

Abstract
The engulfment of apoptotic cells is required for normal metazoan development and tissue remodeling. In Caenorhabditis elegans, two parallel and partially redundant conserved pathways act in cell-corpse engulfment. One pathway, which includes the small GTPase CED-10 Rac and the cytoskeletal regulator ABI-1, acts to rearrange the cytoskeleton of the engulfing cell. The CED-10 Rac pathway is also required for proper migration of the distal tip cells (DTCs) during the development of the C. elegans gonad. The second pathway includes the receptor tyrosine kinase CED-1 and might recruit membranes to extend the surface of the engulfing cell. Cbl, the mammalian homolog of the C. elegans E3 ubiquitin ligase and adaptor protein SLI-1, interacts with Rac and Abi2 and modulates the actin cytoskeleton, suggesting it might act in engulfment. Our genetic studies indicate that SLI-1 inhibits apoptotic cell engulfment and DTC migration independently of the CED-10 Rac and CED-1 pathways. We found that the RING finger domain of SLI-1 is not essential to rescue the effects of SLI-1 deletion on cell migration, suggesting that its role in this process is ubiquitin ligase-independent. We propose that SLI-1 opposes the engulfment of apoptotic cells via a previously unidentified pathway.
Author Summary
Cell death is a normal part of organismal development. When cells die, other cells engulf them. In the roundworm C. elegans, engulfment is facilitated by one pathway that rearranges the actin cytoskeleton and another that recruits membrane. Together they cause the formation of cellular extensions that surround the dead cell. Notably, little is known about how engulfment is inhibited. The cytoskeletal regulatory pathway, which also promotes cell migration, includes CED-10 and ABI-1, homologs of the actin regulators Rac and the Abi proteins, respectively. In mammals, the c-Cbl proto-oncogene interacts with Rac and Abi2 and has been shown to regulate the actin cytoskeleton, so we tested whether the C. elegans homolog of Cbl, SLI-1, regulates engulfment and cell migration. We found that SLI-1 inhibits both processes. Our analysis further showed that SLI-1 does not function by inhibiting other known engulfment proteins. Cbl proteins have ubiquitin ligase domains through which they target proteins for destruction or sequestration. Most of the known functions of Cbl proteins require that domain, but we found that SLI-1 did not require it to block engulfment and cell migration. We propose that SLI-1 inhibits engulfment and cell migration through a previously unidentified pathway.
Introduction
The engulfment of apoptotic cells requires at least two processes to occur in the engulfing cell at the interface with the dying cell. Actin cytoskeletal elements need to be reorganized and membrane needs to be recruited. Together, these two processes result in the engulfing cell surrounding the dying cell. Two conserved molecular pathways were originally identified in Caenorhabditis elegans that are required for apoptotic cell engulfment and regulate these two processes.
In the pathway for membrane recruitment, which we refer to as the CED-1 pathway, four proteins have been identified, CED-7, CED-1, CED-6 and DYN-1 (Figure 1) [1]. These proteins activate DYN-1, a C. elegans dynamin homolog [2], which might recruit membrane for engulfment; in mammalian cells dynamin promotes extension of lamellipodial membrane protrusions [3].
Figure 1. Core molecular pathways required for the engulfment of apoptotic cells.
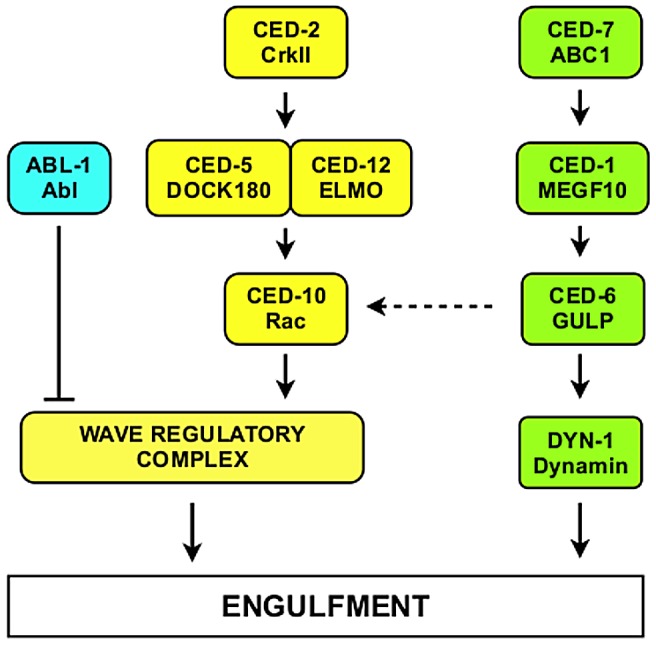
Proteins of the CED-10 Rac pathway are labeled in yellow. Proteins of the CED-1 pathway are labeled in green. C. elegans protein names are indicated above and their mammalian homologs below. The Wave Regulatory Complex (WRC) contains ABI-1, GEX-2, GEX-3, WVE-1 and NUO-3. The dashed arrow from CED-6 to CED-10 indicates that CED-1, CED-6 and CED-7 might also signal through CED-10 [6]. The CED-1 pathway is required for engulfment only, whereas the CED-10 Rac pathway is required for both engulfment and DTC migration.
The pathway for cytoskeletal rearrangement requires the small GTPase CED-10 Rac, the adapter protein CED-2 and the heterodimeric guanine nucleotide exchange factor CED-5/CED-12. CED-2 is thought to activate CED-5/CED-12, which, in turn, activates CED-10 Rac. Rac proteins are members of the Rho family of small GTPases that regulate the cytoskeleton and function in intracellular signaling [4]. CED-10 Rac activation causes actin cytoskeletal rearrangement and promotes engulfment [5], [6].
In addition to the two core engulfment pathways, more recent studies have identified a number of factors that regulate engulfment through these pathways. In C. elegans, MIG-2, the mammalian homolog of RhoG, another Rho family GTPase activates CED-5/CED-12 in parallel to CED-2 [7], [8]. The phosphatidylserine receptor PSR-1, the integrins INA-1 and PAT-3 and a WNT signaling pathway all appear to act upstream of CED-2 [9], [10]. In Drosophila, the Src protein Src42 and the non-receptor tyrosine kinase Shark act through the CED-1 Draper pathway [11]. Furthermore, Calcium release from the endoplasmic reticulum by a junctophilin-containing complex is also required for CED-1 Draper activity [12], [13].
Recently, we reported that the cytoskeletal regulatory protein ABI-1 is also an engulfment protein [14]. The mammalian homolog of ABI-1, Abi2, is found in a number of protein complexes, all of which regulate the actin cytoskeleton. One particular complex, the Wave Regulatory Complex (WRC) causes the formation of actin structures in response to activation by Rac [15], [16]. The WRC is composed of five proteins in C. elegans: WVE-1, GEX-2, GEX-3, ABI-1 and NUO-3. Soto et al. (2002) [17] and Patel et al. (2008) [18] presented evidence that suggested that GEX-2 and WVE-1, respectively, promote engulfment. Our genetic analysis, however, demonstrated that the CED-10 Rac pathway and ABI-1 act at least partially independently of each other. Our current model, based on all of these data is that the CED-10 Rac pathway activates the WRC but that there are other as yet unidentified molecular pathways that activate the WRC in parallel.
Far less studied are proteins that inhibit these two pathways. We showed that the tyrosine kinase and cytoskeletal regulator ABL-1 inhibits engulfment through ABI-1 in parallel to the CED-10 Rac pathway [14]. A small number of other proteins have been shown to inhibit apoptotic cell engulfment (compared to 25 proteins that promote engulfment). In mammalian cell culture, the small GTPase RhoA and its effector Rho-kinase have been shown to inhibit engulfment of apoptotic cells [19], consistent with the fact that RhoA and Rac oppose each other in many cellular processes. How Rho-kinase inhibits engulfment has not been demonstrated. In C. elegans, the Rac GTPase activating protein SRGP-1 inhibits engulfment by inactivating CED-10 [20]. The myotubularin lipid phosphatase MTM-1 and a CED-10 binding protein, SWAN-1, have also been shown to inhibit engulfment in C. elegans [21]–[23]. They are both proposed to act through the CED-10 Rac pathway. Recently, PGRN-1, a C. elegans progranulin has been shown to act in engulfment [24]. Notably, it is unclear how any of these proteins are regulated for their engulfment-inhibitory functions.
Cbl family proteins are E3 ubiquitin ligase and adaptor proteins with multiple cellular functions [25]. Cbl proteins consist of an N-terminal tyrosine kinase binding (TKB) domain followed by a conserved linker, then a RING finger domain and a C-terminal proline rich domain. The TKB domain is comprised of three subdomains: a 4-helix bundle, an EF hand and a modified SH2 domain. The crystal structure of the TKB domain has revealed that the three subdomains act together to bind to phosphotyrosines [26] and orient substrate proteins (usually tyrosine kinases) to allow the RING finger to promote their ubiquitination, targeting them for destruction or sequestration. Thus a major function of Cbl proteins is to downregulate signaling pathways in response to interactions with tyrosine phosphorylated signaling proteins [27]. Recent data show that Abi proteins are activated by epidermal growth factor (EGF) signaling and then in turn activate c-Cbl to polyubiquitinate the EGF receptor in a negative feedback regulatory loop [28]. In C. elegans, the Cbl homolog SLI-1 downregulates EGF signaling by causing ubiquitination of the LET-23 EGFR [29], [30], which decreases signaling from the downstream Ras homolog LET-60. Cbl has also been shown to interact with Rac, the CED-2-related protein Crk and Abl kinase [31]–[33]. We hypothesized that SLI-1 might act in engulfment pathways. In addition, we asked whether it did so by interacting with the C. elegans homologs of the above proteins.
