparing high-MOI infections with wt SFV and SFV-∆789 in wt and eIF2α-AA MEFs, with similar results (Figure 6B). These data showed that although sodium arsenite is not able to induce SGs in cells infected with wt SFV or with SFV-∆789, Pat A can induce a strong SG response, but only in SFV-∆789–infected cells, where G3BP-1 is not recruited to RCs by nsP3. Thus SG dissociation is enhanced by the efficient phospho-eIF2α–independent translation of viral RNAs. Although this effect may be a passive consequence of the particular mechanism used in translation of viral mRNAs, it is clear that SFV also possesses an active mechanism, G3BP-1 sequestration, to dissolve SGs early in infection and prevent their re-formation at late stages of infection.
FIGURE 6:

G3BP-1 sequestration into SFV RCs inhibits Pat A-induced SGs at late times after infection. (A) wt MEFs (left) or eIF2α-AA MEFs (right) were mock infected or infected with SFV-ova or SFV-∆789-ova at an MOI of 0.2 for 7 h before 1-h treatment with sodium arsenite, Pat A or mock treatment. Cells were fixed and stained for G3BP-1, TIA-1, and nsP3. Fifty cells per treatment were scored as SG+ based on G3BP-1 and TIA-1 colocalization. Data are presented as mean ± SD from three or more independent experiments. Student's t test, *p < 0.05, ***p < 0.001. (B) Wild-type MEFs (left) or eIF2α-AA MEFs (right) were mock infected or infected with SFV or SFV-∆789 at an MOI of 50 for 7 h before 1-h treatment with sodium arsenite or Pat A or mock treatment. Cells were fixed and stained for G3BP-1, TIA-1, and nsP3. Cells were counted as in A. Data are presented as mean ± SD from three independent experiments. Student's t test, *p < 0.05, ***p < 0.001.
Sequestration of G3BP-1 by SFV nsP3 blocks SG assembly by multiple inducers in noninfected cells
To determine whether the interaction of nsP3 with G3BP-1 could block SG assembly in the context of a noninfected cell, we assayed the ability of the T-REx-nsP3 and T-REx-nsP3∆30 cells to mount a response to Pat A stress. Cells were treated with tetracycline (tet) for 16 h to induce expression of the viral protein. Mock-treated cells were used as controls. Cells were then stressed with Pat A or mock stressed and fixed and stained for G3BP-1, TIA-1, and nsP3 (Figure 7A). As expected, wt nsP3 colocalized with G3BP-1 but only weakly with TIA-1 in unstressed cells, whereas nsP3∆30 was found in a diffuse staining pattern and did not colocalize with either protein. When cells were stressed with Pat A, however, we found that although wt nsP3 remained colocalized with G3BP-1, both these proteins were colocalized with TIA-1 in SGs. NsP3∆30, on the other hand, remained diffuse in the cytoplasm in Pat A–stressed cells (Figure 7A). These observations suggest that the formation of the nsP3- and G3BP-containing complex alone does not block SG assembly, and, in fact, in those conditions nsP3 is itself found in SGs, a phenomenon not observed in SFV-infected cells. It is possible therefore that apart from nsP3 binding, some other factor, such as the sequestration into viral RCs, is also necessary for blocking G3BP-1 activity. Given that nsP1 is the membrane-anchoring component of the viral RCs (Salonen et al., 2003), and expression of the nsP123 polyprotein leads to assembly of structures that resemble RCs (Varjak et al., 2010), we tested the ability of the nsP123 polyprotein to block SG induction after Pat A treatment in transient expression experiments. The plasmid pcDNA4-nsP123 or pcDNA4-nsP123∆30 was transfected into MEFs, and cells were mock stressed or stressed