

Abstract
The Regulators of G protein signaling (RGS) protein superfamily negatively controls G-protein-coupled receptor (GPCR) signal transduction pathways. RGS16 is enriched in activated/effector T lymphocytes. Here, we show that RGS16 constrains pulmonary inflammation by regulating chemokine-induced T-cell trafficking in response to challenge with Schistosoma mansoni. Naïve Rgs16–/– mice were “primed” for inflammation by accumulation of CCR10+ T cells in the lung. Upon pathogen exposure, these mice developed more robust granulomatous lung fibrosis than wild-type (WT) counterparts. Distinct TH2 or putative TH17 subsets expressing CCR4 or CCR10 accumulated more rapidly in Rgs16–/– lungs following challenge and produced pro-inflammatory cytokines IL-13 and IL-17B. CCR4+ Rgs16–/– TH2 cells migrated excessively to CCL17 and localized aberrantly in challenged lungs. T lymphocytes were partially excluded from lung granulomas in Rgs16–/– mice, instead forming peribronchial/perivascular aggregates. Thus, RGS16-mediated confinement of T cells to Schistosome granulomas mitigates widespread cytokine-mediated pulmonary inflammation.
Keywords: Chemokines, G proteins, RGS proteins, Schistosoma mansoni, fibrosis, TH2, TH17, cytokines
INTRODUCTION
Chemokines dictate coordinated movement of leukocytes through lymphoid organs and sites of inflammation. Naïve and activated leukocyte populations express unique chemokine receptors, and gene-targeting studies have implicated specific chemokines and receptors in leukocyte activation, differentiation, and lymphoid organ development (1). Schistosomiasis, induced by infection with the helminth S. mansoni (among other species) represents a global disease burden due to resultant hepatic fibrosis and portal hypertension (2). S. mansoni cercariae breach host skin and develop into adult male-female worm pairs that generate hundreds of eggs per day (3, 4). Eggs then enter circulation or embolize in host tissues such as liver, intestine, and lung, where a granulomatous reaction and fibrosis develops in an effort to sequester the foreign eggs antigens (Ag) (3). In published mouse models of Schistosomiasis, the pulmonary granulomatous response is initiated by CD4+ TH2 cells and their secreted cytokines, particularly IL-13 (5). Although chemokine factors mediating TH2 recruitment to lungs acutely challenged with S. mansoni eggs have been suggested (e.g. CCL17/CCL22 and CCR4/8), signaling pathways involved in pulmonary inflammation have not been fully defined (6).
Chemokine receptors are G-protein-coupled receptors (GPCRs) linked to Gαi and possibly Gαq to induce chemotaxis (7). The primary signal transducer of GPCRs, the heterotrimeric G protein complex of α, β and γ subunits, induces pathway activation through GDP-GTP exchange on Gα and stimulation of numerous effectors including kinases, phospholipases, and ion channels (8, 9). The intrinsic GTPase activity of the α subunit, which promotes Gα re-association with βγ to form an inactive heterotrimer, terminates ligand-induced signaling. The RGS superfamily, which has more than 30 members in mammalian cells, negatively regulates G protein activity (10). All RGS proteins contain a characteristic 120 amino acid “RGS box”, which facilitates binding to Gα subunits and GTPase accelerating (GAP) activity (11). RGS GAP activity hastens GPCR pathway inactivation by catalyzing the GTPase reaction. Although molecular determinants of RGS activity have been elucidated over the past decade, most physiological functions of RGS proteins in mammals remain unknown. Gαi inactivation by pertussis toxin disrupts physiological hematopoietic cell trafficking including thymic emigration, transendothelial leukocyte migration into lymph nodes, and Ag-induced recruitment of cells to inflamed tissue (7). Because RGS proteins are physiologically relevant inhibitors of Gαi, they are poised to regulate chemokine-mediated responses in vivo (7).
RGS16 was initially discovered as an IL-2-dependent activation gene in human T lymphocytes (12). RGS16 may control TH2 lymphocyte migration in vivo since it is upregulated in activated human TH1 and TH2 cells relative to naïve T cells, and RGS16 overexpression inhibits TH lymphocyte chemotaxis in vitro (13). To explore intracellular regulation of chemokine pathways in pulmonary inflammation, we generated Rgs16–/– mice and studied their response to sensitization with S. mansoni egg antigen followed by an intravenous bolus of live S. mansoni eggs (14). These studies revealed that RGS16 inhibits TH2 chemotaxis to chemokines including CCL17, which constrains T cell localization to Schistosome egg granulomas, thereby reducing the tissue damaging effects of TH2-induced pulmonary inflammation by confining cytokines to specific regions(s) of the lung.
MATERIALS AND METHODS
Generation of Rgs16–/– mice
C57/Bl6 WT mice were purchased from Jackson laboratories. Rgs16–/– mice were generated directly onto a C57/Bl6 background as outlined in Figure 1. All mice were housed in pathogen-free conditions and research performed in accordance with protocols (LAD3e and LPD16e) approved by the National Institutes of Health Institutional Animal Care and Use Committee. Male or female mice between 6 and 12 weeks of age were used for all experiments.
Figure 1. Generation and characterization of Rgs16–/– mice.
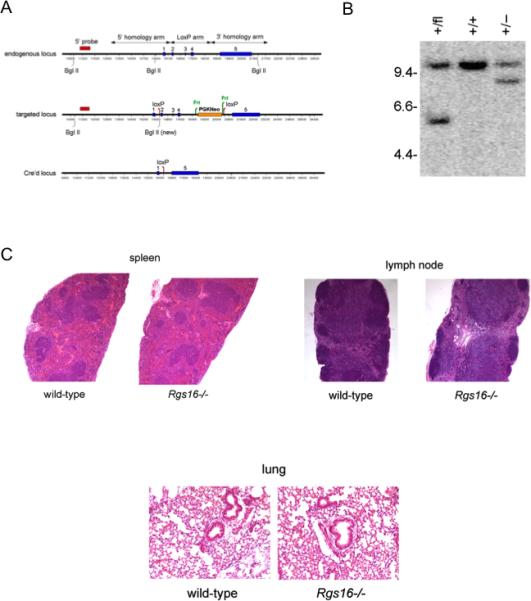
(A) A targeting vector was generated by flanking a floxed-Neor cassette (PGKNeo) by two homologous arms of the endogenous Rgs16 gene. Shown are restriction maps and exons (solid vertical bars) of the endogenous and targeted Rgs16 loci (blue bars). (B) WT and floxed alleles were identified by Southern blotting of Bgl II-digested genomic DNA with a 5’ probe (red), yielding 10.6 kb or 5.5 kb fragments, respectively (middle). Mice homozygous for the floxed allele were crossed with Rosa-Cre strain, resulting in deletion of PGKNeo and exons 2-4, which was identified as an 8.3 kb fragment. (C) Architecture of spleen, lungs, and peripheral lymph nodes (LNs) in naïve WT or Rgs16–/– mice was evaluated by H & E staining.
S. mansoni lung challenge model
Mice were sensitized intraperitoneally with 5000 S. mansoni eggs that were derived previously from sterile, lipopolysaccharide (LPS)-free Balb/c mice harboring a liver infection (3). 14 days later, mice were challenged intravenously with 5000 live eggs of a Puerto Rican strain of S. mansoni (NMRI) as described elsewhere (3). The challenged mice were euthanized at four or seven days post i.v. injection by CO2 inhalation. Lungs, spleens, and mediastinal lymph nodes (MLNs) were harvested from both groups of challenged mice for analysis.