We now present evidence that SLI-1 inhibits apoptotic cell engulfment. Surprisingly, we find that SLI-1 does so in parallel to the two core engulfment pathways and ABL-1 and independent of LET-60 Ras signaling. Lastly, we demonstrate that the ubiquitin ligase domain is partially dispensable for this process demonstrating that its tyrosine kinase-ubiquitinating function is unrelated to its mechanism of action in engulfment.
Results
SLI-1 inhibits apoptotic cell corpse engulfment
In animals with defects in apoptotic cell engulfment, the number of unengulfed corpses in the heads of first larval stage (L1) animals increases with the strength of the engulfment defect and defines a quantitative assay of engulfment defects [34]. L1 wild-type (N2) animals have no unengulfed corpses in their heads. Neither do animals with sli-1 mutations alone. We used two alleles of sli-1 in this study, sy143 and n3538 [35], [36]. sy143 is a C to T transition that changes Gln152 to an amber stop codon; n3538 is a C to T transition that changes Ser305 to Leu. To assess whether sli-1 modulates apoptotic cell engulfment, we tested whether sli-1 mutations suppressed or enhanced the engulfment defects of engulfment pathway genes.
The heads of animals containing a mutation in sli-1 and null mutations in ced-1 or ced-7 or a strong mutation in ced-6 (alleles e1735, n1996 and n2095, respectively) had fewer unengulfed corpses than those with each of the engulfment mutants alone (Table 1). We did not test dyn-1 mutants because they die during embryogenesis. Thus, SLI-1 appears to inhibit the engulfment of apoptotic cells. Alternative explanations for the effect of SLI-1 on these engulfment defects are presented in the next section of the paper. The fact that loss of sli-1 function suppresses the engulfment defects caused by null ced-1, ced-6 and ced-7 mutation demonstrates that SLI-1 acts in parallel to or downstream of the CED-1 pathway.
Table 1. sli-1 mutations suppress the engulfment defects caused by engulfment ced gene mutations.
| Genotype | Corpses ± s.d. | n | p-Value |
| N2 | 0.05±0.23 | 19 | |
| sli-1(sy143) | 0.16±0.37 | 24 | >0.2 |
| sli-1(n3538) | 0.04±0.2 | 25 | >0.2 |
| ced-1(n2091) | 18.7±4.0 | 23 | |
| ced-1(n2091); sli-1(sy143) | 7.7±3.3 | 15 | <0.0001 |
| ced-1(e1735) | 25.3±5.0 | 23 | |
| ced-1(e1735); sli-1(sy143) | 17.8±4.9 | 22 | <0.0001 |
| ced-1(e1735); sli-1(n3538) | 17.5±3.8 | 21 | <0.0001 |
| ced-2(n5101) | 15.8±4.9 | 25 | |
| ced-2(n5101); sli-1(sy143) | 18.5±6.9 | 26 | <0.06 |
| ced-5(n1812) | 30.4±4.9 | 23 | |
| ced-5(n1812); sli-1(sy143) | 34.2±5.9 | 25 | <0.01 |
| ced-5(n1812); sli-1(n3538) | 30.6±4.9 | 18 | >0.2 |
| ced-6(n2095) | 26.5±4.1 | 24 | |
| ced-6(n2095); sli-1(sy143) | 14.2±4.6 | 15 | <0.0001 |
| ced-6(n2095); sli-1(n3538) | 10.2±3.3 | 18 | <0.0001 |
| ced-7(n1996) | 32.1±4.0 | 20 | |
| ced-7(n1996); sli-1(sy143) | 23.5±6.6 | 26 | <0.0001 |
| ced-10(n1993) | 20.0±3.9 | 24 | |
| ced-10(n1993); sli-1(sy143) | 14.1±6.5 | 27 | <0.0001 |
| ced-10(n1993); sli-1(n3538) | 13.8±5.3 | 25 | <0.0001 |
| ced-12(tp2) | 20.8±3.7 | 22 | |
| ced-12(tp2); sli-1(sy143) | 17.3±4.7 | 17 | <0.008 |
| ced-12(tp2); sli-1(n3538) | 20.3±6.0 | 25 | >0.2 |
| ced-12(n3261) | 25.2±5.2 | 20 | |
| ced-12(n3261); sli-1(sy143) | 26.9±5.0 | 21 | >0.2 |
First larval stage (L1) worms were anaesthetized and viewed using DIC microscopy. The numbers of cell corpses in the heads were counted. s.d., standard deviation.
Loss of sli-1 function did not suppress the engulfment defects of null mutations in the CED-10 Rac pathway. Specifically, the engulfment defects of ced-2(n5101), ced-5(n1812) and ced-12(n3261) null mutants were not significantly modified by the presence of sli-1(sy143) or sli-1(n3538) mutations (Table 1). ced-10 null mutants die during embryogenesis but we tested the effect of sli-1 mutations on a partial loss-of-function allele, ced-10(n1993). ced-10(n1993) was suppressed by sli-1(lf) (for sy143, a decrease from 20.0 to 14.1 unengulfed corpses, p<0.0001; for n3538, a decrease to 13.8, p<0.0001). Suppression of a ced-10 partial loss-of-function defect by sli-1 mutations is consistent with a general inhibition of engulfment by sli-1 but suppression of a partial loss-of-function mutation cannot be used to order genes within genetic pathways. In summary, sli-1(lf) was not able to suppress the engulfment defects caused by complete loss-of-function CED-10 pathway mutants. Thus, the CED-10 Rac pathway is unlikely to act by inhibiting SLI-1; rather, SLI-1 acts either parallel to or upstream of the CED-10 Rac pathway.
SLI-1 does not affect the cell-death process directly
sli-1 mutation might decrease the number of unengulfed cell corpses in engulfment mutants in a number of ways other than by suppressing apoptotic cell engulfment. sli-1 mutation could (1) decrease programmed cell death, resulting in fewer cell corpses as is seen in ced-3 caspase mutants [37], (2) alter the timing of corpse appearance during development like the protein CED-8 [38], resulting in fewer corpses at the time of observation, (3) alter cell-corpse morphology so that they could not be identified by DIC microscopy as corpses or, (4) cause the corpses to be unstable and lost rapidly.
To address whether sli-1 normally prevents programmed cell death, we determined whether cells that are known to die by apoptosis normally during development do so in sli-1 mutants. 16 cells undergo programmed cell death in the anterior pharynx during embryogenesis in wild-type animals [39]. The nuclei of these cells are identified easily using DIC microscopy [34]. Mutations in genes that normally cause cell death, such as ced-3 or ced-4, have up to 14 extra recognizable cell nuclei in the anterior pharynx [34], [40]. sli-1(sy143) animals had no more nuclei than wild-type animals in their anterior pharynges (Table 2, sli-1 mutation does not block cell death in the pharynx). To test for apoptosis defects more stringently, we observed whether sli-1 mutation enhanced the death defect of a partial loss-of-function ced-3 mutant (n2427) [41]. We observed no difference between ced-3(n2427) and ced-3(n2427); sli-1(sy143) animals (1.6 vs. 1.1 extra cells, Table 2, sli-1 mutation does not block cell death in the pharynx).
Table 2. sli-1 mutation and programmed cell death.
| sli-1 mutation does not block cell death in the pharynx | |||
| Genotype | Extra cells ± s.d. | n | p-Value |
| N2 | 0.1±0.3 | 13 | >0.2 |
| sli-1(sy143) | 0.1±0.3 | 13 | |
| ced-3(n2427) a | 1.6±1.4 | 13 | >0.2 |
| ced-3(n2427); sli-1(sy143) a | 1.1±0.8 | 13 | |
Third larval stage (L3) worms were anaesthetized and viewed using DIC microscopy. The numbers of extra cell nuclei in the anterior pharynges were counted.
These strains contained nIs96 [lin-11p::gfp] V.
We used time-lapse DIC microscopy to assess whether sli-1 loss-of-function affected the timing, persistence or morphology of cell corpses. The development of wild-type and sli-1(sy143) animals was recorded for approximately 150 minutes. We found that sli-1(sy143) animals developed on average more slowly than wild-type animals. To account for the difference in the rate of development, we counted the number of cell deaths that occurred from the first cell death up to the comma stage. sli-1(sy143) worms take approximately 31 minutes longer than wild-type animals to develop to that stage at 20°C (144 minutes compared to 103 minutes). During this time, approximately 60–65 cell corpses appear in the wild-type animal. The number of cell corpses that appeared and when they appeared in wild-type and sli-1(sy143) embryos did not differ significantly (Figure 2A). However, the timing of appearance approaches statistical significance (p value = 0.053), probably related to the difference in developmental speed. The length of time that corpses persisted was similar in wild-type and sli-1(sy143) animals (Figure 2B). In addition, apoptotic cell corpses in wild-type and sli-1(sy143) animals looked similar (Figure 2C). We conclude that the morphology and time of appearance of apoptotic cell corpses is not affected by sli-1 mutation.
Figure 2. sli-1 does not affect the timing of deaths or morphology of cell corpses.

