

Background: T cells from SLE patients display multiple signaling aberrations, many of which are attributed to increased presence of transcription factor CREMα.
Results: Notch-1 expression is significantly reduced in T cells from active SLE patients. Both epigenetic and transcriptional effects mediated through CREMα contribute to dysregulated Notch-1 expression in SLE T cells.
Conclusion: Notch-1 levels inversely correlate with SLE disease activity.
Significance: Boosting endogenous Notch-1 levels may redirect T cell function in SLE patients.
Keywords: Epigenetics, Notch, Notch Receptor, T Cell, T Cell Biology, Transcription, Transcriptional Regulation, SLE, Lupus
Abstract
Notch signaling constitutes an evolutionarily conserved pathway that transduces signals between neighboring cells and determines major decisions in cell proliferation, survival, and differentiation. Notch signaling has been shown to play a pivotal role during T cell lineage determination. T lymphocytes from patients with systemic lupus erythematosus (SLE) display a severely altered phenotype with several molecular and functional aberrations, including defective capacities to up-regulate Notch-1 receptor expression upon T cell receptor activation. Here, we demonstrate that basal Notch-1 expression is decreased in T cells from active SLE patients at the mRNA and protein levels in various T cell subpopulations. Notch-1 transcript numbers inversely correlate with disease activity in SLE patients. We provide evidence that both enhanced histone H3 methylation and CpG DNA methylation of the human Notch-1 promoter contribute to decreased Notch-1 expression in SLE T cells. Previous data from our group identified cAMP-responsive element modulator α (CREMα), which is up-regulated in SLE T cells, as a key regulator of epigenetic patterns and gene transcription, e.g. that of IL2 and IL17 genes. In this study, we observed increased CREMα binding to the Notch-1 promoter, which eventually resulted in significantly reduced Notch-1 promoter activity and gene transcription. Notably, decreased Notch-1 levels were associated with elevated IL-17A levels. Our data suggest a role for Notch-1 in SLE immunopathogenesis, and for the first time, we present molecular mechanisms that mediate dysregulated Notch-1 expression in SLE T cells.
Introduction
Systemic lupus erythematosus (SLE)3 was initially described as a B cell-dependent disease with an excessive production of autoantibodies; however, it has become evident over the last decades that severe signaling aberrations in T cells also play a major role in SLE pathogenesis (1, 2). This may include defective T cell activation and proliferation, dysregulated T helper cell differentiation, and impaired cytokine production. Previous studies from our group have highlighted the pivotal role of the transcription factor cAMP-responsive element modulator α (CREMα) in the epigenetic and transcriptional regulation of several SLE-relevant target genes in T cells (3–10). The CREM protein family comprises >20 isoforms in humans, and there is broad evidence that the CREMα isoform is robustly overexpressed in SLE T cells. CREMα gene transcription itself is regulated through different promoters (denoted P1 and P2), and activity levels of P1 correlate with disease activity in SLE patients (11, 12). CREMα binds DNA motifs denoted cAMP-responsive elements (CREs), which are defined by the palindromic nucleotide sequence TGACGTCA or its 5′-half-site TGAC. Within the dysbalanced cytokine profile of SLE T cells, CREMα suppresses IL-2 and increases proinflammatory IL-17A production by virtue of its propensities as a transcription factor and “epigenetic modifier” of histones and cytosine-phosphoguanosine (CpG) DNA sequences (5–7).
T cell activation through the canonical T cell receptor complex can be significantly influenced by various co-stimulatory signals, including CD28, CTLA-4, and signaling lymphocyte activation molecules. Coactivation of these surface proteins may transmit inhibitory or stimulatory messages for T cell function (13–16). The Notch receptors constitute an evolutionarily conserved family of transmembrane molecules (in mammals, Notch-1–4) that transduce signals between neighboring cells. They are involved in short-distance cell-cell communication processes during organogenesis, cell fate decisions, and cell polarity. Notch receptors are synthesized as heterodimers and anchored in the cell membrane (4, 17). The extracellular domain of the Notch receptors comprises multiple epidermal growth factor-like repeats that can be bound by both canonical (in humans, those of the Jagged or Delta-like family) and non-canonical (e.g. secreted NOV (nephroblastoma overexpressed gene), MAGP-1 (microfibril-associated glycoprotein-1) and MAGP-2, or secreted Y-box protein-1) ligands (18, 19). Classical Notch ligands exist either as membrane-bound proteins or as soluble factors that are shed from the membrane. Receptor occupation by ligands promotes two proteolytic steps within the receptor protein, exerted by an ADAM metalloprotease and a γ-secretase, the latter belonging to the presenilin family (20). Proteolytic cleavage of the receptor releases the intracellular domain, which is targeted to the nucleus. Here, it associates with other transcriptional regulators, most importantly, CSL/RBP-Jκ, and modifies transcription of the HES (hairy enhancer of split) and Hey gene families that code for transcription factors themselves (21, 22).