Histopathology and immunohistochemistry
Organs were fixed in 10% neutral buffered formalin (EMD Chemicals) and embedded in paraffin. Tissue sections (5 μm) were evaluated for granuloma volume and diameter by H & E staining, and lung fibrosis was scored based on Picrosirius red stain for collagen using published scoring methods (15). Giemsa stains were utilized to quantify eosinophil numbers. The individual who scored all histological features was blinded to the experimental design. Lungs harvested at 7 days post intravenous challenge were stained for the following markers: anti-CD3 (1:100) (Dako) or anti-CCR10 (1:500) (Abcam). A polyconal anti-RGS16 antibody (Dru-4) was generated by immunizing rabbits with an N-terminal RGS16 peptide (MCRTLATFPNTC-amide) and collection of serum. The antibody was affinity purified using a peptide-conjugated resin (Spring Valley Laboratories). Slides were deparaffinized and washed twice in distilled water for 5 mins at room temperature (RT). After pre-treatment for antigen retrieval (20-40 mins, 75-90°C), the slides were washed again, and endogenous peroxidase activity was inhibited with H2O2 for 10 min at RT. Non-specific binding was blocked using 5% bovine serum albumin (BSA) in (Sigma Aldrich) in PBS for 20 min at RT. Sections were incubated with primary antibody or matched isotype controls (1:500) overnight at RT, washed and incubated with the biotinylated secondary antibody (1:500, 30 mins, RT) and finally incubated with streptavidin-horseradish peroxidase (HRP) (1:400, 30 mins, RT) (Alpha Diagnostics Ltd.). Signal was detected with 3,3'-diaminobenzidine (DAB) (1-20 mins, RT). Sections were counterstained in Carazzi hematoxylin (Histoserv, Inc.) for 2 min. at RT followed by dehydration in graded alcohols (75%, 95%, 100%) for 1 min each at RT and xylene (Sigma). Images were obtained with a Leica DMI 4000B microscope and quantified using Image Pro Plus software (Media Cybernetics).
Real-time PCR and gene expression arrays
Total RNA was isolated using the RNeasy mini kit (Qiagen), followed by DNase I treatment (Invitrogen). cDNA was synthesized from 0.25-1 μg RNA using the Superscript III First-Strand Synthesis kit (Invitrogen). Real-time reverse transcriptase-quantitative PCR (RT-QPCR) was performed using the TaqMan™ strategy (Applied Biosystems). No RT controls were included to verify DNAse digestion. Gene expression [(Rgs16 (NM_011267.3), Socs2 (NM_007706.3), Ccr10 (NM_007721.4), Ccl28 (NM_020279.3), Il13 (NM_016971.2), Il17b (NM_019508.1) or the internal reference control β-actin (NM_007393.3)] was measured by multiplex PCR using probes labeled with 6-carboxy-fluorescein (FAM). The simultaneous measurement of target gene-FAM and β-actin-FAM allowed for normalization of the amount of cDNA added per sample. Duplicate PCR reactions were performed using the TaqMan Universal PCR Master Mix and the ABI 7500 Standard™ or Step One Plus™ sequence detection systems (Applied Biosystems) according to the following thermal cycle routine: 95°C for 10 mins followed by 40 cycles of 95°C for 1 min and 60°C for 20 mins. A comparative threshold cycle (CT) method was used to determine gene expression relative to the no-sample control (calibrator). Steady-state mRNA levels were expressed as an n-fold difference relative to the calibrator. For each sample, the RGS16 CT value was normalized with the formula: ΔCT = CT RGS16 - CT β-ACTIN. To determine relative expression levels, the following formula was used: ΔΔ CT = ΔCT sample -ΔCT calibrator. Relative expression is presented as 2 -ΔΔ CT. Multiplex qPCR was performed using a mouse chemokines and receptors qPCR array (SA Biosciences) according to manufacturer's instructions.
Cytokine analysis
Single cell suspensions were prepared from mediastinal LNs harvested 7 days following i.v. challenge; 0.5 × 106 cells/well were plated in triplicate wells of 96-well round polystyrene plates (Corning) in RPMI plus 10% fetal bovine serum (Invitrogen). Cell were left untreated or stimulated with Schistosome egg antigen (SEA) from S. mansoni in PBS (10 μg/mL final concentration) for 3 days at 37°C. Cell supernatants were collected and cytokine levels were measured with a fluorescence-based Bio-Plex™ Pro mouse cytokine TH1/TH2 assay (Bio-rad) according to manufacturer's protocols. Cells were fixed by the addition of PBS containing 2% BSA and 4% paraformaldehyde (PFA) (Electron Microscopy Sciences). ELISAs were performed to quantify secretion of mouse cytokines IL-4, IL-13, IFNγ, IL-17A, or IL-17B (R&D Systems) in accordance with manufacturer's protocols.
Flow cytometry and intracellular staining
Organs were harvested 4 or 7 days post challenge and converted to single cell suspensions followed by fixation in PBS containing 2% BSA and 4% PFA. Surface markers were analyzed with the following antibodies: CD3 (17A2), B220 (RA3-6B2), CD11c (N418) (eBioscience), CD4 (RM4-5), CD8 (53-6.7), CCR3 (83103), CXCR4 (2B11) CXCR3 (CXCR3-173), CCR6 (C34-3448) (BD Biosciences), CCR4 (2G12) (Biolegend), or CCR10 (248918) (R&D Systems) and cytokines were detected with anti-IL-5 (TRFK5) (BD Biosciences), anit-IL-13 (ebio13A), anti-interferon γ (IFN-γ) (XMG1.2) (eBioscience), or anti-IL-17B (R&D Systems). For intracellular cytokine staining, cells were activated with phorbol 12-myristate 13-acetate (PMA) (50 ng/ mL) and ionomycin (0.5 μM) (Sigma) for 6 h at 37°C in the presence of brefeldin A (BD Biosciences). Cells were permeabilized with PBS/0.1% saponin (Sigma) and blocked with 5% PBS/milk (Santa Cruz Biotechnology) (15 mins, 4°C) and Fc block (BD Biosciences). Fixed, permeabilized cells were stained in the dark with fluorescently conjugated antibodies (1 h, 4°C) and washed 2X with FACS buffer. Samples were analyzed by flow cytometry using an LSR Fortessa (BD Biosciences), and data analyzed using FlowJo™ (Tree Star Inc).
In vitro TH1, TH2 or TH17 culture
Single cell suspensions were generated from peripheral LNs from naive mice. Naïve T cells (CD4+CD62L+) were isolated using the naïve T cell isolation kit II (Miltenyi Biotec). 3 × 105 cells per well were plated in 6-well plates pre-coated with anti-CD3 (1 μg/ mL) and anti-CD28 (3 μg/ mL) containing RPMI medium plus 10% FBS, recombinant mouse IL-4 (10 ng/mL) (Peprotech), anti-mouse IFNγ (10 μg/ mL) (BD Biosciences) and 50 μM 2-mercaptoethanol for 3 days at 37°C. For differentiation into a TH1 phenotype, naïve T cells were cultured in media containing IL-2 (10 ng/ mL), IL-12 (10 ng/ mL) (R&D Systems) and anti-IL-4 (10 μg/ mL) (eBioscience), referred to as the TH1 cocktail. For differentiation into a TH17 phenotype, naïve T cells were cultured in media containing IL-23 (5 ng/ mL), IL-21 (100 ng/ mL), IL-6 (10 ng/ mL), hTGFβ (5 ng/ mL), anti-IL-4 (10 μg/ mL), anti-IFNγ (10 μg/mL) (R&D Systems) and anti-IL-12 (10 μg/ mL) (BD Biosciences), referred to as the TH17 cocktail. The differentiated, activated TH1 or TH2 cells were then expanded in medium containing recombinant mouse IL-2 (10 ng/mL) and IL-7 (5 ng/ mL) (Peprotech) or in IL-2 alone for TH17 cells.