(A) The number and time of appearance of apoptotic cell corpses that occurred from the first cell death to the comma stage were recorded at 3-min intervals in wild-type and sli-1(sy143) animals using time-lapse DIC microscopy (see Materials and Methods). Mean numbers of corpses at each time point were calculated from three embryos for both wild-type and sli-1(sy143) animals. Statistical analysis was by the Wilcoxon rank-sum test. The curves are similar (p>0.05). (B) The duration of cell-corpse visibility is similar in wild-type and sli-1(sy143) embryos. The percentage of cell corpses visible for a given period was recorded. The duration of appearance of all cell corpses recorded from three wild-type (n = 159 cell corpses) and three sli-1(sy143) (n = 154) embryos was analyzed. Statistical analysis was by the Wilcoxon rank-sum test. The curves are similar (p>0.5). (C) The morphology of cell corpses in wild-type and sli-1(sy143) embryos are similar. Arrowheads, apoptotic corpses. Embryos were at similar stages of development, approximately six minutes prior to the comma stage. Bar = 5 microns.
sli-1 is broadly expressed during embryogenesis
To study the expression pattern of SLI-1, we expressed gfp under control of the sli-1 promoter. Specifically, we fused the 5000 bp 5′ of the sli-1 ATG to gfp and injected that construct into wild type (N2) worms. Fluorescence was seen broadly throughout the embryo beginning prior to gastrulation and continuing through the L1 stage (Figure S1). Higher levels of expression were seen in cells that would form the head beginning at approximately the 1½ fold stage of embryonic development. This pattern continued through the first larval stage with L1 animals showing GFP expression at high levels in the head and at lower levels throughout the body, including in body wall muscles, hypodermis, intestine, anal depressor muscles, and several neurons. During later larval development expression is seen in the distal tip cells (DTCs) (Figure S1 panel ix). In adults, GFP was found in the head, body wall muscles, hypodermis, DTCs and some neurons. This expression pattern is consistent with our results; we observe sli-1-dependent phenotypes in the heads and DTCs (see next section in results).
We also generated a translational fusion with sli-1 containing a C-terminal gfp expressed under control of the sli-1 promoter. This transgene was injected as an extrachromosomal array into ced-10(n1993); sli-1(sy143) animals. In animals in which high levels of GFP were observed, the animals invariably died during embryogenesis with bizarre morphological defects, indicating that overexpression of sli-1 is toxic to worms (Figure 3B). However, in animals with low levels of SLI-1::GFP expression, morphological abnormalities were not seen. We found that in these low level SLI-1::GFP expressing animals, the sli-1 mutant engulfment phenotype was rescued (data not shown).
Figure 3. Expression pattern of SLI-1::GFP under control of the sli-1 promoter.

SLI-1::GFP was expressed under the control of its own promoter in an extrachromosomal array. (A) Images of morphologically normal animals expressing the SLI-1::GFP transgene. i, iii and v show fluorescence images and ii, iv and vi show accompanying DIC images (i–iv, embryos; v–vi, L4 larvae). Arrowheads in i–iv indicate positions of unengulfed cell corpses. In image iii corpses are not visible. Arrowheads in v and vi indicate the DTC. (B) Images of morphologically abnormal embryos expressing the SLI-1::GFP transgene. i, iii and v show fluorescence images and ii, iv and vi show accompanying DIC images. Arrowheads indicate positions of unengulfed cell corpses. Bar = 5 microns.
We analyzed the expression pattern of Psli-1sli-1::gfp in animals that were morphologically normal. The expression profile of this transgene was quite similar to that of the transcriptional fusion but the expression level was far lower (Figure 3A). Interestingly, SLI-1::GFP was observed surrounding cell corpses in most transgenic animals though in a small minority of cell corpses in each animal (Figure 3A, panel i). While this finding suggests that SLI-1 normally is found at the cell-cell interface at some point during the engulfment process, there are several caveats, some of which argue against and others for this interpretation. SLI-1 might not normally surround cell corpses and only does so in animals in which the SLI-1::GFP transgene is overexpressed (although we suspect that it is not overexpressed at that high a level in morphologically normal animals, as we noted above). Another fact that appears inconsistent with SLI-1 normally being present at the interface between the engulfing and engulfed cell is that most corpses seen on DIC were not surrounded by GFP haloes. However, at least two phenomena could account for the lack of more GFP haloes. First, the embryos where we could analyze unengulfed cells had comparatively low levels of SLI-1::GFP expression, which would decrease the sensitivity of the assay. Second, since SLI-1 inhibits engulfment, it might need to be removed from the cell-cell interface for engulfment to occur. Thus, SLI-1 might only surround cell corpses briefly before being relocated within the engulfing cell.
In mammals, the SLI-1 homolog Cbl is found primarily in the cytoplasm, but also at the plasma membrane and bound to the cytoskeleton [25]. In our transgenic lines in which SLI-1::GFP was overexpressed at high levels, GFP was seen preferentially at the cell periphery and less so in the cytoplasm (Figure 3B) though it is unclear if this localization is physiological given the overexpression. Furthermore, these embryos were very abnormal morphologically so conclusions regarding subcellular localization in these animals should be made very cautiously.
Because high levels of SLI-1::GFP cause embryonic lethality we were unable to test whether unengulfed cell corpses accumulated in animals with high levels of SLI-1::GFP. However we did analyze animals with low levels of SLI-1::GFP. We observed the number of unengulfed cell corpses in N2 embryos that were morphologically normal at the two-fold stage and contained the sli-1::gfp transgene and compared them to N2 animals without the transgene. Embryos that contained the transgene had 9.2 unengulfed corpses vs. 8.1 for animals without the transgene (Table S1). While this difference is statistically significant (p<0.04), it is unclear if this represents a biologically significant difference; this is not surprising since the amount of overexpression appears to be low. At hatching there was no significant difference in the number of unengulfed cell corpses between animals with or without the transgene.
sli-1 mutation suppresses other defects associated with engulfment pathway genes
Studies of C. elegans mutants partially defective in programmed cell death (such as partial loss-of-function ced-3 mutants) demonstrated that engulfment dysfunction can enhance apoptotic defects [41], [42]. These studies concluded that engulfment of dying cells promotes their apoptosis. Similar promotion of cell death by engulfment has been observed in Drosophila [43], indicating that the cell-killing effect of engulfment is evolutionarily conserved.
In partial loss-of-function ced-3 mutants, such as n2427, some of the cells fated to die will begin the dying process (based on morphological appearance) but then recover and survive. However, in animals with engulfment gene mutations as well as partial ced-3 loss-of-function mutations a much larger percentage of cells normally fated to die survive.
We compared the number of extra nuclei in the pharynges of ced-12(tp2); ced-3(n2427) and ced-12(tp2); ced-3(n2427); sli-1(sy143) animals to determine if sli-1 loss-of-function could suppress the apoptotic defect of an engulfment pathway mutation. Fewer extra nuclei were seen in animals that contained the sli-1(sy143) mutation (Table 2, sli-1 suppresses the cell-killing effect of an engulfment gene), demonstrating that SLI-1 suppresses the cell-killing promoted by engulfment genes, consistent with it engulfment suppression role.
The two distal tip cells (DTCs) migrate during development from the center of the animal outward and then back again, meeting approximately in the center of the animal. As they move, the gonads form behind them, resulting in two U-shaped gonads [44], [45]. In ced-10 Rac pathway mutants, the gonads often have extra turns or arms caused by abnormal DTC migration [46]. We tested whether sli-1 mutation suppressed the DTC migration defects of ced-10 Rac pathway mutants. Mutation of sli-1 decreased the percentage of gonadal morphology defects in all ced-10 Rac pathway mutants tested, including null ced-5 and ced-12 mutants (Figure 4). 48% of the gonads of ced-5(n1812) animals were abnormal whereas only 29% of the gonads of ced-5(n1812); sli-1(sy143) animals were abnormal (p<0.008), while the percentages of abnormal gonads in ced-12(n3261) and ced-12(n3261); sli-1(sy143) animals were 40% and 12%(p<4.9×10−6). These data demonstrate that SLI-1 inhibits DTC migration and that it does so independent of the CED-10 Rac pathway. Notably, since the CED-1 pathway has no role in DTC migration, SLI-1 appears to act in parallel to both engulfment pathways.
Figure 4. sli-1 mutation suppresses the DTC migration defects caused by all CED-10 Rac pathway gene mutations.

The gonads of young adult animals mutant for an engulfment gene with or without sli-1 mutation were observed and scored for morphology using DIC microscopy. Scoring was as described in Materials and Methods. Percentages of abnormal gonad arms are shown. More than 80 gonad arms were scored for all genotypes. p values derived using Fisher's exact test are shown.
In summary, loss of sli-1 suppresses ced-10 Rac pathway DTC migration defects but does not suppress ced-10 Rac pathway engulfment defects. At least two models could account for these findings. SLI-1 could act through one molecular pathway to inhibit apoptotic cell engulfment (e.g. the CED-10 Rac pathway) and through another molecular pathway to inhibit DTC migration. Alternatively, SLI-1 might act in a common pathway to inhibit both engulfment and migration but the relative importance of that pathway might be much greater in DTC migration than in engulfment. This difference would account for the ability of sli-1 mutation to suppress CED-10 Rac pathway DTC migration defects but not CED-10 Rac pathway engulfment defects.
For example, the CED-10 Rac pathway and another SLI-1-inhibited pathway might both promote DTC migration and either pathway alone is sufficient for normal DTC migration. If this were the case, loss of SLI-1 function would derepress the SLI-1-regulated pathway and suppress DTC migration defects even if the CED-10 Rac pathway were completely non-functional, as we observed. Engulfment, however, might be totally dependent on the CED-10 Rac pathway. In this case, even if sli-1 loss-of-function derepressed the other parallel pathway, the defect caused by loss of the CED-10 Rac pathway might not be able to be overcome by derepression of the SLI-1-regulated pathway. We favor this model (i.e. that SLI-1 acts on the same parallel pathway in both engulfment and DTC migration) both because of its parsimony and because of data we will present later in the paper (See last paragraph of the section titled SLI-1 acts independently of ABL-1).