Notch signaling is involved in various peripheral T cell responses, e.g. polarization of T helper (Th) cells. Both receptors and ligands can be regulated by post-translational protein modifications, proteolytic processing, endocytosis, and membrane trafficking. Overall, the cytokine milieu, expression pattern of the receptors and their ligands on peripheral T and B cells, and the specificity of the ligand-receptor interaction may determine specific cell fate decisions (23). Notably, chemical inhibition of all four Notch receptors by nonspecific γ-secretase inhibitors inhibits Th1- and Th17-type differentiation and ameliorates signs of autoimmunity and renal damage in lupus-prone MRL-lpr mice (24). γ-Secretase inhibitors have also been shown to be beneficial in experimental autoimmune encephalomyelitis, which is another Th17-dependent disease model and has features that resemble multiple sclerosis in humans (25). γ-Secretase inhibitors have already entered the stage of clinical trials in humans; however, they are associated with major side effects (mainly gut toxicity), which have been attributed to the pan-Notch blockade (26). More specific interventions targeting individual Notch receptors, e.g. by application of monoclonal antibodies, appear to be more tolerable (27). In the experimental autoimmune encephalomyelitis model, selective inhibition of individual Notch receptors using specific antibodies abrogated Th1- and Th17-type responses (25).
In this study, we observed significantly decreased amounts of Notch-1 in T cells from clinically active SLE patients at both the mRNA and protein levels. We provide evidence that Notch-1 gene expression is highly controlled through changes in the epigenetic conformation of the Notch-1 promoter, including histone and CpG DNA methylation and transcriptional repression mediated by CREMα. Eventually, reduced Notch-1 levels in human T cells are associated with increased IL-17A production, as observed in SLE patients.
EXPERIMENTAL PROCEDURES
Primary Human T Cells and Human T Cell Line
The SLE patients included in our analyses were female and diagnosed according to the American College of Rheumatology classification criteria (28). They were recruited from the Division of Rheumatology at the Beth Israel Deaconess Medical Center after written informed consent under protocol 2006-P-0298. Healthy individuals were chosen as controls. Peripheral venous blood was collected in heparin lithium tubes, and total human T cells were purified as described previously (18). All primary human T cells and human Jurkat T cells were kept in RPMI 1640 medium supplemented with 10% fetal bovine serum.
mRNA Extraction and Quantitative RT-PCR
Total RNA was isolated from purified human T cells using an RNeasy mini kit (Qiagen). Residual genomic DNA contamination was removed by DNase I (Qiagen). RNA was reverse-transcribed into cDNA using a reverse transcription system (Promega). Sequences for real-time quantitative RT-PCR (qRT-PCR) primers were as follows: Notch-1, 5′-ctgcctgtctgaggtcaatg-3′ (forward) and 5′-tcacagtcgcacttgtaccc-3′ (reverse); IL17A, 5′-cgaaatccaggatgccc-3′ (forward) and 5′-gacaccagtatcttctccag-3′ (reverse); and 18 S rRNA, 5′-actcaacacgggaaacctca-3′ (forward) and 5′-aaccagacaaatcgctccac-3′ (reverse). Real-time qPCR was performed on an ABI OneStepPlus real-time PCR system.
Flow Cytometry
Cells (0.5 million) from each blood donor were stained ex vivo with FITC-labeled anti-CD3, Pacific Blue-labeled anti-CD4, phycoerythrin-labeled anti-CD8, and allophycocyanin-labeled anti-Notch-1 antibodies. Samples were incubated at room temperature for 30 min, washed twice with PBS, and fixed in a 4% formaldehyde solution. Expression of cell surface markers was assessed on a BD Biosciences LSRII flow cytometer, and data were gated and displayed on FlowJo Version 7.6.5 (TreeStar Inc., San Carlos, CA).