Generation and purification of TAT fusion proteins
The coding region of human RGS16 was amplified by PCR using pcDNA3.1-RGS16 as a template and subcloned in-frame into pTat-H6HA-GFP, which results in an amino-terminally tagged GFP-RGS16 (16). Recombinant TAT fusion proteins were expressed in Escherichia coli and purified by nickel-affinity chromatography as described (17). TAT proteins (TAT-GFP-RGS16 or TAT-GFP control) were added directly to T cell cultures for 2 h prior to stimulus addition.
Chemotaxis assay
Differentiated TH2 cells (0.5 × 106 cells/well) were plated in upper wells of 5 μm pore polycarbonate membrane Transwell migration chambers containing RPMI plus 0.5% BSA (Corning). The bottom wells contained chemokines (CXCL12, CCL21, CCL17, CXCL9 or CCL20) (R&D Systems). Control wells, in which upper and lower chambers had equivalent chemokine concentrations, were used to determine chemokinesis. Cells migrated to the lower chamber were counted after 3 h by flow cytometry using a FACSCalibur (BD Biosciences).
CFSE assay for analysis of cell proliferation
Cells were labeled using the CellTrace™ carboxyfluorescein diacetate succinimidyl ester (CFSE) cell proliferation kit (Invitrogen) plated in 96 well plates left untreated or pre-coated with anti-CD3/anti-CD28. Cells were harvested 4 days later and CSFE dilution profiles evaluated by flow cytometry.
Statistical analysis
Data sets were compared by student's t test for 2 groups or 1 way ANOVA for multiple groups using Graph Pad Prism software. Differences with a P value ≤ 0.05 were considered significant.
RESULTS
Generation of Rgs16–/– mice
Rgs16 was targeted by flanking exons 2 to 4 with loxP sites (Fig. 1A). We generated knockouts by crossing mice with floxed Rgs16 allele with Rosa-Cre mice, which have the gene encoding Cre recombinase inserted into the ubiquitous Rosa26 locus. Correct targeting of the Rgs16 allele was confirmed by Southern blot (Fig. 1B). Homozygous mice were viable and fertile and exhibited no gross phenotypic abnormalities. Architecture of spleen, lungs, and peripheral lymph nodes (LNs) in naïve Rgs16–/– mice was comparable to WT mice by light microscopy (Fig. 1C). Percentages of CD4+ and CD8+ T cells, B cells, and dendritic cells (DCs) in, spleen, lungs and LNs were similar in WT and Rgs16–/– mice (Table 1).
Table 1.
Cellular composition of spleens, lymph nodes or lungs of naive WT or Rgs16-/- mice.
| Tissue | Mice | CD3+ (%) | CD3+ CD4+ (%) | CD3+ CD8+ (%) | B220+ (%) | CD11c+ (%) |
|---|---|---|---|---|---|---|
| Spleen | WT* | 41.32±15.42 | 59.13±5.6 | 5.06±6.33 | 49.04±5.51 | 8.24±8.17 |
| Rgs16-/- | 48.69±5.47 | 58.39±1.55 | 6.31±6.56 | 47.8±6.59 | 4.90±4.05 | |
| Lymph node | WT | 60.27±6.32 | 55.52±4.84 | N.D. | 30.53±4.54 | N.D. |
| Rgs16-/- | 63.23±4.27 | 54.06±1.4 | N.D. | 32.08±4.62 | N.D. | |
| Lung | WT | 18.14±3.81 | 24.12±10.99 | 0.105±0.06 | 6.99±2.10 | 6.54±2.18 |
| Rgs16-/- | 23.79±10.17 | 21.06±18.24 | N.D. | 7.29±5.85 | 6.92±3.99 |
B lymphocytes, T lymphocytes, and dendritic cell percentages in lymphoid organs and lung were determined by means of flow cytometry using the indicated markers. Results are mean ± S.E.M. from 6 mice of each genotype. N.D., not done.
RGS16 inhibits TH1 and TH2 chemotaxis
To investigate function(s) of RGS16 in T-cell dependent inflammation, we examined its expression pattern in mouse effector T cells and their chemotactic responses. Compared to naïve (CD4+CD62L+) T cells isolated from peripheral LNs, activated T cells polarized to a TH2 phenotype by IL-4 and anti-IFNγ antibody expressed 5-fold more Rgs16 mRNA while those polarized to TH1 phenotype by a TH1 cocktail or TH17 phenotype by a TH17 cocktail expressed 2-fold more Rgs16 mRNA (Fig. 2A). We measured chemotaxis of naïve CD4 T cells to gradients of CCL21 and CXCL12 as these chemokines have an important function in homeostatic lymphocyte trafficking in lymphoid organs (18). Consistent with relatively low Rgs16 expression in naïve CD4 T cells, CXCL12 and CCL21 induced equivalent chemotaxis of naïve splenic CD4 T cells from unchallenged WT and Rgs16–/– mice (Fig. 2B). TH2 cells upregulate several “inflammatory” chemokine receptors not typically expressed by naïve cells including CCR4 and CCR8, which mediate trafficking to inflamed tissues containing CCL17 (19). Accordingly, in vitro differentiated TH2 cells from mice lacking RGS16 migrated much more towards CCL17 gradients than WT counterparts (Fig. 2C). At the CCL17 concentration inducing a peak response (50 nM), nearly double the number of RGS16-deficent TH2 cells than WT cells migrated to the lower chamber (~ 80% v. 40%).
Figure 2. RGS16 inhibits TH2 chemotaxis.

(A) Naïve T cells were isolated from peripheral LNs and differentiated into TH1, TH2, or TH17 cells as outlined in the Methods. Relative Rgs16 expression was measured by real-time PCR (**P = 0.005, *P < 0.01, unpaired t-test). (B) Whole splenocytes or splenic CD4 T cells C57/Bl6 and Rgs16–/– mice were exposed to chemokine gradients in transwell plates for 3 h at 37°C, followed by enumeration of migrated cells by flow cytometry. (C) Migration of TH1 or TH2 cells towards CXCL9 or CCL17 gradients, respectively, at the indicated concentrations was measured as in (B) (*P = 0.04, **P < 0.003, unpaired t-test). (D) Chemokinesis was measured by incubating cells with equimolar concentrations of CCL17 (50 nM) in the upper and lower chambers of transwell plates followed by enumeration of cells by flow cytometry. (E) Rgs16 expression in cells migrated to the lower chamber of CCL17-containing transwell plates (“migratory”) was compared to cells retained in the upper chamber (“non-migratory”) by real-time PCR (*P = 0.04, paired t-test). (F) TH2 cells from Rgs16–/– mice were left untreated or pre-incubated with TAT-GFP or TAT-RGS16 (60-500 nM) for 1 h prior to exposure to CCL17 gradients in transwell assays (***P < 0.001, 1 way ANOVA, TAT-RGS16 compared to untreated or TAT-GFP). All data are mean ± S.E.M of 3-4 independent experiments using cells from 1 mouse/group in each.