SLI-1 probably acts in engulfing cells
To determine whether SLI-1 function is required in the dying cell or the engulfing cell, we used sli-1 mutant animals containing a sli-1 transgene that was expressed under the control of heat shock promoters (protocol adapted from Wu and Horvitz (1998) [46]). Specifically, the number of cell corpses in the heads of newly hatched worms was counted within 300 minutes of heat shock. Since all apoptotic deaths in the heads occur prior to 300 minutes before hatching, sli-1 could not be expressed in the dying cells.
Expression of sli-1 in ced-10(n1993); sli-1(sy143) animals increased the number of unengulfed corpses in L1 heads from 13.4 to 23.7 (p<1×10−4) (for comparison, ced-10(n1993) animals had 20.0 corpses (Table 1)), whereas expression of a gfp-only control transgene did not increase the number of unengulfed corpses (17.3 vs. 16.4; p>0.2) (Table 3). Notably, in the gfp-expressing animals, GFP was not seen in the cell corpses, in support of our hypothesis that the engulfed cell did not make new proteins (data not shown). Thus, expressing sli-1 outside of the engulfed cell rescues the sli-1 mutant phenotype, indicating that sli-1 acts in the engulfing cell.
Table 3. Overexpression of sli-1 reverses the effect of sli-1(sy143) on engulfment in ced-10(n1993); sli-1(sy143) animals.
| Transgene | Heat Shock | Corpses ± s.d. | n | p-Value |
| Phspgfp | − | 17.3±6.1 | 25 | >0.2 |
| Phspgfp | + | 16.4±6.7 | 23 | |
| Phspsli-1wt | − | 13.4±4.2 | 34 | <1×10−4 |
| Phspsli-1wt | + | 23.7±6.2 | 27 | |
| Phspsli-1ΔRING | − | 13.9±4.7 | 20 | <0.006 |
| Phspsli-1ΔRING | + | 20.0±6.9 | 15 |
ced-10(n1993); sli-1(sy143) embryos containing the transgenes indicated above were heat shocked at 33°C for 1 hour and then allowed to recover for 3–3.5 hours at 20°C. The numbers of cell corpses in the heads of anaesthetized first larval stage (L1) worms were counted using DIC microscopy within 0.5 h of hatching. The number of persistent corpses was determined from two independent transgenic lines for each transgene except for Phspsli-1ΔRING (see text for explanation). s.d., standard deviation.
SLI-1 acts independently of ABL-1
Five proteins have been identified in C. elegans that inhibit the engulfment of apoptotic cells. Three of them, the myotubularin lipid phosphatase MTM-1, the adapter SWAN-1 and the RacGAP SRGP-1, act through the CED-10 Rac pathway [20]–[23]. It is unknown how the C. elegans progranulin, PGRN-1, suppresses engulfment defects [24]. Genetic and biochemical data indicate that ABL-1 inhibits ABI-1 in parallel to the CED-10 Rac pathway [14].
Since abl-1 and sli-1 both act independently of the ced-10 Rac pathway, we asked whether sli-1 and abl-1 act in the same pathway. We generated triple mutant strains containing mutations in an engulfment gene and in abl-1 and sli-1 and compared the engulfment defects and DTC migration defects to those of double mutant strains containing mutations in engulfment genes and either abl-1 or sli-1. We found that the engulfment defect of the null mutant ced-1(e1735) was suppressed to a greater degree by the combination of abl-1(ok171) and sli-1(n3538) than by either mutation alone (Table 4). The same phenomenon was observed for the partial loss of function ced-10(n1993) allele. The engulfment defect of the null mutant ced-5(n1812) was not suppressed by the abl-1 or sli-1 mutations together or alone, consistent with our prior results that neither sli-1 nor abl-1 loss-of-function can suppress null defects in the ced-10 Rac pathway. The ced-6(n2095) engulfment defect was suppressed by both the abl-1(ok171) and the sli-1(n3538) alleles, but they did not enhance each other. The ced-6(n2095); sli-1(n3538) strain had 10.2 unengulfed corpses while the ced-6(n2095); abl-1(ok171) sli-1(n3538) strain had 10.4 unengulfed corpses. While it is not clear why these mutations did not enhance each other in the ced-6 mutant background, the suppression by sli-1(n3538) is very strong and we suspect that we are near the threshold of the sensitivity of the engulfment assay so that further enhancement cannot be detected despite independent effects on engulfment.
Table 4. Losses of sli-1 and abl-1 enhance each other's suppression of some engulfment gene defects.
| Genotype | Corpses ± s.d. | n | p-Value |
| ced-1(e1735) | 25.3±5.0 | 23 | |
| ced-1(e1735); sli-1(n3538) | 17.5±3.8 | 21 | 0.0049 |
| ced-1(e1735); abl-1(ok171) | 21.3±5.0 | 20 | <0.0001 |
| ced-1(e1735); abl-1(ok171) sli-1(n3538) | 14.2±4.4 | 24 | |
| ced-5(n1812) | 30.4±4.9 | 23 | |
| ced-5(n1812); sli-1(n3538) | 30.6±4.9 | 18 | >0.2 |
| ced-5(n1812); abl-1(ok171) | 28.4±4.4 | 20 | >0.2 |
| ced-5(n1812); abl-1(ok171) sli-1(n3538) | 29.7±4.8 | 22 | |
| ced-6(n2095) | 26.5±4.1 | 24 | |
| ced-6(n2095); sli-1(n3538) | 10.2±3.3 | 18 | >0.2 |
| ced-6(n2095); abl-1(ok171) | 20.2±3.5 | 20 | <0.0001 |
| ced-6(n2095); abl-1(ok171) sli-1(n3538) | 10.4±2.3 | 21 | |
| ced-10(n1993) | 20.0±3.9 | 24 | |
| ced-10(n1993); sli-1(n3538) | 13.8±5.3 | 25 | 0.0011 |
| ced-10(n1993); abl-1(ok171) | 12.1±4.4 | 20 | 0.016 |
| ced-10(n1993); abl-1(ok171) sli-1(n3538) | 8.9±4.6 | 19 |
First larval stage (L1) worms were anaesthetized and viewed using DIC microscopy. The numbers of cell corpses in the heads were counted. s.d., standard deviation. p-Values refer to comparisons between the double mutant and accompanying triple mutant strains.
For the DTC migration defect, ced-5(n1812) was suppressed by both abl-1(ok171) and sli-1(n3538) and was significantly more suppressed by the combination of the two mutations (Figure 5). By contrast, the DTC migration defect of ced-10(n1993) was suppressed so effectively by sli-1(n3538) that the addition of the abl-1(ok171) mutation did not enhance the suppression, similar to what was observed in engulfment with the ced-6(n2095)-containing strains. However, it appears that there is a trend towards increased suppression with sli-1 and abl-1 mutations together though the difference does not reach statistical significance (Figure 5).
Figure 5. Losses of sli-1 and abl-1 enhance each other's suppression of some engulfment gene DTC defects.
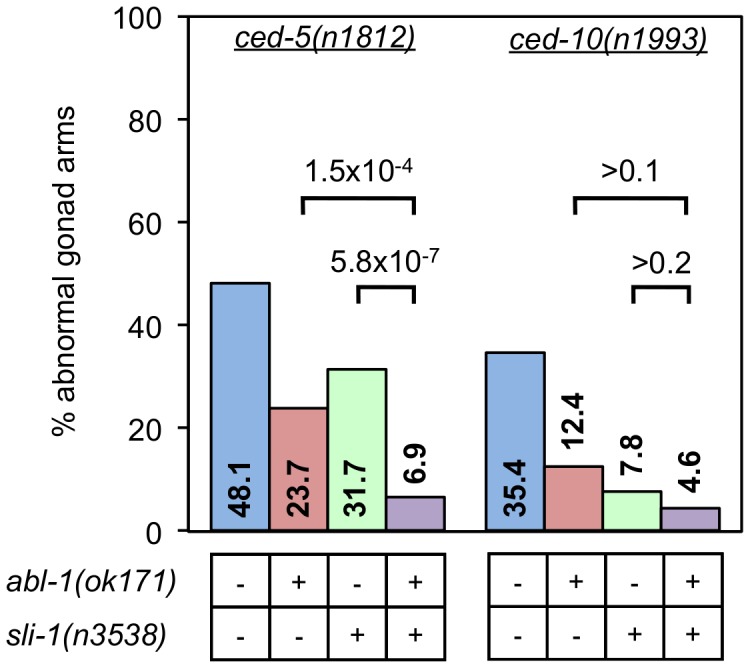
The gonads of ced-5(n1812) and ced-10(n1993) young adult worms with and without mutations in abl-1(ok171) and/or sli-1(n3538) were scored for morphology using DIC microscopy. More than 80 gonad arms were scored for all genotypes. p values derived using Fisher's exact test are shown.