by the addition of Pat A for 1 h and fixed and stained for eIF3 (to identify SGs), G3BP-1, and nsP3 (Figure 7B). Mock-transfected cells displayed diffuse cytoplasmic staining for eIF3 and G3BP-1, both of which colocalized into puncta in stressed cells, representing SGs. In contrast, in cells expressing wt nsP123, nsP3 colocalized in puncta with G3BP-1, which generally did not contain eIF3 and thus represented RC-like structures. When such cells were stressed, this localization was maintained such that very few, if any, eIF3- and G3BP-1–positive puncta were detected, indicating that the cells were unable to induce SGs upon Pat A stress. As expected, cells expressing nsP123∆30 did not display any colocalization of the viral protein with G3BP-1 under any conditions. Much of the viral protein was found in puncta, which may be RC-like structures, but it was also found diffuse in the cytoplasm. In addition, the nsP123∆30-expressing cells were able to respond to Pat A treatment by the induction of eIF3- and G3BP-1–positive SGs (Figure 7B). The number of SG-positive cells in mock-transfected and pBK-CMV-EGFP–, pcDNA4-nsP123–, and pcDNA4-nsP123∆30–transfected cultures in the presence or absence of Pat A treatment was quantified by manual counting (Figure 7C). In mock-transfected cells and in cells transfected with pBK-CMV-EGFP and pcDNA4-nsP123∆30, Pat A induced a strong SG response. Although 13% of pcDNA4-nsP123–transfected cells were able to respond to Pat A treatment, this number was significantly reduced compared with the other treatments. Of interest, transfection of pBK-CMV-EGFP or pcDNA4-nsP123∆30 but not pcDNA4-nsP123 induced an SG response in the absence of Pat A treatment, indicating that nsP123 was able to block transfection-induced SGs also. Similar results were obtained in transfection experiments when 2-deoxy-d-glucose (2-DG) was used as an SG inducer (Figure 7D). Taken together, these data show that the binding of G3BP-1 by the C-terminal domain of nsP3 and its recruitment to viral RC-like structures efficiently blocked the assembly of SGs induced by phospho-eIF2α–independent stress pathways.
FIGURE 7:

Sequestration of G3BP-1 by SFV nsP3 blocks SG assembly by multiple inducers in noninfected cells. (A) T-REx-nsP3 and T-REx-nsP3∆30 cells were induced with tet for 16 h and mock stressed or stressed with Pat A for 1 h. Cells were fixed and stained for TIA-1, G3BP-1, and nsP3 and analyzed by confocal microscopy. Data are representative of three independent experiments. Bar, 10 μm. (B) MEFs were transfected with pcDNA4-nsP123 or pcDNA4-nsP123∆30 or mock transfected. Transfected cells were mock treated or treated with Pat A for 1 h and fixed and stained for eIF3, G3BP-1, and nsP3 and analyzed by confocal microscopy. Cells containing predominantly SGs or RC-like structures are indicated with yellow or cyan arrowheads, respectively. Bar 10 μm. (C) Cells treated as in B were scored as SG+ based on G3BP-1 and eIF3 colocalization. Data are presented as mean ± SD from three independent experiments in which 50 cells per treatment were counted. Student's t test, *p < 0.05 and ***p < 0.001. (D) MEFs were mock transfected or transfected with pBK-CMV-EGFP, pcDNA4-nsP123, or pcDNA4-nsP123∆30. Transfected cells were mock treated or treated with 2-DG for 1.5 h and fixed and stained for eIF3, G3BP-1, and nsP3. Cells were scored as SG+ based on G3BP-1 and eIF3 colocalization. Data are presented as mean ± SD from three independent experiments in which 50 cells per treatment were counted. Unpaired t test, *p < 0.05 and ***p < 0.001.