In contrast to TH2 cells, TH1 and TH17 cells express a distinct set of chemokine receptors including CXCR3 and CCR5 (TH1) or CCR6 (TH17) (20-22). Similar to the behavior of RGS16-deficient TH2 cells, TH1 cells differentiated from Rgs16–/– mice migrated significantly more towards a gradient of CXCL9 (CXCR3 ligand) than did WT counterparts (Fig. 2C). For unclear reasons, we did not observe significant chemotaxis of murine TH17 cells towards CCL20 gradients in transwell assays, independent of genotype (Supplemental Fig. 1). WT and Rgs16–/– T cells migrated to a similar extent in the presence of equivalent chemokine concentrations in the upper and lower chambers, indicating that the loss of RGS16 affected chemotaxis rather than chemokinesis (Fig. 2D). Rgs16 expression correlated with chemokine resistance. WT T cells retained in the upper chamber in the presence of a CCL17 gradient for 3 h (“non-migratory”) expressed significantly more Rgs16 than cells migrating to the lower chamber during that time period (“migratory”) (Fig. 2E). Finally, because WT and Rgs16–/– TH polarized subsets expressed similar levels of surface chemokine receptors including CXCR3 for TH1 cells, CXCR4, CCR10, or CCR4 for TH2 cells, and CCR6 for TH17 cells, these results indicated that RGS16 inhibits chemotaxis downstream of receptors (Supplemental Fig. 1). Consistent with this interpretation, reconstitution of RGS16-deficient TH2 cells with TAT-RGS16 reduced CCL17-evoked chemotaxis compared to untreated cells or cells incubated with a control TAT protein (GFP) (Fig. 2F). Collectively, these results suggest that RGS16 directs trafficking of TH1 or TH2 lymphocytes by curbing their response to chemokine.
Rgs16–/– mice have increased granulomatous lung fibrosis after challenge with S. mansoni
To evaluate TH2 trafficking in vivo in Rgs16–/– mice and its impact on an acute inflammatory response, we sensitized mice with S. mansoni eggs intraperitoneally, followed by intravenous injection of eggs 14 days later (Fig. 3A). Mice were sacrificed 4 and 7 days after the intravenous inoculation, at which time S. mansoni eggs embolize in the lungs, resulting in synchronous pulmonary inflammation characterized by granulomas, infiltration of neutrophils, eosinophils, and TH2 lymphocytes, and collagen deposition/fibrosis (23). Consistent with a role for RGS16 in the regulation of host responses to S. mansoni, Rgs16 mRNA expression was increased in lungs of sensitized, challenged mice 7 days following intravenous Schistosome challenge (Fig. 3B). We also detected RGS16 in lungs of challenged mice by immunohistochemistry (Fig. 3C). RGS16+ cells localized predominantly in granulomas surrounding lodged S. mansoni eggs. Lungs of Rgs16–/– mice developed significantly more inflammation and fibrosis than those of WT mice (Fig. 3D). Fibrosis scores (Fig. 3E), granuloma volumes (Fig. 3F), and eosinophil scores (Fig. 3G) were all higher in lungs of knockout mice compared to WT C57/Bl6 mice. These results indicated that RGS16 constrains acute, granulomatous pulmonary fibrosis induced by S. mansoni infection.
Figure 3. Enhanced pulmonary inflammation in Rgs16–/– challenged with Schistosoma mansoni.

(A) Experiment layout. (B) Rgs16 expression in naïve or S. mansoni-challenged lungs was measured by real-time PCR (*P = 0.04, unpaired t-test). (C) RGS16 expression detected by immunohistochemistry using an RGS16 antibody in lungs of WT or knockout mice as indicated. Images are representative of 3-5 mice/group. (D) Picrosirius red staining for collagen in lungs of WT or Rgs16–/– mice 7 days following inoculation with S. mansoni eggs. (E-G) Fibrosis scores (E), granuloma volumes (F), and eosinophil scores (G) were evaluated in WT and Rgs16–/– lungs 7 days after helminth challenge. Data represent 8 mice/group evaluated in 2 independent experiments (*P < 0.04, unpaired t-test).
RGS16 deficiency induces anomalous lymphocyte trafficking in vivo
To determine which lymphocyte subsets mediated the atypical fibrotic response of Rgs16–/– mice lungs to S. mansoni, we immunophenotyped pulmonary inflammatory infiltrates by means of flow cytometry. Although we detected no substantial differences in percentages or total numbers of B and T cells in lungs of challenged WT and Rgs16–/– mice, there was a skewed TH2 response in knockout mice as evidenced by increased percentages of IL-13+ T cells in lungs 4 days following helminth challenge. The ratio of IL-13+ (TH2)/ IFNγ+ (TH1) frequencies was significantly increased in Rgs16–/– lungs relative to WT (Figs. 4A-B). As TH cells from WT and Rgs16–/– mice exhibited comparable rates of differentiation, proliferation and survival in vitro (Supplemental Figure 2 and data not shown), aberrant cell growth and/or death probably did not account for the differences in cellular composition observed.
Figure 4. Aberrant TH2 migration in Rgs16–/– lungs following S. mansoni challenge.
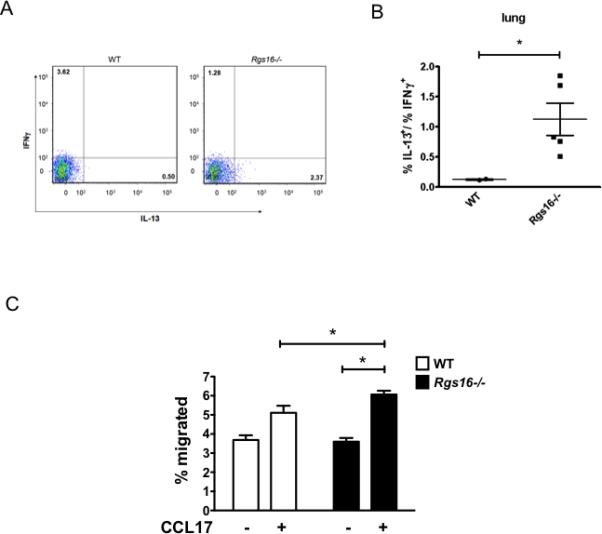
(A-B) Frequencies of IL-13+ or INFγ+ T cells (CD3+) (A) or ratio IL1-3+/IFNγ+ T cell percentages (B) in lungs 4 days following S. mansoni challenge were quantified by flow cytometry (*P = 0.04, unpaired t-test). Numbers in each quadrant represent percentage of total T cells. (C) Chemotaxis of splenic CD4+ T cells collected from spleens of WT or Rgs16–/– mice 4 days following helminth challenge (mean ± S.E.M. of 4-5 mice/group, *P = 0.03, unpaired t-test).