abi-1 encodes the only C. elegans homolog of Abi, a member of the Wave Regulatory Complex (WRC). A combination of genetic and biochemical data suggest that ABL-1 and the CED-10 Rac pathway both act on the WRC through ABI-1 in parallel to each other: CED-10 Rac activates ABI-1 and ABL-1 inhibits it. Since SLI-1 acts in parallel to ABL-1, we asked whether it also acts on ABI-1. The only abi-1 mutations in existence (and abi-1 feeding RNAi) are quite weak and have no effect on engulfment alone but do enhance the engulfment defects of mutations in other engulfment genes. Therefore, we analyzed the effects of abi-1 mutation in combination with another engulfment mutation. Specifically, ced-1(e1735) null mutant animals containing combinations of mutations of abi-1 and/or sli-1 were assessed for the magnitude of their engulfment defects. sli-1(sy143) suppressed the engulfment defect of ced-1(e1735) animals in the presence or absence of the abi-1(tm494) mutation (Figure 6A). ced-1(e1735) L1 animals had 25.3 unengulfed corpses and ced-1(e1735); abi-1(tm494) animals had 35.0 corpses. ced-1(e1735); abi-1(tm494); sli-1(sy143) animals had 30.1 corpses. Similar findings were found for ced-5(n1812) mutants (Figure 6B). We also tested the effect of abi-1 on DTC migration using the ced-5(n1812) null mutation (Figure 6C). Similar to the findings with ced-1 in engulfment, sli-1(sy143) suppressed the DTC migration defect of ced-5(n1812) (48% vs. 29%) and sli-1(sy143) suppressed the DTC migration defect of an abi-1(tm494); ced-5(n1812) double mutant (49% vs. 26%). Thus, mutation of abi-1 did not completely suppress the effect of sli-1 on engulfment or DTC migration. abi-1(tm494) abolishes the ability of abl-1 null mutations to suppress defects in engulfment and DTC migration [14]. While these results do not prove that sli-1 acts in a different pathway from abi-1, the findings are in stark contrast to those for abl-1, since abi-1 mutation does not abrogate the effects of a sli-1 null mutation on engulfment and DTC migration. Thus, abi-1 might act independently of the WRC.
Figure 6. Loss of sli-1 function suppresses engulfment and DTC migration defects in an abi-1 loss-of-function mutant.

(A) The heads of ced-1(e1735) L1 animals with or without the sli-1(sy143) mutation and with or without treatment with abi-1 feeding RNAi were scored for engulfment defects using DIC microscopy. The mean ± standard deviation (s.d.) is indicated within each bar. Error bars are sd. p values derived using Student's t test are shown. At least 15 animals were analyzed for each genotype. (B) The heads of ced-5(n1812) L1 animals with or without the sli-1(sy143) and/or abi-1(tm494) mutations were scored as in (A). At least 15 animals were analyzed for each genotype. (C) The gonads of ced-5(n1812) young adult worms with and without mutations in sli-1(sy143) and/or abi-1(tm494) were scored for morphology using DIC microscopy. More than 80 gonad arms were scored for all genotypes. p values derived using Fisher's exact test are shown.
The finding that sli-1(sy143) suppresses the abi-1(tm494) engulfment defect in the presence of a ced-5(n1812) null mutation (Figure 6B) supports our model that sli-1 acts in parallel to the ced-10 Rac pathway rather than upstream of the ced-10 Rac pathway in engulfment. The ced-5(n1812) mutation totally inactivates the ced-10 Rac pathway. If sli-1 acted upstream of the ced-10 Rac pathway, the ced-5(n1812) mutation would block the ability of sli-1(sy143) to suppress the abi-1(tm494) engulfment defect, which we did not observe.
SLI-1 acts independently of LET-60 Ras
SLI-1 inhibits the LET-23 EGFR/LET-60 Ras pathway and is thought to do so by ubiquinating the LET-23 protein, targeting it either for destruction or sequestration [29], [30]. Mammalian Ras activates Rac. Therefore, it was plausible that SLI-1 might inhibit engulfment by suppressing the LET-23/LET-60 pathway and consequently decreasing activation of CED-10 Rac by LET-60. To test this possibility, we generated strains doubly mutant for engulfment genes and the gain-of-function mutation let-60(n1046gf). We would expect gain-of-function mutations in this pathway to suppress engulfment defects if sli-1 normally inhibits this pathway. We found no consistent effect on the number of unengulfed apoptotic cells in animals with or without the let-60(n1046gf) mutation (Figure 7). One allele of ced-12 was slightly enhanced while another allele of ced-12 and an allele of ced-2 were slightly suppressed. The only significantly modulated mutation was ced-6(n2095), which was suppressed. Possibly this effect reflects a gene- or allele-specific interaction with let-60. Regardless, this pattern does not phenocopy either sli-1 mutation. Thus, sli-1 does not appear to act through the let-23 EGFR/let-60 Ras pathway to inhibit engulfment.
Figure 7. A let-60 Ras gain-of-function mutation does not affect apoptotic cell engulfment.

SLI-1 uses the TKB domain to affect engulfment and migration
To determine which domain of SLI-1 is required for its suppression of engulfment and DTC migration defects, we ectopically expressed truncated forms of SLI-1 under control of the C. elegans heat-shock promoters in sli-1 mutant animals. The SLI-1 protein contains three domains, an N-terminal domain that binds tyrosine kinases (and several other proteins), a RING finger, which mediates its E3 ubiquitin ligase function and a C-terminal domain, which contains several proline-rich regions. Minigenes encoding wild-type sli-1 and truncation mutants of sli-1 lacking each of the three domains expressed under heat-shock promoter control were injected into ced-10(n1993); sli-1(sy143) worms. These constructs were generated previously [30] and generously provided to us by Paul Sternberg. ced-10(n1993); sli-1(sy143) larvae harboring extrachromosomal arrays were incubated for one hour at 33°C, and their gonadal morphologies were analyzed 30 hours later in young adults. The arrays contained sli-1 minigenes encoding full-length sli-1 or sli-1 lacking the N-terminus, RING finger or C-terminus (sli-1wt, sli-1ΔN, sli-1ΔRING or sli-1ΔC, respectively). Figure 8 shows that the sli-1wt construct rescued the defect completely, while sli-1ΔRING and sli-1ΔC both partially rescued the defect and the sli-1ΔN did not rescue the defect at all. We also tested the sli-1ΔRING transgene in engulfment and found that it partially rescued the engulfment suppression defect (Table 3).
Figure 8. The TKB domain of sli-1 is required for its cell migration function.

(A) Truncation minigene sli-1 constructs used in this study. Red lines represent proline-rich areas. L, conserved linker domain between the TKB domain (composed of the 4H, EF hand and SH2 domains) and the RING finger. (B) The gonads of ced-10(n1993); sli-1(sy143) young adult animals with or without transgenic arrays containing the genes shown in (A) were scored for abnormal morphology after heat shock. 200 gonad arms in two independent strains were analyzed per genotype. p values derived using Fisher's exact test are shown.
Thus, the N-terminal tyrosine kinase binding domain was strictly required for the function of sli-1 in DTC migration, whereas the RING finger and C-terminus were at least partially dispensable, suggesting that the ubiquitin ligase activity is unlikely to be central to the role of sli-1 in DTC migration. Consistent with our findings in DTC migration, the RING finger was also partially dispensable in engulfment.
Discussion
We have demonstrated that SLI-1 negatively regulates the engulfment of apoptotic cells. sli-1 inhibits the engulfment process as well as the migration of distal tip cells during gonadogenesis and the engulfment-related cell-killing process. Our genetic analysis suggests that SLI-1 acts in a manner that does not require the known engulfment pathways. Ectopic expression experiments indicate that SLI-1 acts in engulfing cells and that its function is dependent on its N-terminal tyrosine kinase binding domain. Interestingly, these experiments demonstrate that the ubiquitin ligase function of SLI-1 is at least partially dispensable.
In mammals, the SLI-1 homolog Cbl interacts physically interacts with the CED-2-related protein Crk, Abl, Abi2 and regulates the activity of the CED-10 homolog Rac [28], [31]. In addition, in both mammals and worms, SLI-1 Cbl downregulates LET-23 EGFR by ubiquitination [29]. These interactions provided the rationale for our study of SLI-1 in engulfment initially. However, we found that the effects of SLI-1 on engulfment were independent of all of these proteins (with the possible exception of ABI-1; we were unable to test an abi-1 null mutant). This finding highlights the multiple roles signaling proteins play in the regulation of complex cell biological processes. Also, these data emphasize the value of genetic analyses in discerning the physiological relevance of physical interactions discovered in vitro for a particular process.
Like many other genetic suppressors, sli-1 mutation has no effect on normal engulfment. Specifically, only two engulfment suppressors, srgp-1 and pgrn-1, have been shown to increase the rate of clearance of apoptotic cells in wild-type animals whereas abl-1, swan-1 and mtm-1 do not do so [14], [20]–[24]. Notably, the srgp-1 and pgrn-1 effects are subtle ones seen in early embryos. Possibly, the engulfment process is so efficient that derepressing it by removing inhibitors has little or no demonstrable effect. Similarly, only srgp-1 and mtm-1 cause engulfment defects when overexpressed. However, overexpression of a protein does not always result in increased activity; activation of the protein might be required, explaining the lack of overexpression phenotypes. In the case of sli-1, overexpression is toxic to worms so our ability to discern whether overexpression caused increased cell corpse accumulation was limited.
The discovery of SLI-1 as an inhibitor of engulfment adds to the small list of engulfment inhibitory proteins. Moreover, our genetic analysis puts SLI-1 into a new genetic pathway. Specifically, sli-1 loss-of-function mutations suppress the engulfment defects of ced-1 pathway null mutations and the DTC migration defects of ced-10 Rac pathway null mutations. Thus, SLI-1 could act in a molecular pathway in parallel to both the ced-1 and ced-10 Rac pathways or it might act downstream of one or both pathways. However, the ced-1 pathway has no role in DTC migration, so it is unlikely that sli-1 acts downstream of the ced-1 pathway given its effect on that process. Also, sli-1 loss-of-function mutations do not suppress the engulfment defects of ced-10 Rac pathway null mutations and therefore cannot be downstream of the ced-10 Rac pathway. Thus, the simplest model consistent with the data is that sli-1 acts in parallel to both ced-10 Rac and ced-1 pathways.
abl-1, another inhibitor of engulfment and DTC migration defects, has a very similar pattern of interactions with the two core engulfment pathways, demonstrating that it, too, acts in parallel to the ced-1 and ced-10 Rac pathways. We show that abl-1 and sli-1 act in parallel to each other in these processes as well. Thus, SLI-1 defines a new pathway of inhibition of engulfment and DTC migration.