DISCUSSION
In this work we showed that SFV nsP3 forms a complex with the SG proteins G3BP via the C-terminal region of nsP3 containing seven–amino acid repeat motifs, which we recently identified as having an important function in virus replication (Varjak et al., 2010). These repeats are well conserved in the Old World but not in the New World or piscine alphaviruses. The association of nsP3 with nsP1 and the other components of viral RCs sequesters G3BP such that SG assembly is blocked. By comparing the numbers of SG-containing cells in cultures infected with wt SFV and with SFV-∆789, which lacks the G3BP-interacting motifs, we observed a higher number of cells responding by induction of SGs to infection with SFV-∆789 than with SFV. In both infections, however, the number of cells with SGs diminished with time, most likely because of the efficient translation of viral mRNAs. This is confirmed by similar experiments with SFV-ova and SFV-∆789-ova, viral vectors that lack the translational enhancer element and direct less efficient translation of viral mRNAs. SGs persist in cells infected with these vectors for longer times than in cells infected with enhancer-containing viruses. We thus propose that the nsP3/G3BP interaction is important at very early times in infection to reduce the numbers of SGs or otherwise weaken the cell's ability to form SGs on viral mRNAs. This effect is especially pronounced in regions of the cell where viral replication is first initiated. At later times, the efficient initiation of translation of enhancer-containing 26S mRNAs, coupled with the unavailability of G3BP, leads to total disruption of SGs in infected cells. This is confirmed by our secondary stress experiments using Pat A. This treatment, in contrast to sodium arsenite, inhibits 26S mRNA translation and leads to significant increases in SG numbers in cells infected with SFV-∆789 and SFV-∆789-ova but not wt SFV or SFV-ova.
Transient SG induction is a feature common to several RNA-virus infections (Beckham and Parker, 2008; Lloyd, 2012; White and Lloyd, 2012). For many viruses, early events in viral replication, such as the generation of double-stranded RNA replication intermediates, lead to the activation of PKR, the phosphorylation of eIF2α, and SG assembly on abortive initiation complexes. For others, such as poliovirus, the signals for SG induction are less clear but may be linked with virus-induced alterations in the translation initiation complex. A number of viruses have been shown to use specific mechanisms to disrupt the formation of SGs on their mRNAs. Poliovirus 3Cpro cleaves G3BP-1 with kinetics correlating to the disruption of SGs in infected cells (White et al., 2007). Confirming the role for this cleavage in the block of SGs, a noncleavable mutant G3BP-1 variant conferred upon infected cells the ability to respond to arsenite treatment by forming SGs and led to significant reductions in viral titers. The flaviviruses, West Nile virus, and dengue virus both recruit the SG proteins TIA-1/R to viral replication sites in a manner that contributes to the inability of infected cells to respond to sodium arsenite treatment by the induction of SGs (Emara and Brinton, 2007). Sendai virus trailer RNA binds to TIAR, and a virus lacking this sequence induces more SGs in infected cells (Iseni et al., 2002). Similarly, the trailer RNA of RSV also acts to block SG assembly, although this effect was shown to be independent of TIAR (Hanley et al., 2010). Because there are numerous other examples of transient or blocked SG formation in viral infections (Beckham and Parker, 2008; Lloyd, 2012; White and Lloyd, 2012), it is likely that there exist other specific mechanisms for SG disruption encoded by these viruses. However, since we have also found that the efficient translation of viral RNAs in the normally restrictive milieu of a cell containing high levels of phospho-eIF2α can itself contribute to the disruption of SG, it might be worth considering that such a passive mechanism might operate in other viral infections also.
G3BP-1 has been shown to form a complex with alphaviral nsP3 proteins (Cristea et al., 2006; Frolova et al., 2006; Gorchakov et al., 2008; Fros et al., 2012) but also with nsP2 (Atasheva et al., 2007) and nsP4 (Cristea et al., 2010), whereas other studies of the host protein components of the alphaviral RCs failed to detect this (Bourai et al., 2012). Our results show that the formation of a complex with G3BP is dependent only on the C-terminal seven–amino acid repeat motifs of nsP3. Without this interaction, G3BP is not recruited to the RCs, and we conclude that, at least for SFV, any interactions between G3BP and nsP2 or 4 are indirect or must depend on initial sequestration to the complexes by nsP3. Recently it was proposed that Chikungunya virus inhibits SG assembly by interaction between G3BP and the SH3-domain–binding region of nsP3 (Fros et al., 2012), previously shown to bind to amphiphysin-1 and -2 in infected cells (Neuvonen et al., 2011). In contrast, our results with SFV strongly suggest a direct interaction between G3BP and the nsP3 C-terminal seven–amino acid repeat motifs that does not require the SH3-domain–binding region. Furthermore, SFV-∆789, which contains an unaltered SH3-domain–binding motif, induced a more persistent SG response than wt SFV and was unable to block infected cells from responding to further stress with Pat A by induction of SGs. Sindbis virus protein production was increased in the absence of G3BP (Cristea et al., 2010), indicating that the proteins have some antiviral role in reducing viral gene expression, possibly through their role in SG assembly. SFV-∆789, which does not sequester G3BP into its RCs and induces a stronger SG response than wt SFV, replicates to lower titers than wt SFV in all cell lines tested, further supporting an antiviral role for SGs. Of interest, a direct antiviral role for SG-like antiviral granules was recently shown in vaccinia virus (VV)–infected cells (Simpson-Holley et al., 2011). In cells infected with a VV mutant lacking the PKR antagonist E3L, it was shown that G3BP-1–, TIA-1–, and eIF3-positive granules were recruited to viral factories and had a direct role in restriction of viral replication. Moreover the increasing number of viruses that use specific mechanisms to inhibit SGs suggests a general antiviral role for these granules.