Splenic CD4 T cells isolated from challenged Rgs16–/– mice migrated more towards CCL17 gradients than cells from WT mice (Fig. 4C). Thus, irregular trafficking patterns of fibrosis-inducing IL-13+ TH2 cells might contribute to the enhanced inflammation in Rgs16–/– mice following helminth challenge. Consistent with this hypothesis, we observed starkly atypical T lymphocyte localization patterns in lungs of RGS16-deficient mice following S. mansoni inoculation compared to WT. Whereas T cells were distributed uniformly within fibrotic granulomas surrounding embolized eggs in WT lungs, they were restricted to the periphery of granulomas in lungs of Rgs16–/– mice (Fig. 5A). In addition, we detected dense perivascular/peribronchial T cell aggregates that were largely absent in WT mice (Figs. 5B-C). These data suggest that RGS16 attenuates inflammation in Schistosome egg-challenged lungs by retaining T lymphocytes within granulomas.
Figure 5. Anomalous lymphocyte localization in helminth-challenged Rgs16–/– lungs.
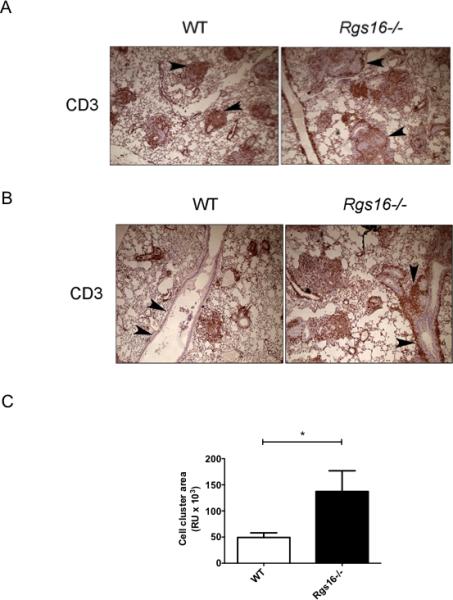
(A-B) T cell localization in helminth-challenged lungs evaluated by immunohistochemistry with a CD3 antibody. Images in (A) show parenchymal T cell accumulation in granulomas while those in (B) demonstrate perivascular/peribronchial cell aggregates (arrowheads). Total area containing cellular aggregrates around airways and vessels was quantified using Image Pro Plus software (*P = 0.04, unpaired t-test) as indicated in (C). Images represent 8 mice/group evaluated in 2 independent experiments.
RGS16 regulates lymphocyte trafficking in vivo mediated by CCR4 and CCR10
Because TH2 cells express CCR4, and RGS16-deficient T cells migrated excessively towards CCL17, we hypothesized that the CCL17/22-CCR4 pathway mediated uncharacteristic migration of a subset of TH2 cells (CCR4+IL-13+) in challenged lungs of Rgs16–/– mice. Consistent with this hypothesis, we detected CCR4+IL-13+ T cells in the lungs of Rgs16–/– much earlier than in WT mice (four days after challenge with S. mansoni) (Fig. 6A). To determine whether other chemokine/receptor pairs contributed to T lymphocyte mis-localization in lungs of helminth-challenged Rgs16–/– mice, we analyzed differential gene expression by qPCR array (full gene list in Supplemental Table). Notably, we found selectively increased expression of Il17b and Ccr10 mRNA in lungs of Rgs16–/– mice compared to WT. CCR10 is expressed by effector/memory T cells, Langerhans cells and plasma cells, among others (24-26), and its ligands CCL27/28 are produced by epidermal keratinocytes (27) and displayed on the surface of dermal endothelial cells (28). CCR10 expression on skin-homing TH2 cells (29) has been implicated in the pathogenesis of T-cell mediated inflammatory skin diseases including atopic dermatitis and contact hypersensitivity (16, 17). We detected CCR10 in lungs of challenged mice by immunohistochemistry, and its staining pattern mirrored T cell localization in WT and Rgs16–/– mice (predominantly in granulomas or peribronchial/perivascular areas, respectively) (Fig. 6B). This result suggests a role for CCR10 in S. mansoni-evoked inflammation and in the aberrant trafficking of cytokine-producing T cells we observed in Rgs16–/– mice.
Figure 6. Lung lymphocyte populations in Rgs16–/– mice following helminth challenge.

(A) CCR4+ TH2 cells (IL-13+) in lungs of challenged mice were enumerated by flow cytometry. (B) CCR10 expression in helminth-challenged lungs evaluated by immunohistochemical staining with a CCR10 antibody. (C-D) Ccl28 (C) or Ccr10 (D) expression in naïve or S. mansoni-challenged lungs evaluated by real-time PCR (*P = 0.04, unpaired t-test). (E-G) CCR10+ T cells in naïve lungs (E), or cytokine+ CCR10+ (F) or CCR4+ (G) T cells in helminth-challenged lungs were quantified by means of flow cytometry. (H) Frequencies of IL-17B+ T cells (CD3+) in lungs 7 days following S. mansoni challenge were quantified by means of flow cytometry. Numbers represent percentage of total T cells. All data was generated using 3 naïve mice/ group, 3-5 challenged WT mice or 4-5 challenged Rgs16-/- mice.
Although we found decreased or comparable expression of Ccl17 and CCR10 ligands Ccl27 and Ccl28 in lungs of WT and Rgs16–/– mice (in the presence or absence of S. mansoni challenge) (Fig. 6C, Supplemental Table, and data not shown), naïve lungs of knockout mice had significantly increased Ccr10 expression (Fig. 6D). We detected CCR10+ T lymphocytes in naïve lungs of Rgs16–/– mice but not in WT mice (Fig. 6E), suggesting that anomalous migration of RGS16-deficient, CCR10+ T cells to the lungs accounted for the increased Ccr10 gene expression. Moreover, lungs from Rgs16–/– mice, but not from WT mice, contained cytokine-producing CCR4+ or CCR10+ T cells (IL-13 or IL-17B) 4-7 days following S. mansoni challenge (Figs. 6F-H). Collectively, these results indicate that ectopic trafficking of CCR4+ and CCR10+ T effector cells to the lungs of Rgs16–/– mice may exacerbate fibrogenesis in response to pulmonary helminth challenge through enhanced cytokine production.
Cytokine abnormalities in S. mansoni-challenged Rgs16–/– mice
In addition to increased Ccr10 expression, array analysis also revealed upregulation of cytokines in helminth-challenged Rgs16–/– lungs compared to WT—specifically Il13 and Il17b. We confirmed these results by real-time PCR (Fig. 7A-B) and investigated the source(s) of cytokines by re-stimulating MLN cells from WT or Rgs16–/– mice with Schistosome egg antigen (SEA). LN supernatants from Rgs16–/– mice had significantly more IL-5, IL-10, and IL-13 than WT (Fig. 7C). As effector T cells are generally considered to be the most common sources of these cytokines, we analyzed quantities of LN cytokine-producing CD4 T cells in WT and knockout mice. In contrast to the increased numbers of cytokine+ T cells present in lungs of S. mansoni-challenged Rgs16–/– mice relative to WT at the earlier time point (4 days post-challenge), frequencies of IL-13+ or IL-17B+ T cells and intensity of IL-13 staining were similar in WT and Rgs16–/– MLNs at the 7 day timepoint (Figs. 7D-E). These results suggested that further characterization of these cell populations is required to determine the source of increased cytokines in MLN supernatants of Rgs16–/– mice relative to WT following re-exposure to S. mansoni.
Figure 7. Cytokine abnormalities in Rgs16–/– mice inoculated with S. mansoni.