Plasmids and Luciferase Assays
An expression plasmid for human CREMα (in the pcDNA3.1/V5-His-TOPO vector, Invitrogen) was kindly provided by G. N. Europe-Finner (Faculty of Medical Sciences, Newcastle upon Tyne, United Kingdom) (29). A 2.1-bp spanning Notch-1 reporter construct (in pGL3-Basic vector, Promega) was generated and kindly donated by M. Ruppert (University of Alabama at Birmingham) (30). An expression plasmid encoding the constitutively active intracellular Notch-1 domains was kindly provided by K. Sakamoto (Tokyo Medical and Dental University, Tokyo, Japan) (31). All plasmid DNA preparations were carried out with DNA purification kits (Qiagen) and sequence-verified (GENEWIZ, Inc., Cambridge, MA). Jurkat T cells (3 million) were transfected with a total amount of 3 μg of plasmid DNA at a molar effector:reporter transfection ratio of 3:1 using an Amaxa human T cell Nucleofector kit (Lonza) and an Amaxa Nucleofector II device (program U014, Lonza). Each reporter experiment included 10 ng of Renilla luciferase construct as an internal control. Five hours after transfection, cells were collected and lysed, and luciferase activity was quantified using the Promega Dual-Luciferase assay system according to the manufacturer's instructions. Luciferase experiments were repeated three times, and values in the bar diagrams are given as mean ± S.D.
ChIP Assays
Anti-H3K27me3 antibody and nonspecific normal rabbit IgG were obtained from Upstate Biotechnology. Polyclonal anti-CREMα antibody detecting human CREMα was designed in our group and has been described previously (5). ChIP-grade ChIP assay was carried out according to the manufacturer's instructions (Upstate Biotechnology). Briefly, 1–2 million total T cells were cross-linked with 1% formaldehyde, washed with cold PBS, and lysed in buffer containing protease inhibitors (Roche Applied Science). Cell lysates were sonicated to shear DNA and sedimented, and diluted supernatants were immunoprecipitated with the indicated antibodies and ChIP-grade Protein A/G Plus-agarose (Thermo Scientific). A proportion (20%) of the diluted supernatants were kept as “input” (input represents PCR amplification of the total sample). Protein-DNA complexes were eluted in 1% SDS and 0.1 m NaHCO3 and reverse-cross-linked at 65 °C. DNA was recovered using a QIAamp DNA mini kit (Qiagen) and subjected to PCR analysis using an ABI OneStepPlus real-time PCR system. The real-time qPCR primer sequences used to detect the CRE site within the human Notch-1 promoter were as follows: 3′-aaatcagcggaaggagcac-5′ (forward) and 3′-tgattgcccgagcacttgac-5′ (reverse). The amount of immunoprecipitated DNA was substracted by the amplified DNA that was bound by the nonspecific normal IgG and subsequently calculated relative to the respective input DNA.
Methylated CpG DNA Immunoprecipitation
A methylated CpG DNA immunoprecipitation assay was carried out according to the manufacturer's instructions (Zymo Research). Briefly, genomic DNA from T cells obtained from SLE patients and healthy control individuals was purified using the AllPrep RNA/DNA/protein mini kit (Qiagen) and sheared to fragments of ∼200 bp with DNA Shearase (Zymo Research). Subsequently, 100 ng of sheared genomic DNA were used for methylated CpG DNA immunoprecipitation. Methylated DNA was recovered and subjected to PCR analysis with an ABI OneStepPlus real-time PCR system using the same Notch-1 promoter primers used in the ChIP experiments (see sequences above). Equal amounts (100 ng) of completely (100%) methylated human DNA and demethylated human DNA (Zymo Research) were included as the input and negative control.
CREMα Transgenic Mice
Transgenic mice on a FVB background with T cell-specific CREMα overexpression (under the control of the CD2 promoter) have recently been described (33). Naive T cells from these mice were obtained from spleens by negative isolation using magnetic cell separation with CD4+ MACS® kits (Miltenyi) according to the manufacturer's instructions. T cells (5 million) were used for extracting RNA (RNeasy mini kit). cDNA was synthesized using RevertAid H minus transcriptase (Fermentas GmbH, St. Leon-Rot, Germany). Real-time qPCR was performed using a SYBR Green PCR kit (Eurogentec, Cologne, Germany). The following primers were used: Notch-1, 5′-actatctcggcggcttttc-3′ (forward) and 5′-ggcactcgttgatctcctct-3′ (reverse); and β-actin, 5′-actattggcaacgagcggttc-3′ (forward) and 5′-ttacggatgtcaacgtcacacttc-3′ (reverse).
siRNA Experiments
Jurkat T cells were transfected with 10 nm irrelevant control siRNA and Notch-1-specific siRNA (OriGene) using Lipofectamine 2000 (Invitrogen) as described previously (6). Cells were collected 24 h after transfection and processed for mRNA analysis as indicated above.