Although we cannot exclude a role for G3BP in the RCs, we believe it unlikely that G3BP is required for assembly of RCs or for efficient production of viral RNA. Infection with SFV-∆789, which does not recruit G3BP into RCs, leads to the appearance of SGs at similar times as wt SFV, suggesting that the time of production of early replicative intermediates that lead to the activation of PKR and phosphorylation of eIF2α is equal for the two viruses. Moreover, viral RCs in cells infected with SFV, SFV-∆8, SFV-∆78, and SFV-∆789 are similar in number and appearance, despite markedly different levels of G3BP recruitment. G3BP-1 was found to accumulate with single-stranded RNA of rubella virus, a noncytopathic togavirus, suggesting a role for the protein in a late event in viral replication (Matthews and Frey, 2012). G3BP-1 also accumulates in viral RCs of HCV (Yi et al., 2006; Jones et al., 2010) and in VV factories (Katsafanas and Moss, 2007). Whether G3BP has specific role(s) in the replication and gene expression of these diverse viruses or whether its sequestration by viral factors acts mainly to modulate or inhibit SG assembly remains to be elucidated. In summary, we provide further evidence that SGs function in innate antiviral defense and describe the mechanism by which SFV, and most likely many other alphaviruses, block the formation of SGs and ensure efficient gene expression and replication.
MATERIALS AND METHODS
Cell culture, plasmids, and virus propagation
Wild-type and eIF2α-AA SV129 MEFs were maintained in DMEM (Sigma-Aldrich, St. Louis, MO) with 10% fetal calf serum (FCS; Sigma-Aldrich), 2 mM l-glutamine, and penicillin–streptomycin. BHK-21 cells (American Type Culture Collection, Manassas, VA) were cultured in Glasgow minimal essential medium (Sigma-Aldrich) supplemented with 10% FCS, 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 10% tryptose phosphate broth, 2 mM l-glutamine, and penicillin–streptomycin (Invitrogen, Carlsbad, CA). BHK-EGFP-G3BP-1 cells were generated by transfection of BHK-21 cells with pEGFP-G3BP-1 (a kind gift from Nancy Kedersha, Harvard Medical School, Cambridge, MA), followed by selection in Geneticin (1 mg/ml; Invitrogen). Previously published T-REx-nsP3, T-REx-nsP3∆10, and T-REx-nsP3∆30 cells (Varjak et al., 2010) were maintained in Iscove's modified DMEM (Sigma-Aldrich) with 10% FCS, 2 mM l-glutamine, and penicillin–streptomycin supplemented with blasticidin (5 μg/ml; Invitrogen) and Zeocin (30 μg/ml; Invitrogen). Expression of nsP3 and variants was induced by treatment with tetracycline (1 μg/ml; Sigma-Aldrich) for 16 h. pcDNA4-nsP123 and pcDNA4-nsP123∆30 plasmids were published previously (Varjak et al., 2010). pEGFP-31 was generated by ligating a sequence encoding residues 447–477 of SFV4 nsP3 between the BglII and EcoRI sites of pEGFP-C1 (Clontech, Mountain View, CA). For transfection, MEFs were grown until 50% confluence and then transfected with 1 μg of plasmid using Lipofectamine 2000 (Invitrogen) reagent. Cells were stressed by addition of sodium arsenite (0.5 μM) or Pat A (100 nM; a kind gift from Jerry Pelletier, McGill University, Montreal, Canada) in respective complete medium for 60 min or 2-DG (500 mM, Sigma-Aldrich) in PBS for 90 min before fixation. Wild-type SFV working stocks were derived from the SFV4 infectious clone (pSP6–SFV4) as described previously (Liljeström et al., 1991). SFV-∆8, ∆78, and ∆789 mutant stocks were generated