The genetic interactions between abl-1 and abi-1 and sli-1 and abi-1 differ considerably. Whereas even very weak loss-of-function of abi-1 completely suppresses the effects of abl-1 mutations on engulfment and DTC migration, the same abi-1 mutation only minimally suppresses the effect of sli-1 on these processes. These findings are consistent with a model in which sli-1 acts independently of the Wave Regulatory Complex in engulfment and DTC migration though we cannot conclude that since abi-1 null mutants were not used in the analysis.
Most of our understanding of the function of SLI-1 comes from mammalian studies of its homolog c-Cbl in cell culture. These studies have demonstrated a large number of protein-protein interactions. To discover which of these interactions might be relevant to the engulfment inhibitory function of sli-1, we tested which domains were required to rescue SLI-1 function. The only essential domain was the N-terminal TKB domain. While our studies do not preclude a role for the C-terminal proline-rich or RING finger domains, they do indicate that these domains are not central to the engulfment and cell migration functions of SLI-1.
The TKB domain includes three motifs: a four helix bundle, a Ca++ binding EF hand and an SH2 domain. These three motifs together define a unique domain that binds phosphotyrosines of protein tyrosine kinases [26]. This binding, in turn, allows the E3 ubiquitin ligase function of the RING finger of Cbl to ubiquitinate and target these tyrosine kinases for destruction or sequestration. However, since the RING finger domain, which is required for ubiquitination, is partially dispensable for inhibition of cell migration by SLI-1, the above mechanism cannot explain our results.
In addition to tyrosine kinases, several other proteins have been shown to interact with the N-terminal TKB domain. One of them is APS, an adapter protein that is involved in insulin signaling [47]. However there is no obvious APS homolog in C. elegans. Furthermore, APS signaling requires the C-terminus of Cbl in mammals and the phenotypes we describe only partially require the C-terminal domain. Another interactor, SLAP, the Src-like adapter protein, also binds to the N-terminus of Cbl [48]. It, too, has no obvious homolog in C. elegans.
A third TKB domain interactor is tubulin. Alpha and beta tubulin bind to the Cbl N-terminus [49], [50], and Cbl co-purifies with tubulin in B-cell lysates [51]. The idea that an interaction between SLI-1 and tubulin is involved in engulfment suppression is intriguing for several reasons. First, it would support a role for microtubules in apoptotic cell engulfment, which until now has been shown to be regulated solely by actin cytoskeletal rearrangement. Second, it would fit with our genetic findings concerning sli-1. Specifically, sli-1 inhibits both engulfment and DTC migration, two processes totally dependent on appropriate cytoskeletal regulation. Third, sli-1 appears to act in parallel to all known engulfment genes and engulfment inhibitors. That, too, would be consistent with sli-1 action affecting an entirely different molecular pathway, namely one regulating microtubules.
The discovery that sli-1 acts through a pathway in parallel to the two core engulfment pathways (ced-10 Rac and ced-1) suggests that there are still other cell biological processes involved in apoptotic cell engulfment yet to be discovered. Since the two core pathways were discovered over 20 years ago, it begs the question of why these processes were not identified previously. Possibly, defects in the unidentified processes result in embryonic lethality so they were not identified in genetic screens. Alternatively, these pathways are redundant with the core pathways and, therefore, would only be discovered in the absence of one or both of them. Regardless of the answer, the existence of other pathways suggests that very tight control of engulfment is required during development.
Much of the work on engulfment has been aimed at identifying which signals from the dying cell activate the ced-10 Rac and ced-1 pathways. Our findings suggest that in addition to the need for positive signals, engulfing cells require multiple inhibitory signals to prevent inappropriate engulfment. As discussed earlier, engulfment of dying cells promotes their programmed cell deaths. Potentially there are circumstances during development when cells are particularly susceptible to engulfment-mediated death, which, unless prevented, would result in excess cell death and developmental errors. Perhaps these inhibitory pathways exist as a failsafe mechanism to prevent such errors.
Materials and Methods
Strains and genetics
C. elegans strains were maintained at 22°C as described [52]. The N2 Bristol strain was used as the wild-type strain. Animals were grown on NGM plates and fed OP50 bacteria [4], [53]. The mutations and integrants used were: LGI: ced-1(e1735, n2091), ced-12(n3261, tp2); LGIII: abi-1(tm494), ced-6(n2095), ced-7(n1996); LGIV: ced-2(n5101), ced-3(n2427), ced-5(n1812), ced-10(n1993), dpy-13(e184sd), let-60(n1046gf); LGV: unc-76(e911), nIs96 [41]; LGX: abl-1(ok171), nIs106 [41], sli-1(n3538, sy143). Mutant alleles for which no citation is given were described previously [54]. Information about ok and tm alleles can be found at www.wormbase.org (tm alleles were kindly provided by S. Mitani, Tokyo Women's Medical University, Japan). The following balancer chromosomes were used: LGI; LGIII: hT2[qIs48], LGII: mIn1[mIs14], LGIV; LGV: nT1[qIs51].
We isolated ced-2(n5101) from a C. elegans deletion library; genomic DNA pools from the progeny of EMS or UV-TMP mutagenized animals were screened for deletions using PCR as described [55]. ced-2(n5101) removes 637 nucleotides from chromosome IV, 242 base pairs 5′ to the ced-2 ATG, the entire first exon (439 bp) and 12 bp of the first intron.
Quantitation of engulfment defects
Unengulfed apoptotic corpses were visualized in the heads of young larvae as refractile discs directly using Nomarski differential interference contrast (DIC) microscopy [56], [57]. Apoptotic cell corpses were counted in the heads of first larval stage (L1) animals within 30 min of hatching, except for animals treated with RNAi (see below). Animals were anaesthetized in 30 mM sodium azide in M9 [53] and viewed using DIC optics on a Zeiss Inverted Axio Observer compound microscope (Thornwood, NY, USA). For animals treated with feeding RNAi, L1 animals were picked, and those with gonads that had not passed the 4-cell stage (all within 60 minutes of hatching) were viewed as described above. p values for pairwise comparisons were calculated using the Student's t test.
Quantitation of cell-death defects
For quantitation of cell-death defects in the anterior pharynx, animals in the third larval stage (L3) were anaesthetized and viewed with DIC microscopy as described above. Briefly, the locations of the nuclei of the 16 cells that undergo programmed cell death in the anterior pharynx are known [39]. In wild-type animals by the L3 stage, all of those nuclei have disappeared; any nuclei in these locations in the animals examined at the L3 stage were scored as extra cells. p values for pairwise comparisons in the pharynges were calculated using Student's t test.
Time-lapse microscopy
Single embryos were placed on agar pads, sealed with petroleum jelly and viewed at 20°C using a Zeiss Inverted Axio Observer compound microscope equipped with Nomarski DIC accessories, a Zeiss AxioCam HRm digital camera and Zeiss Axiovision image acquisition software. Pictures were taken every 3 min for 200 min, and images were analyzed beginning with the appearance of the first cell corpse and ending at the comma stage. The time of appearance of each corpse was recorded. For each time point, 60–80 serial z sections at 0.4 µm/section were recorded. Images were analyzed with ImageJ64 1.45 s (http://imagej.nih.gov/ij) using the plugin Cell Counter. p values for comparisons between strains were calculated using the Wilcoxon rank-sum test.
Quantitation of DTC defects
Adult animals 18 h after the mid-fourth larval stage (L4) were anaesthetized and viewed as described above in Quantitation of engulfment defects and gonads were visualized [44], [58]. Only gonads that were completely visualized were scored. Specifically, gonads that were partially occluded by other structures were not scored. DTC migration was scored as defective when the gonad was morphologically abnormal (extra turn, two arms or bizarre twists) or when the gonad was short or long. Gonadal length was defined as abnormal when the gonad tip was distal to the ipsilateral spermatheca (short) or distal to the contralateral spermatheca (long). The vast majority of abnormalities were in morphology rather than in length. p values for pairwise comparisons were calculated using Fisher's exact test.