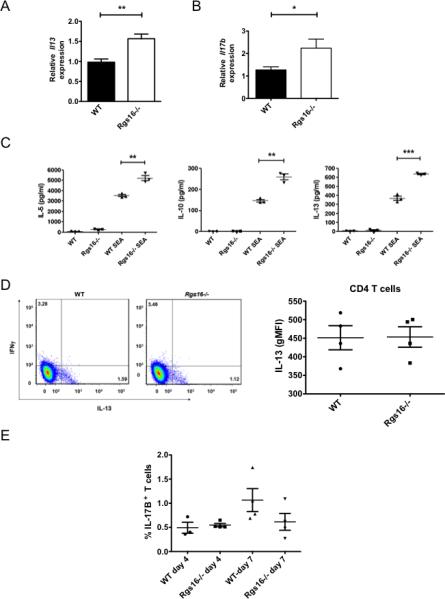
(A-B) Expression of Il13 (A) or Il17b (B) was evaluated in naïve or helminth-challenge lungs by real-time PCR (*P = 0.03; **P = 0.005, unpaired t-test). (C) Cells were harvested from lung-draining mediastinal LNs 7 days following inoculation with S. mansoni. Cells were left untreated or re-stimulated with Schistome egg Ag (SEA) for 3 days followed by measurement of the indicated cytokine levels in supernatants (*P = 0.005; ***P =0.0001, unpaired t-test). (D) Frequencies of IL-13+ or INFγ+ T cells (CD3+) in LNs 7 days following S. mansoni challenge were quantified by means of flow cytometry Numbers in each quadrant represent percentage of total T cells. Graph on the right shows intensity of IL-13 staining (geometric mean fluorescence intensity, MFI) in the CD4 T cell population. (E) Frequencies of IL-17B+ T cells (CD3+) in LNs 4 or 7 days following S. mansoni challenge were quantified by means of flow cytometry. All data was generated using 3 naïve mice/ group, 3-5 challenged WT mice or 4-5 challenged Rgs16-/- mice. For (C), data is representative of 7-8 mice/group evaluated in 2 independent experiments.
DISCUSSION
We have elucidated a function for a modifier of GPCR signaling, RGS16, in a TH2-mediated murine pulmonary inflammatory response to helminth challenge—specifically, through regulation of lung T cell trafficking and cytokine production. The loss of RGS16 in mice triggered an enhanced granulomatous reaction in the lung following challenge with S. mansoni eggs, resulting in more fibrosis and eosinophil influx, anomalous localization of T cells, and increased cytokine production. This study also highlights an unanticipated function of the CCR10-CCL27/8 chemokine axis and IL-17B in the pathogenesis of S. mansoni-associated inflammation.
Several lines of evidence suggest that RGS16 directly controls differentiated T helper/effector T cell migration patterns but does not regulate trafficking of quiescent, naïve T lymphocytes. RGS16 expression is highly upregulated in differentiated mouse and human TH1, TH2, and TH17 cells compared to naïve CD4 T cells [(13) and Fig. 2A]. Chemokines involved prominently in maintenance of lymphoid compartments through induction of lymphocyte extravasation through high endothelial venules of spleen and LNs (CCL21 and CXCL12) induced comparable chemotaxis of naïve WT and RGS16-deficient T cells. In contrast, Rgs16–/– effector TH2 lymphocytes differentiated in vitro or cells extracted from S. mansoni-challenged lungs had exaggerated chemotaxis towards a TH2-associated chemokine (CCL17). The degree of RGS16 expression correlated inversely with the extent of migration, and chemotaxis of Rgs16–/– T cells was reduced by reconstitution with RGS16. Spleen and LN lymphocyte populations and organ architecture in the absence of immune challenge were unchanged in either RGS16-Tg or Rgs16–/– mice [(13) and this study] whereas acute inoculation with S. mansoni accentuated trafficking of differentiated TH2 cells to these sites.
A surprising finding of this study is the anomalous collection of CCR10+ TH2 cells in the lungs of Rgs16–/– mice. Although the CCR10-CCL27 axis has been previously associated with TH2-mediated inflammation in the skin, its role in pulmonary pathology induced by S. mansoni challenge was unknown (20). CCL27 is produced in skin epidermal keratinocytes (27) and presented by dermal endothelial cells (28). CCL17 also promotes CCL27 induction by keratinocytes in the presence of TNFα (30). CCL28, another CCR10 ligand, has been implicated in CCR10-mediated leukocyte homing to the respiratory tract in a murine asthma model (31). The presence of similar or reduced amounts of CCL27/28 ligands in lungs of Rgs16–/– mice compared to WT suggests that aberrant chemotactic responses of RGS16-deficient T cells to these ligands underlies their accumulation in lungs in the absence of immune challenge. However, for unknown reasons we did not observe chemotaxis of TH2 cells (WT or knockout) towards these chemokines in Transwell assays in vitro despite expression of CCR10.
We also found increased Il13 expression in challenged lungs of Rgs16–/–mice relative to WT, consistent with its central contribution to fibrosis induced by helminth infection (32, 33) (34). Although previous work has also defined a role for IL-17A in helminth-associated inflammation, we observed increased Il17b expression in lungs of S. mansoni challenged, Rgs16–/– mice relative to WT but did not detect Il17a. These results suggest an unanticipated function of IL-17B in helminth immunity. Among other IL-17 family members, neutralization of IL-17B in a collagen-induced model of murine arthritis suppressed disease progression by reducing cell infiltration and production of pro-inflammatory cytokine such as IL-1 β, TNF- α (35), factors also known to induce CCL28 in airway epithelial cells (36, 37). Although we detected IL-17B+ T cells in challenged lungs, it is unclear whether these cells are conventional TH17 cells. Indeed, in vitro differentiated TH17 cells produced IL-17A but not IL-17B (Supplemental Fig. 3). Taken together, these findings suggest the presence of a unique pro-inflammatory environment downstream of IL-17B in lungs of Rgs16–/– mice challenged with S. mansoni. The absence of RGS16 may promote T cell chemotaxis to CCL27/28 displayed on the surface of endothelial cells, which could account for the peribronchial/perivascular accumulation of T cells we observed in challenged lungs of Rgs16–/– mice.
How RGS16 regulates Ag-induced cytokine production requires further study. We detected unique populations of IL-13 or IL-17B cytokine-producing cells expressing CCR10 or CCR4 much earlier in lungs of Rgs16–/– mice following helminth challenge. Although quantities of cytokine+ T cells in lungs were equivalent in the two strains by day 7, overall lung cytokine expression was increased in lungs of knockout mice at this time point relative to WT. Additional gene array analysis revealed reduced expression of suppressor of cytokine signaling 2 (Socs2) in lungs of naïve Rgs16–/– mice compared to WT counterparts but not in RGS16-deficient TH2 cells differentiated in vitro (Supplemental Fig. 3 and data not shown). These data suggest that lung T cells in Rgs16–/– mice are primed for increased TH2 cytokine production due to reduced Socs2 expression (38).
On the other hand, although levels of immunoreactive cytokines in supernatants of Ag-restimulated MLN cells extracted from Rgs16–/– mice 7 days post S. mansoni challenge were significantly higher than those from WT mice, flow cytometric analysis demonstrated equivalent cytokine+ T cell frequencies and intensity of cytokine staining in these same LN T cell populations. Polarized TH cells from WT and Rgs16–/– mice secreted roughly equivalent cytokine amounts (IFNγ for TH1; IL-4 or IL-13 for TH2; IL-17A for TH17) in response to T-cell receptor stimulation with anti-CD3 and anti-CD28 in vitro (Supplemental Fig. 3 and data not shown). Increased accumulation of specific populations of cytokine-producing TH cells in MLNs of Rgs16–/– mice compared to WT (presumably due to altered trafficking patterns) could also account for the overall increases in secreted cytokines observed. Thus, based on these data alone we cannot yet determine whether Ag-stimulated, RGS16-deficient T lymphocytes generate more cytokine than WT cells on a per cell basis.