as described (Varjak et al., 2010). pSFV3-ova was constructed by ligation of the ova sequence with flanking BamHI and XmaI sites into similarly cut pSP6SFV3 (Liljeström and Garoff, 1991). pSFV-∆789-ova was constructed by ligating the XhoI–BglII fragment from pCMV-SFV∆789, containing the deletion mutation into the corresponding position in pSP6-SFV3-ova. rSFV working stocks were produced and packaged as described previously (Liljeström and Garoff, 1991; Smerdou and Liljeström, 1999). Virus titration was performed by quantification of plaque numbers, and rSFV stocks were titrated using immunofluorescence in MEFs or BHK cells. SFV stocks were used for infection as follows: cell monolayers were washed with phosphate-buffered saline (PBS) and virus or rSFV was added in DMEM (Sigma-Aldrich) with periodic shaking for 1 h at 37°C. Virus solutions were then removed and cells washed with PBS before adding prewarmed complete medium.
Immunofluorescence
Cells grown on coverslips were fixed by incubation in 3.7% formaldehyde (Sigma-Aldrich) in PBS for 10 min at room temperature, followed by incubation in ice-cold methanol for 10 min. Antibodies used for immunofluorescence include mouse anti-nsP1, rabbit anti-nsP3 (Tamberg et al., 2007), mouse anti–G3BP-1 (BD Transduction Laboratories, Lexington, KY), rabbit anti–G3BP-1 (Aviva Systems Biology, San Diego, CA), rabbit anti–G3BP-2 (Assay Biotech, Sunnyvale, CA), mouse anti-TIA1/R (3E6; Taupin et al., 1995), goat anti–TIA-1, goat anti-eIF3η, and mouse anti-eIF4E (Santa Cruz Biotechnology, Santa Cruz, CA), and mouse anti-dsRNA (English & Scientific Consulting Bt., Szirák, Hungary). Antibodies were diluted in block buffer (5% horse serum [Sigma-Aldrich], 0.1% sodium azide in PBS). Washed coverslips were then incubated in block buffer containing secondary antibodies and 1 μg/ml Hoechst 33258 (Invitrogen) or DRAQ5 (Biostatus, Leicestershire, United Kingdom) for 1 h for identification of cell nuclei. Washed coverslips were then mounted in vinol mounting medium, and images were captured using a supercontinuum confocal TCS SP5 X (Leica, Wetzlar, Germany) with a pulsed white light laser. Images were processed and compiled using Photoshop (Adobe, San Jose, CA).
Immunoprecipitations and immunoblotting
For immunoprecipitations (IPs) cells were lysed on ice in lysis buffer (20 mM HEPES, pH 7.4, 110 mM potassium acetate, 2 mM MgCl2, 0.1% Tween 20, 1% Triton X-100, 0.5% sodium deoxycholate, 0.5 M NaCl) and clarified by centrifugation at 10,000 × g for 10 min at 4ºC. The supernatants were incubated with rabbit anti–G3BP-1, mouse anti–G3BP-1, rabbit anti-nsP3, mouse anti-GFP (Abcam, Cambridge, MA), rabbit nonimmune serum immunoglobulin G (IgG), or goat anti–mouse IgG (Southern Biotech, Birmingham, AL) for 15 min at room temperature. Washed Protein G Mag Sepharose beads (GE Healthcare, Piscataway, NJ) were added to the supernatants and incubated at 4ºC overnight. The IPs were washed with lysis buffer, eluted in Laemmli buffer, and heated for 10 min at 96ºC. IPs or whole-cell lysates were separated in a NuPAGE Bis-Tris polyacrylamide gel (Invitrogen) and transferred to polyvinylidene fluoride membranes (GE Healthcare). Antibodies used were rabbit anti-nsP3, rabbit anti–G3BP-1, mouse anti–G3BP-1, rabbit anti–G3BP-2, mouse anti-GFP (BD Biosciences, San Diego, CA), and goat anti-actin (Santa Cruz Biotechnology). Chemiluminescence was detected using the ECL reagents (GE Healthcare).