Expression analysis of sli-1
For the transcriptional GFP fusion, a PCR product encoding the 5 kb genomic fragment upstream of the M02A10.3a (sli-1) start site was made with SalI/XbaI ends. The product was then digested with SalI and XbaI and ligated to pPD95.75 from the Fire Lab C. elegans kit (Addgene). The resulting plasmid contained the 5 kb upstream of M02A10.3a adjacent to gfp (GFP[S65C]). The plasmid was injected into gonads of N2 animals with the coinjection marker Punc-122::rfp (50 ng/µl for each with 50 ng/µl 1 Kb Plus DNA Ladder (Invitrogen) to a total concentration 150 ng/µl). Three independent transgenic lines were observed and photographed using fluorescence and DIC microscopy. For the translational GFP fusion, we used in vivo recombination (http://wormbook.org/chapters/www_reportergenefusions/reportergenefusions.html). Fosmid WRM0611cB12 was digested with MscI and SpeI, generating a 9 kb fragment which includes 5 kb of sequence upstream of M02A10.3a and 4 kb of the M02A10.3a sequence. To make the second fragment, a 5 kb full length M02A10.3a sequence was PCR amplified from fosmid WRM0611cB12 and then inserted into vector pDEST-MB14 using the Gateway method (Invitrogen), resulting in an in-frame fusion of M02A10.3a with GFP at its C-terminus. Then this plasmid was cut with PstI and SacII, making a 6 kb fragment including the C-terminal 4.5 kb of M02A10.3a fused with gfp and some additional sequence from pDEST-MB14. The 2 fragments were mixed with the co-injection marker Pmyo-2::rfp (50 ng/µl for each with 50 ng/µl 1 Kb Plus DNA Ladder to a total concentration 200 ng/µl) and injected into the gonads of ced-10(n1993);sli-1(sy143) animals. Three independent transgenic lines were analyzed. All lines demonstrated rescue of the sli-1 engulfment suppression defect.
sli-1 rescue
Phspsli-1wt, Phspsli-1ΔN, Phspsli-1ΔRING, and Phspsli-1ΔC were described previously [30]. Briefly, sli-1ΔN encodes the first 64 amino acids of SLI-1 followed by a short linker (Leu Ala Leu) and then amino acid (aa) 350 through the end of the protein (aa 583). sli-1ΔRING encodes the first 393 amino acids followed by the following linker (Glu Thr Gly Thr Thr Phe Glu) and then amino acid 432 through the end of the protein. sli-1ΔC encodes the first 447 amino acids of the protein. The Phspgfp plasmids have been described previously [59]. Each minigene was expressed under the control of the hsp16/2 and hsp16/41 promoters. Phsp plasmids were injected into ced-10(n1993); sli-1(sy143) animals at a concentration of 20 ng/µl with a plasmid containing myo-2::rfp as a coinjection marker at 5 ng/µl and with 35 ng/µl of 1 Kb Plus DNA Ladder for a total concentration of 80 ng/µl per injection. The pharynges of transgenic animals were RFP-positive. For quantification of unengulfed apoptotic cell corpses, embryos were grown at 20°C, heat-shocked for one hour at 33°C, placed at 20°C for up to four hours after which cell corpses in the heads of newly hatched first larval stage (L1) animals were counted. For quantification of DTC migration defects, animals were heat shocked for one hour at 33°C and placed at 22°C for 30 hours. DTC morphology in young adults was then analyzed in an equal number of animals with and without the transgenic arrays. 200 gonad arms were analyzed per genotype. Two independent transgenic lines were analyzed for each transgene combination except for the engulfment analysis of Phspsli-1ΔRING, in which only one line was used. This was because only one of the lines produced viable L1 larvae after heat shock during embryogenesis. Attempts were made with five separate lines. We presume this line had lower expression levels based on the fact that high expression levels of SLI-1 proteins are toxic to worms. Also, the line that produced viable larvae had comparatively faint GFP staining.
RNA interference by feeding
Animals were fed bacteria that contained either the RNAi empty feeding vector L4440 [60] or an RNAi feeding vector with part of the abi-1 gene, B0336.6, cloned into it. We obtained the abi-1 feeding plasmid from Open Biosystems (Huntsville, AL, USA). The DNA sequence of the clone was determined to verify its accuracy. Feeding RNAi was performed as described [60], [61]. Briefly, bacteria were grown in liquid culture overnight and then transferred to NGM plates containing 1 mM isopropyl-D-β-thiogalactopyranoside (IPTG). Fourth-larval stage (L4) animals were placed on these plates and 24 h later were transferred to fresh plates. Progeny were tested for engulfment or DTC migration defects.
Supporting Information
Expression pattern of Psli-1::gfp. gfp was expressed under control of the sli-1 promoter. i, iii, v, vii and ix show fluorescence images and ii, iv, vi, viii and x show accompanying DIC images. i and ii, embryo at gastrulation; iii and iv, embryo at 1½-fold stage; v and vi, L1 head; vii and viii, L1 body; ix and x, L4 gonad with arrowheads showing DTC. Bar = 5 microns.
(TIF)
Overexpression of sli-1 might cause an engulfment defect in twofold embryos. Twofold embryos were viewed using DIC microscopy. The numbers of cell corpses were counted. s.d., standard deviation.
(DOCX)
Acknowledgments
We thank M. Hammarlund for suggestions concerning the manuscript.
Funding Statement
CA, SZ, ES, and MEH were supported by direct funding from the Yale Cancer Center to MEH. MEH was supported by Award Number K08CA104890 from the National Cancer Institute. HRH is an Investigator of the Howard Hughes Medical Institute. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Mangahas P, Zhou Z (2005) Clearance of apoptotic cells in Caenorhabditis elegans . Semin Cell Dev Biol 16: 295–306. [DOI] [PubMed] [Google Scholar]
- 2. Yu X, Odera S, Chuang C, Lu N, Zhou Z (2006) C. elegans Dynamin mediates the signaling of phagocytic receptor CED-1 for the engulfment and degradation of apoptotic cells. Developmental Cell 10: 743–757. [DOI] [PubMed] [Google Scholar]
- 3. Kruchten AE (2006) Dynamin as a mover and pincher during cell migration and invasion. Journal of Cell Science 119: 1683–1690. [DOI] [PubMed] [Google Scholar]
- 4. Etienne-Manneville S, Hall A (2002) Rho GTPases in cell biology. Nature 420: 629–635. [DOI] [PubMed] [Google Scholar]
- 5. Brugnera E, Haney L, Grimsley C, Lu M, Walk SF, et al. (2002) Unconventional Rac-GEF activity is mediated through the Dock180–ELMO complex. Nat Cell Biol 4: 574–582. [DOI] [PubMed] [Google Scholar]
- 6. Kinchen JM, Cabello J, Klingele D, Wong K, Feichtinger R, et al. (2005) Two pathways converge at CED-10 to mediate actin rearrangement and corpse removal in C. elegans . Nature 434: 93–99. [DOI] [PubMed] [Google Scholar]
- 7. Lundquist EA, Reddien PW, Hartwieg E, Horvitz HR, Bargmann CI (2001) Three C. elegans Rac proteins and several alternative Rac regulators control axon guidance, cell migration and apoptotic cell phagocytosis. Development 128: 4475–4488. [DOI] [PubMed] [Google Scholar]
- 8. deBakker CD, Haney LB, Kinchen JM, Grimsley C, Lu M, et al. (2004) Phagocytosis of apoptotic cells is regulated by a UNC-73/TRIO-MIG-2/RhoG signaling module and armadillo repeats of CED-12/ELMO. Curr Biol 14: 2208–2216. [DOI] [PubMed] [Google Scholar]
- 9. Wang X, Wu Y-C, Fadok VA, Lee M-C, Gengyo-Ando K, et al. (2003) Cell corpse engulfment mediated by C. elegans phosphatidylserine receptor through CED-5 and CED-12. Science 302: 1563–1566. [DOI] [PubMed] [Google Scholar]
- 10. Hsu T-Y, Wu Y-C (2010) Engulfment of apoptotic cells in C. elegans is mediated by Integrin α/SRC signaling. Current Biology 20: 477–486 doi:10.1016/j.cub.2010.01.062. [DOI] [PubMed] [Google Scholar]
- 11. Ziegenfuss JS, Biswas R, Avery MA, Hong K, Sheehan AE, et al. (2008) Draper-dependent glial phagocytic activity is mediated by Src and Syk family kinase signalling. Nature 453: 935–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cuttell L, Vaughan A, Silva E, Escaron C, Lavine M, et al. (2008) Undertaker, a Drosophila junctophilin, links Draper-mediated phagocytosis and calcium homeostasis. cell 135: 524–534. [DOI] [PubMed] [Google Scholar]
- 13. Gronski MA, Kinchen JM, Juncadella IJ, Franc NC, Ravichandran KS (2009) An essential role for calcium flux in phagocytes for apoptotic cell engulfment and the anti-inflammatory response. Cell Death Differ 16: 1323–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hurwitz ME, Vanderzalm PJ, Bloom L, Goldman J, Garriga G, et al. (2009) Abl Kinase inhibits the engulfment of apopotic cells in Caenorhabditis elegans . PLoS Biol 7: e99 doi:10.1371/journal.pbio.1000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Miki H, Suetsugu S, Takenawa T (1998) WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. EMBO J 17: 6932–6941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eden S, Rohatgi R, Podtelejnikov AV, Mann M, Kirschner MW (2002) Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature 418: 790–793. [DOI] [PubMed] [Google Scholar]
- 17. Soto MC, Qadota H, Kasuya K, Inoue M, Tsuboi D, et al. (2002) The GEX-2 and GEX-3 proteins are required for tissue morphogenesis and cell migrations in C. elegans . Genes Dev 16: 620–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Patel F, Bernadskaya Y, Chen E, Jobanputra A, Pooladi Z, et al. (2008) The WAVE/SCAR complex promotes polarized cell movements and actin enrichment in epithelia during C. elegans embryogenesis. Developmental Biology 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tosello-Trampont A-C, Nakada-Tsukui K, Ravichandran KS (2003) Engulfment of apoptotic cells is negatively regulated by Rho-mediated signaling. J Biol Chem 278: 49911–49919. [DOI] [PubMed] [Google Scholar]