Although our work and that of others have shown that RGS proteins inhibit chemokine-mediated lymphocyte chemotaxis and adhesive responses in vitro (39, 40), function(s) of RGS proteins in T-cell mediated immunity have not been explored in detail. Surprisingly, Rgs2–/– mice had reduced footpad swelling following local inoculation with lymphocytic choriomeningitis virus (LCMV) compared to WT, which correlated with impaired proliferation of, and IL-2 production by, RGS2-deficent T cells. Taken together, these results and our studies suggest unique, context-dependent functions of individual RGS proteins in immune cells that may or may not be predictable based on their shared biochemical (GAP) activity. Further exploration of RGS16 in specific T cell populations and in the setting of immune challenges will be needed to fully clarify its function(s) in adaptive immunity. Although we present abundant evidence that anomalous TH2 trafficking and increased T-cell derived cytokines contribute to the enhanced pulmonary inflammation in S. mansoni-challenged, RGS16-deficient mice relative to WT (Figs. 4-7), non-TH-dependent factors may also mediate fibrosis in this setting. Additional models of inflammation and approaches such as adoptive T cell transfer and/or generation of bone marrow chimeras will be needed to determine the relative importance of T-cell intrinsic- and T-cell-independent factors to the immune responses of mice lacking RGS16.
Supplementary Material
ACKNOWLEDGEMENTS
None
This study was supported by the Intramural Research Program of the NIAID, NIH (Grant AI000746 to K.M.D.)
REFERENCES
- 1.Cyster JG. Lymphoid organ development and cell migration. Immunol Rev. 2003;195:5–14. doi: 10.1034/j.1600-065x.2003.00075.x. [DOI] [PubMed] [Google Scholar]
- 2.Wynn TA, Thompson RW, Cheever AW, Mentink-Kane MM. Immunopathogenesis of schistosomiasis. Immunol Rev. 2004;201:156–167. doi: 10.1111/j.0105-2896.2004.00176.x. [DOI] [PubMed] [Google Scholar]
- 3.Wilson MS, Mentink-Kane MM, Pesce JT, Ramalingam TR, Thompson R, Wynn TA. Immunopathology of schistosomiasis. Immunol Cell Biol. 2007;85:148–154. doi: 10.1038/sj.icb.7100014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pearce EJ, MacDonald AS. The immunobiology of schistosomiasis. Nat Rev Immunol. 2002;2:499–511. doi: 10.1038/nri843. [DOI] [PubMed] [Google Scholar]
- 5.Dessein A, Kouriba B, Eboumbou C, Dessein H, Argiro L, Marquet S, Elwali NE, Rodrigues V, Li Y, Doumbo O, Chevillard C. Interleukin-13 in the skin and interferon-gamma in the liver are key players in immune protection in human schistosomiasis. Immunol Rev. 2004;201:180–190. doi: 10.1111/j.0105-2896.2004.00195.x. [DOI] [PubMed] [Google Scholar]
- 6.Jakubzick C, Wen H, Matsukawa A, Keller M, Kunkel SL, Hogaboam CM. Role of CCR4 ligands, CCL17 and CCL22, during Schistosoma mansoni egg-induced pulmonary granuloma formation in mice. Am J Pathol. 2004;165:1211–1221. doi: 10.1016/S0002-9440(10)63381-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kehrl JH. Chemoattractant receptor signaling and the control of lymphocyte migration. Immunol Res. 2006;34:211–227. doi: 10.1385/IR:34:3:211. [DOI] [PubMed] [Google Scholar]
- 8.Gilman AG. G proteins: transducers of receptor-generated signals. Annu Rev Biochem 1. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- 9.Marinissen MJ, Gutkind JS. G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol Sci. 2001;22:368–376. doi: 10.1016/s0165-6147(00)01678-3. [DOI] [PubMed] [Google Scholar]
- 10.Bansal G, Druey KM, Xie Z. R4 RGS proteins: regulation of G-protein signaling and beyond. Pharmacol Ther. 2007;116:473–495. doi: 10.1016/j.pharmthera.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tesmer JJ, Berman DM, Gilman AG, Sprang SR. Structure of RGS4 bound to AlF4--activated G(i alpha1): stabilization of the transition state for GTP hydrolysis. Cell. 1997;89:251–261. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- 12.Beadling C, Druey KM, Richter G, Kehrl JH, Smith KA. Regulators of G protein signaling exhibit distinct patterns of gene expression and target G protein specificity in human lymphocytes. J Immunol. 1999;162:2677–2682. [PubMed] [Google Scholar]
- 13.Lippert E, Yowe DL, Gonzalo JA, Justice JP, Webster JM, Fedyk ER, Hodge M, Miller C, Gutierrez-Ramos JC, Borrego F, Keane-Myers A, Druey KM. Role of regulator of G protein signaling 16 in inflammation-induced T lymphocyte migration and activation. J Immunol. 2003;171:1542–1555. doi: 10.4049/jimmunol.171.3.1542. [DOI] [PubMed] [Google Scholar]
- 14.Townsend MJ, Fallon PG, Matthews DJ, Jolin HE, McKenzie AN. T1/ST2-deficient mice demonstrate the importance of T1/ST2 in developing primary T helper cell type 2 responses. J Exp Med. 2000;191:1069–1076. doi: 10.1084/jem.191.6.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramalingam TR, Pesce JT, Sheikh F, Cheever AW, Mentink-Kane MM, Wilson MS, Stevens S, Valenzuela DM, Murphy AJ, Yancopoulos GD, Urban JF, Jr., Donnelly RP, Wynn TA. Unique functions of the type II interleukin 4 receptor identified in mice lacking the interleukin 13 receptor alpha1 chain. Nat Immunol. 2008;9:25–33. doi: 10.1038/ni1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bansal G, Xie Z, Rao S, Nocka KH, Druey KM. Suppression of immunoglobulin E-mediated allergic responses by regulator of G protein signaling 13. Nat Immunol. 2008;9:73–80. doi: 10.1038/ni1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagahara H, Vocero-Akbani AM, Snyder EL, Ho A, Latham DG, Lissy NA, Becker-Hapak M, Ezhevsky SA, Dowdy SF. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat Med. 1998;4:1449–1452. doi: 10.1038/4042. [DOI] [PubMed] [Google Scholar]
- 18.Stein JV, N.-A. C. Chemokine control of lymphocyte trafficking: a general overview. Immunology. 2005;116:1–12. doi: 10.1111/j.1365-2567.2005.02183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D'Ambrosio D, Iellem A, Bonecchi R, Mazzeo D, Sozzani S, Mantovani A, Sinigaglia A. Cutting Edge: Selective Up-Regulation of Chemokine Receptors CCR4 and CCR8 upon Activation of Polarized Human Type 2 Th Cells. J Immunol. 1998;161:5111–5115. [PubMed] [Google Scholar]