Supplementary Material
Acknowledgments
We thank Marcus Ström and Max Riess for technical assistance. pEGFP-G3BP-1 and Pat A were kindly provided by Nancy Kedersha (Harvard Medical School) and Jerry Pelletier (McGill University), respectively. M.P. gratefully acknowledges a Karolinska Institutet Doktorand studentship. This work was supported by grants from the Swedish Research Council to G.M.M. and to G.K.H. and by Estonian Science Foundation Grant 9400 and the European Union through the European Regional Development Fund via the Center of Excellence in Chemical Biology to A.M.
Abbreviations used:
- BHK
baby hamster kidney cells
- eIF
eukaryotic initiation factor
- G3BP
Ras-GAP SH3 domain-binding protein
- MOI
multiplicity of infection
- MEF
mouse embryonic fibroblasts
- nsP
nonstructural protein
- Pat A
pateamine A
- RC
replication complex
- SG
stress granule
- SFV
Semliki Forest virus
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-08-0619) on October 19, 2012.
REFERENCES
- Ahola T, Lampio A, Auvinen P, Kaariainen L. Semliki Forest virus mRNA capping enzyme requires association with anionic membrane phospholipids for activity. EMBO J. 1999;18:3164–3172. doi: 10.1093/emboj/18.11.3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhrymuk I, Kulemzin SV, Frolova EI. Evasion of innate immune response: the Old World alphavirus nsP2 protein induces rapid degradation of Rpb1, a catalytic subunit of RNA polymerase II. J Virol. 2012;86:7180–7191. doi: 10.1128/JVI.00541-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P, Kedersha N. Stress granules. Curr Biol. 2009;19:R397–R398. doi: 10.1016/j.cub.2009.03.013. [DOI] [PubMed] [Google Scholar]
- Atasheva S, Gorchakov R, English R, Frolov I, Frolova E. Development of Sindbis viruses encoding nsP2/GFP chimeric proteins and their application for studying nsP2 functioning. J Virol. 2007;81:5046–5057. doi: 10.1128/JVI.02746-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckham CJ, Parker R. P bodies, stress granules, and viral life cycles. Cell Host Microbe. 2008;3:206–212. doi: 10.1016/j.chom.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordeleau ME, Cencic R, Lindqvist L, Oberer M, Northcote P, Wagner G, Pelletier J. RNA-mediated sequestration of the RNA helicase eIF4A by pateamine A inhibits translation initiation. Chem Biol. 2006;13:1287–1295. doi: 10.1016/j.chembiol.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Bourai M, et al. Mapping of Chikungunya virus interactions with host proteins identified nsP2 as a highly connected viral component. J Virol. 2012;86:3121–3134. doi: 10.1128/JVI.06390-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breakwell L, Dosenovic P, Karlsson Hedestam GB, D'Amato M, Liljeström P, Fazakerley J, McInerney GM. Semliki Forest virus nonstructural protein 2 is involved in suppression of the type I interferon response. J Virol. 2007;81:8677–8684. doi: 10.1128/JVI.02411-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristea IM, Carroll JW, Rout MP, Rice CM, Chait BT, MacDonald MR. Tracking and elucidating alphavirus-host protein interactions. J Biol Chem. 2006;281:30269–30278. doi: 10.1074/jbc.M603980200. [DOI] [PubMed] [Google Scholar]
- Cristea IM, Rozjabek H, Molloy KR, Karki S, White LL, Rice CM, Rout MP, Chait BT, MacDonald MR. Host factors associated with the Sindbis virus RNA-dependent RNA polymerase: role for G3BP1 and G3BP2 in virus replication. J Virol. 2010;84:6720–6732. doi: 10.1128/JVI.01983-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang Y, Kedersha N, Low WK, Romo D, Gorospe M, Kaufman R, Anderson P, Liu JO. Eukaryotic initiation factor 2alpha-independent pathway of stress granule induction by the natural product pateamine A. J Biol Chem. 2006;281:32870–32878. doi: 10.1074/jbc.M606149200. [DOI] [PubMed] [Google Scholar]