- 20. Neukomm LJ, Frei AP, Cabello J, Kinchen JM, Zaidel-Bar R, et al. (2010) Loss of the RhoGAP SRGP-1 promotes the clearance of dead and injured cells in Caenorhabditis elegans . Nat Cell Biol 13: 79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zou W, Lu Q, Zhao D, Li W, Mapes J, et al. (2009) Caenorhabditis elegans myotubularin MTM-1 negatively regulates the engulfment of apoptotic cells. PLoS Genet 5: e1000679 doi:10.1371/journal.pgen.1000679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Neukomm LJ, Nicot A-S, Kinchen JM, Almendinger J, Pinto SM, et al. (2011) The phosphoinositide phosphatase MTM-1 regulates apoptotic cell corpse clearance through CED-5-CED-12 in C. elegans . Development 138: 2003–2014. [DOI] [PubMed] [Google Scholar]
- 23. Yang Y, Lu J, Rovnak J, Quackenbush SL, Lundquist EA (2006) SWAN-1, a Caenorhabditis elegans WD repeat protein of the AN11 family, is a negative regulator of Rac GTPase function. Genetics 174: 1917–1932 doi:10.1534/genetics.106.063115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kao AW, Eisenhut RJ, Martens LH, Nakamura A, Huang A, et al. (2011) A neurodegenerative disease mutation that accelerates the clearance of apoptotic cells. Proc Natl Acad Sci USA 108: 4441–4446 doi:10.1073/pnas.1100650108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Swaminathan G, Tsygankov AY (2006) The Cbl family proteins: Ring leaders in regulation of cell signaling. J Cell Physiol 209: 21–43. [DOI] [PubMed] [Google Scholar]
- 26. Meng W, Sawasdikosol S, Burakoff SJ, Eck MJ (1999) Structure of the amino-terminal domain of Cbl complexed to its binding site on ZAP-70 kinase. Nature 398: 84–90. [DOI] [PubMed] [Google Scholar]
- 27. Schmidt MHH, Dikic I (2005) The Cbl interactome and its functions. Nat Rev Mol Cell Biol 6: 907–918. [DOI] [PubMed] [Google Scholar]
- 28. Tanos B, Pendergast A (2007) Abi-1 forms an epidermal growth factor-inducible complex with Cbl: Role in receptor endocytosis. Cell Signal 19: 1602–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yoon CH, Lee J, Jongeward GD, Sternberg PW (1995) Similarity of sli-1, a regulator of vulval development in C. elegans, to the mammalian proto-oncogene c-cbl. Science 269: 1102–1105. [DOI] [PubMed] [Google Scholar]
- 30. Yoon CH, Chang C, Hopper NA, Lesa GM, Sternberg PW (2000) Requirements of multiple domains of SLI-1, a Caenorhabditis elegans homologue of c-Cbl, and an inhibitory tyrosine in LET-23 in regulating vulval differentiation. Mol Biol Cell 11: 4019–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Scaife RM, Langdon WY (2000) c-Cbl localizes to actin lamellae and regulates lamellipodia formation and cell morphology. Journal of Cell Science 113 Pt 2: 215–226. [DOI] [PubMed] [Google Scholar]
- 32. Scaife RM, Job D, Langdon WY (2003) Rapid microtubule-dependent induction of neurite-like extensions in NIH 3T3 fibroblasts by inhibition of ROCK and Cbl. Mol Biol Cell 14: 4605–4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Soubeyran P, Barac A, Szymkiewicz I, Dikic I (2003) Cbl-ArgBP2 complex mediates ubiquitination and degradation of c-Abl. Biochem J 370: 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ellis RE, Jacobson DM, Horvitz HR (1991) Genes required for the engulfment of cell corpses during programmed cell death in Caenorhabditis elegans . Genetics 129: 79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jongeward GD, Clandinin TR, Sternberg PW (1995) sli-1, a negative regulator of let-23-mediated signaling in C. elegans . Genetics 139: 1553–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ceol CJ, Stegmeier F, Harrison MM, Horvitz HR (2006) Identification and classification of genes that act antagonistically to let-60 Ras signaling in Caenorhabditis elegans vulval development. Genetics 173: 709–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ellis HM, Horvitz HR (1986) Genetic control of programmed cell death in the nematode C. elegans . Cell 44: 817–829. [DOI] [PubMed] [Google Scholar]
- 38. Stanfield GM, Horvitz HR (2000) The ced-8 gene controls the timing of programmed cell deaths in C. elegans . Mol Cell 5: 423–433. [DOI] [PubMed] [Google Scholar]
- 39. Sulston JE, Schierenberg E, White JG, Thomson JN (1983) The embryonic cell lineage of the nematode Caenorhabditis elegans . Developmental Biology 100: 64–119. [DOI] [PubMed] [Google Scholar]
- 40. Shaham S, Reddien PW, Davies B, Horvitz HR (1999) Mutational analysis of the Caenorhabditis elegans cell-death gene ced-3 . Genetics 153: 1655–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reddien PW, Cameron S, Horvitz HR (2001) Phagocytosis promotes programmed cell death in C. elegans . Nature 412: 198–202. [DOI] [PubMed] [Google Scholar]
- 42. Hoeppner DJ, Hengartner MO, Schnabel R (2001) Engulfment genes cooperate with ced-3 to promote cell death in Caenorhabditis elegans . Nature 412: 202–206. [DOI] [PubMed] [Google Scholar]
- 43. Li W, Baker N (2007) Engulfment is required for cell competition. Cell 129: 1215–1225. [DOI] [PubMed] [Google Scholar]
- 44. Hirsh D, Oppenheim D, Klass M (1976) Development of the reproductive system of Caenorhabditis elegans . Developmental Biology 49: 200–219. [DOI] [PubMed] [Google Scholar]
- 45. Kimble JE, White JG (1981) On the control of germ cell development in Caenorhabditis elegans . Developmental Biology 81: 208–219. [DOI] [PubMed] [Google Scholar]
- 46. Wu YC, Horvitz HR (1998) C. elegans phagocytosis and cell-migration protein CED-5 is similar to human DOCK180. Nature 392: 501–504. [DOI] [PubMed] [Google Scholar]
- 47. Hu J, Hubbard SR (2005) Structural characterization of a novel Cbl phosphotyrosine recognition motif in the APS family of adapter proteins. J Biol Chem 280: 18943–18949. [DOI] [PubMed] [Google Scholar]
- 48. Tang J, Sawasdikosol S, Chang JH, Burakoff SJ (1999) SLAP, a dimeric adapter protein, plays a functional role in T cell receptor signaling. Proc Natl Acad Sci USA 96: 9775–9780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fernandez JA, Keshvara LM, Peters JD, Furlong MT, Harrison ML, et al. (1999) Phosphorylation- and activation-independent association of the tyrosine kinase Syk and the tyrosine kinase substrates Cbl and Vav with tubulin in B-cells. J Biol Chem 274: 1401–1406. [DOI] [PubMed] [Google Scholar]
- 50. Teckchandani AM, Panetti TS, Tsygankov AY (2005) c-Cbl regulates migration of v-Abl-transformed NIH 3T3 fibroblasts via Rac1. Experimental Cell Research 307: 247–258. [DOI] [PubMed] [Google Scholar]
- 51. Feshchenko EA, Smirnova EV, Swaminathan G, Teckchandani AM, Agrawal R, et al. (2004) TULA: an SH3- and UBA-containing protein that binds to c-Cbl and ubiquitin. Oncogene 23: 4690–4706. [DOI] [PubMed] [Google Scholar]
- 52. Brenner S (1974) The genetics of Caenorhabditis elegans . Genetics 77: 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wood W and the C. elegans Research Community (1988) The nematode Caenorhabditis elegans. Plainview (New York): Cold Spring Harbor Laboratory Press. 667 p.
- 54.Riddle DL, Blumenthal T, Meyer BJ, Priess JR (1997) C. elegans II: Plainview (New York): Cold Spring Harbor Laboratory. 1222 p.
- 55. Ceol CJ, Horvitz HR (2001) dpl-1 DP and efl-1 E2F act with lin-35 Rb to antagonize Ras signaling in C. elegans vulval development. Mol Cell 7: 461–473. [DOI] [PubMed] [Google Scholar]
- 56. Sulston JE, Horvitz HR (1977) Post-embryonic cell lineages of the nematode, Caenorhabditis elegans . Developmental Biology 56: 110–156. [DOI] [PubMed] [Google Scholar]
- 57. Hedgecock EM, Sulston JE, Thomson JN (1983) Mutations affecting programmed cell deaths in the nematode Caenorhabditis elegans . Science 220: 1277–1279. [DOI] [PubMed] [Google Scholar]
- 58. Nishiwaki K (1999) Mutations affecting symmetrical migration of distal tip cells in Caenorhabditis elegans . Genetics 152: 985–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Reddien PW, Horvitz HR (2000) CED-2/CrkII and CED-10/Rac control phagocytosis and cell migration in Caenorhabditis elegans . Nat Cell Biol 2: 131–136. [DOI] [PubMed] [Google Scholar]
- 60. Timmons L, Fire A (1998) Specific interference by ingested dsRNA. Nature 395: 854. [DOI] [PubMed] [Google Scholar]
- 61. Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J (2001) Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans . Genome Biol 2: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expression pattern of Psli-1::gfp. gfp was expressed under control of the sli-1 promoter. i, iii, v, vii and ix show fluorescence images and ii, iv, vi, viii and x show accompanying DIC images. i and ii, embryo at gastrulation; iii and iv, embryo at 1½-fold stage; v and vi, L1 head; vii and viii, L1 body; ix and x, L4 gonad with arrowheads showing DTC. Bar = 5 microns.
(TIF)
Overexpression of sli-1 might cause an engulfment defect in twofold embryos. Twofold embryos were viewed using DIC microscopy. The numbers of cell corpses were counted. s.d., standard deviation.
(DOCX)