- 20.Stanford MM, Issekutz TB. The relative activity of CXCR3 and CCR5 ligands in T lymphocyte migration: concordant and disparate activities in vitro and in vivo. J Leukoc Biol. 2003;74:791–799. doi: 10.1189/jlb.1102547. [DOI] [PubMed] [Google Scholar]
- 21.Hirata T, Osuga Y, Takamura M, Kodama A, Hirota Y, Koga K, Yoshino O, Harada M, Takemura Y, Yano T, Taketani Y. Recruitment of CCR6-expressing Th17 cells by CCL 20 secreted from IL-1 beta-, TNF-alpha-, and IL-17A-stimulated endometriotic stromal cells. Endocrinology. 2010;151:5468–5476. doi: 10.1210/en.2010-0398. [DOI] [PubMed] [Google Scholar]
- 22.Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, Yamaguchi T, Nomura T, Ito H, Nakamura T, Sakaguchi N, Sakaguchi S. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. The Journal of experimental medicine. 2007;204:2803–2812. doi: 10.1084/jem.20071397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takatsu K, Nakajima H. IL-5 and eosinophilia. Curr. Opin. Immunol. 2008;20:288–294. doi: 10.1016/j.coi.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 24.Kagami S, Saeki H, Tsunemi Y, Nakamura K, Kuwano Y, Komine M, Nakayama T, Yoshie O, Tamaki K. CCL27-transgenic mice show enhanced contact hypersensitivity to Th2 but not Th1 stimuli. Eur J Immunol. 2008;38:647–657. doi: 10.1002/eji.200737685. [DOI] [PubMed] [Google Scholar]
- 25.Homey B, Wang W, Soto H, Buchanan ME, Wiesenborn A, Catron D, Muller A, McClanahan TK, Dieu-Nosjean MC, Orozco R, Ruzicka T, Lehmann P, Oldham E, Zlotnik A. Cutting edge: The orphan chemokine receptor G protein-coupled receptor-2 (GPR-2, CCR10) binds the skin-associated chemokine (CTACK/ALP/ILC). J Immunol. 2000;164:3465–3470. doi: 10.4049/jimmunol.164.7.3465. [DOI] [PubMed] [Google Scholar]
- 26.Nakayama T, Hieshima K, Izawa D, Tatsumi Y, Kanamaru A, Yoshie O. Cutting edge: Profile of chemokine receptor expression on human plasma cells accounts for their efficient recruitment to target tissues. J Immunol. 2003;170:1136–1140. doi: 10.4049/jimmunol.170.3.1136. [DOI] [PubMed] [Google Scholar]
- 27.Morales J, Homey B, Vicari AP, Hudak S, Oldham E, Hedrick J, Orozco R, Copeland NG, Jenkins NA, McEvoy LM, Zlotnik A. CTACK, a skin-associated chemokine that preferentially attracts skin-homing memory T cells. Proc. Natl. Acad. Sci. USA. 1999;96:14470–14475. doi: 10.1073/pnas.96.25.14470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Homey B, Alenius H, Muller A, Soto H, Bowman EP, Yuan W, McEvoy L, Lauerma AI, Assmann T, Bünemann E, Lehto M, Wolff H, Yen D, Marxhausen H, To W, Sedgwick J, Ruzicka T, Lehmann P, Zlotnik A. CCL27-CCR10 interactions regulate T cell-mediated skin inflammation. Nat. Med. 2002;8:157–165. doi: 10.1038/nm0202-157. [DOI] [PubMed] [Google Scholar]
- 29.Bansal G, DiVietro JA, Kuehn HS, Rao S, Nocka KH, Gilfillan AM, Druey KM. RGS13 controls g protein-coupled receptor-evoked responses of human mast cells. J Immunol. 2008;181:7882–7890. doi: 10.4049/jimmunol.181.11.7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vestergaard C, Johansen C, Christensen U, Just H, Hohwy T, Deleuran M. TARC augments TNF-alpha-induced CTACK production in keratinocytes. Exp Dermatol. 2004;13:551–557. doi: 10.1111/j.0906-6705.2004.00202.x. [DOI] [PubMed] [Google Scholar]
- 31.English K, Brady C, Corcoran P, Cassidy JP, Mahon BP. Inflammation of the respiratory tract is associated with CCL28 and CCR10 expression in a murine model of allergic asthma. Immunol Lett. 2006;103:92–100. doi: 10.1016/j.imlet.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 32.Chiaramonte MG, Donaldson DD, Cheever AW, Wynn TA. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. J. Clin Invest. 1999;104:777–785. doi: 10.1172/JCI7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McDermott JR, Humphreys NE, Forman SP, Donaldson DD, Grencis RK. Intraepithelial NK cell-derived IL-13 induces intestinal pathology associated with nematode infection. J Immunol. 2005;175:3207–3213. doi: 10.4049/jimmunol.175.5.3207. [DOI] [PubMed] [Google Scholar]
- 34.Simonian PL, Roark CL, Wehrmann F, Lanham AK, Diaz del Valle F, Born WK, O'Brien RL, Fontenot AP. Th17-polarized immune response in a murine model of hypersensitivity pneumonitis and lung fibrosis. J Immunol. 2009;182:657–665. [PMC free article] [PubMed] [Google Scholar]
- 35.Yamaguchi Y, Fujio K, Shoda H, Okamoto A, Tsuno NH, Takahashi K, Yamamoto K. IL-17B and IL-17C are associated with TNF-alpha production and contribute to the exacerbation of inflammatory arthritis. J Immunol. 2007;179:7128–7136. doi: 10.4049/jimmunol.179.10.7128. [DOI] [PubMed] [Google Scholar]
- 36.O'Gorman MT, Jatoi NA, Lane SJ, Mahon BP. IL-1beta and TNF-alpha induce increased expression of CCL28 by airway epithelial cells via an NFkappaB-dependent pathway. Cell Immunol. 2005;238:87–96. doi: 10.1016/j.cellimm.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 37.Scanlon KM, Hawksworth RJ, Lane SJ, Mahon BP. IL-17A Induces CCL28, Supporting the Chemotaxis of IgE-Secreting B Cells. Int Arch Allergy Immunol. 2011;156:51–61. doi: 10.1159/000322178. [DOI] [PubMed] [Google Scholar]
- 38.Knosp CA, Carroll HP, Elliott J, Saunders SP, Nel HJ, Amu S, Pratt JC, Spence S, Doran E, Cooke N, Jackson R, Swift J, Fitzgerald DC, Heaney LG, Fallon PG, Kissenpfennig A, Johnston JA. SOCS2 regulates T helper type 2 differentiation and the generation of type 2 allergic responses. The Journal of experimental medicine. 2011;208:1523–1531. doi: 10.1084/jem.20101167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Han JI, Huang NN, Kim DU, Kehrl JH. RGS1 and RGS13 mRNA silencing in a human B lymphoma line enhances responsiveness to chemoattractants and impairs desensitization. J Leukoc Biol. 2006;79:1357–1368. doi: 10.1189/jlb.1105693. [DOI] [PubMed] [Google Scholar]
- 40.Garcia-Bernal D, Dios-Esponera A, Sotillo-Mallo E, Garcia-Verdugo R, Arellano-Sanchez N, Teixido J. RGS10 restricts upregulation by chemokines of T cell adhesion mediated by alpha4beta1 and alphaLbeta2 Integrins. J Immunol. 2011;187:1264–1272. doi: 10.4049/jimmunol.1002960. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.