- Emara MM, Brinton MA. Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc Natl Acad Sci USA. 2007;104:9041–9046. doi: 10.1073/pnas.0703348104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolova E, Gorchakov R, Garmashova N, Atasheva S, Vergara LA, Frolov I. Formation of nsP3-specific protein complexes during Sindbis virus replication. J Virol. 2006;80:4122–4134. doi: 10.1128/JVI.80.8.4122-4134.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fros JJ, Domeradzka NE, Baggen J, Geertsema C, Flipse J, Vlak JM, Pijlman GP. Chikungunya virus nsP3 blocks stress granule assembly by recruitment of G3BP into cytoplasmic foci. J Virol. 2012;86:10873–10879. doi: 10.1128/JVI.01506-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garmashova N, Gorchakov R, Frolova E, Frolov I. Sindbis virus nonstructural protein nsP2 is cytotoxic and inhibits cellular transcription. J Virol. 2006;80:5686–5696. doi: 10.1128/JVI.02739-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorchakov R, Garmashova N, Frolova E, Frolov I. Different types of nsP3-containing protein complexes in Sindbis virus-infected cells. J Virol. 2008;82:10088–10101. doi: 10.1128/JVI.01011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley LL, McGivern DR, Teng MN, Djang R, Collins PL, Fearns R. Roles of the respiratory syncytial virus trailer region: effects of mutations on genome production and stress granule formation. Virology. 2010;406:241–252. doi: 10.1016/j.virol.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iseni F, Garcin D, Nishio M, Kedersha N, Anderson P, Kolakofsky D. Sendai virus trailer RNA binds TIAR, a cellular protein involved in virus-induced apoptosis. EMBO J. 2002;21:5141–5150. doi: 10.1093/emboj/cdf513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CT, et al. Real-time imaging of hepatitis C virus infection using a fluorescent cell-based reporter system. Nat Biotechnol. 2010;28:167–171. doi: 10.1038/nbt.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsafanas GC, Moss B. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe. 2007;2:221–228. doi: 10.1016/j.chom.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Chen S, Gilks N, Li W, Miller IJ, Stahl J, Anderson P. Evidence that ternary complex (eIF2-GTP-tRNA(i)(Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol Biol Cell. 2002;13:195–210. doi: 10.1091/mbc.01-05-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Cho MR, Li W, Yacono PW, Chen S, Gilks N, Golan DE, Anderson P. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J Cell Biol. 2000;151:1257–1268. doi: 10.1083/jcb.151.6.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liljeström P, Garoff H. A new generation of animal cell expression vectors based on the Semliki Forest virus replicon. Biotechnology (NY) 1991;9:1356–1361. doi: 10.1038/nbt1291-1356. [DOI] [PubMed] [Google Scholar]
- Liljeström P, Lusa S, Huylebroeck D, Garoff H. In vitro mutagenesis of a full-length cDNA clone of Semliki Forest virus: the small 6,000-molecular-weight membrane protein modulates virus release. J Virol. 1991;65:4107–4113. doi: 10.1128/jvi.65.8.4107-4113.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd RE. How do viruses interact with stress-associated RNA granules. PLoS Pathog. 2012;8:e1002741. doi: 10.1371/journal.ppat.1002741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lulla A, Lulla V, Merits A. Macromolecular assembly-driven processing of the 2/3 cleavage site in the alphavirus replicase polyprotein. J Virol. 2012;86:553–565. doi: 10.1128/JVI.05195-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews JD, Frey TK. Analysis of subcellular G3BP redistribution during rubella virus infection. J Gen Virol. 2012;93:267–274. doi: 10.1099/vir.0.036780-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInerney GM, Kedersha NL, Kaufman RJ, Anderson P, Liljeström P. Importance of eIF2α phosphorylation and stress granule assembly in alphavirus translation regulation. Mol Biol Cell. 2005;16:3753–3763. doi: 10.1091/mbc.E05-02-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuvonen M, Ahola T. Differential activities of cellular and viral macro domain proteins in binding of ADP-ribose metabolites. J Mol Biol. 2009;385:212–225. doi: 10.1016/j.jmb.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuvonen M, Kazlauskas A, Martikainen M, Hinkkanen A, Ahola T, Saksela K. SH3 domain-mediated recruitment of host cell amphiphysins by alphavirus nsP3 promotes viral RNA replication. PLoS Pathog. 2011;7:e1002383. doi: 10.1371/journal.ppat.1002383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Griffin DE. Interaction of Sindbis virus non-structural protein 3 with poly(ADP-ribose) polymerase 1 in neuronal cells. J Gen Virol. 2009;90:2073–2080. doi: 10.1099/vir.0.012682-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubach JK, Wasik BR, Rupp JC, Kuhn RJ, Hardy RW, Smith JL. Characterization of purified Sindbis virus nsP4 RNA-dependent RNA polymerase activity in vitro. Virology. 2009;384:201–208. doi: 10.1016/j.virol.2008.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salonen A, Vasiljeva L, Merits A, Magden J, Jokitalo E, Kaariainen L. Properly folded nonstructural polyprotein directs the Semliki forest virus replication complex to the endosomal compartment. J Virol. 2003;77:1691–1702. doi: 10.1128/JVI.77.3.1691-1702.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson-Holley M, Kedersha N, Dower K, Rubins KH, Anderson P, Hensley LE, Connor JH. Formation of antiviral cytoplasmic granules during orthopoxvirus infection. J Virol. 2011;85:1581–1593. doi: 10.1128/JVI.02247-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöberg EM, Suomalainen M, Garoff H. A significantly improved Semliki Forest virus expression system based on translation enhancer segments from the viral capsid gene. Biotechnology (NY) 1994;12:1127–1131. doi: 10.1038/nbt1194-1127. [DOI] [PubMed] [Google Scholar]
- Smerdou C, Liljeström P. Two-helper RNA system for production of recombinant Semliki Forest virus particles. J. Virol. 1999;73:1092–1098. doi: 10.1128/jvi.73.2.1092-1098.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamberg N, Lulla V, Fragkoudis R, Lulla A, Fazakerley JK, Merits A. Insertion of EGFP into the replicase gene of Semliki Forest virus results in a novel, genetically stable marker virus. J Gen Virol. 2007;88:1225–1230. doi: 10.1099/vir.0.82436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taupin JL, Tian Q, Kedersha N, Robertson M, Anderson P. The RNA-binding protein TIAR is translocated from the nucleus to the cytoplasm during Fas-mediated apoptotic cell death. Proc Natl Acad Sci USA. 1995;92:1629–1633. doi: 10.1073/pnas.92.5.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourriere H, Chebli K, Zekri L, Courselaud B, Blanchard JM, Bertrand E, Tazi J. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J Cell Biol. 2003;160:823–831. doi: 10.1083/jcb.200212128. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Varjak M, Zusinaite E, Merits A. Novel functions of the alphavirus nonstructural protein nsP3 C-terminal region. J Virol. 2010;84:2352–2364. doi: 10.1128/JVI.01540-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JP, Cardenas AM, Marissen WE, Lloyd RE. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe. 2007;2:295–305. doi: 10.1016/j.chom.2007.08.006. [DOI] [PubMed] [Google Scholar]
- White JP, Lloyd RE. Regulation of stress granules in virus systems. Trends Microbiol. 2012;20:175–183. doi: 10.1016/j.tim.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi Z, Fang C, Pan T, Wang J, Yang P, Yuan Z. Subproteomic study of hepatitis C virus replicon reveals Ras-GTPase-activating protein binding protein 1 as potential HCV RC component. Biochem Biophys Res Commun. 2006;350:174–178. doi: 10.1016/j.bbrc.2006.09.027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.