Statistical Analyses
Student's paired two-tailed t test was used for statistical analysis. The Pearson product moment correlation coefficient (r) was used to determine the correlation between Notch-1 mRNA levels and individual SLE disease activity indices (SLEDAIs).
RESULTS
Notch-1 mRNA Expression Is Decreased in T Cells from Active SLE Patients
Notch-1 mRNA expression was analyzed by real-time qRT-PCR in total T cells obtained from a cohort of 61 SLE patients, with 32 of them being classified as active patients and 24 as healthy control individuals (Fig. 1). A composite SLEDAI, which reflects clinical symptoms and the degree of laboratory alterations in SLE patients, was used to define active patients (SLEDAI > 3). Notably, active patients displayed significantly lower Notch-1 mRNA levels (as assessed by normalized Ct values using the second derivative maximum method) than both inactive SLE patients (SLEDAI = 1–3) and healthy controls. Expression levels were not significantly different between healthy controls and inactive patients. mRNA expression analyses for another member of the Notch receptor family, i.e. Notch-2, did not yield major differences between these groups (data not shown). To further prove that Notch-1 mRNA expression varies with SLE disease activity levels, we performed longitudinal analyses in four SLE individuals and compared Notch-1 mRNA expression with the corresponding SLEDAIs over time (Fig. 2). Indeed, we observed strong negative correlations between Notch-1 mRNA magnitudes and SLEDAIs in the patients analyzed (Pearson's r between −0.56 and −0.90). Thus, the extent of Notch-1 mRNA expression mirrors disease activity in SLE patients.
FIGURE 1.

Notch-1 mRNA expression in T cells from SLE patients and healthy controls. Total T cell mRNAs from 24 healthy control individuals (CON) and 61 SLE patients (subgrouped according SLEDAIs) were analyzed for relative Notch-1 expression by real-time qRT-PCR. Crossing points (Ct) were calculated using the second derivative maximum method, an algorithm that requires minimal user input. Normalized Ct values (ΔCt = Ct(target gene) − Ct(average of control genes GAPDH and CD3ϵ)) were used to compare Notch-1 expression levels between different sample groups. To better visualize trends in relative Notch-1 mRNA expression, the y axis is displayed in an inverse manner. Horizontal lines indicate the mean ± S.D. ns, not significant.
FIGURE 2.

Longitudinal analyses of Notch-1 mRNA expression and corresponding SLEDAIs. Total T cell mRNAs from four SLE patients (SLE 1 to SLE 4) were collected every other month, and both relative Notch-1 mRNA expression in total T cells (normalized Ct values; left y axis, ▴) and SLEDAIs (right y axis, ♦) were determined at each time point. Individual Pearson's correlation coefficients (r) are given in the upper right corner of each panel.
Surface Expression of Notch-1 Protein Is Also Reduced in Active SLE Individuals
Next, we determined Notch-1 protein expression at the membrane surface of primary T cells purified from 12 SLE patients and 6 healthy controls by flow cytometry using an allophycocyanin-labeled anti-Notch-1 antibody (Fig. 3). These studies were performed in total T cells; however, co-staining for CD4 and CD8 surface markers allowed for more specific conclusions with regard to T cell subpopulations. Notch-1 protein expression was markedly decreased in active SLE individuals compared with inactive patients and healthy individuals. This difference reached the level of statistical significance in total T cells (Fig. 3, A and B) and CD4+ T helper cells (Fig. 3C), whereas the difference was almost significant in the CD8+ cytotoxic T cell subset (Fig. 3D).
FIGURE 3.

Notch-1 protein expression at the surface of T cells from SLE patients and healthy controls. A, CD3+ T cells from healthy control individuals (CON) and SLE patients were analyzed for Notch-1 protein expression by flow cytometry. Percentages of Notch-1+ cells are given in the diagram. B, a representative staining pattern is shown. T helper (CD3+CD4+; C) and cytotoxic (CD3+CD8+; D) T cells were analyzed for Notch-1 protein expression by flow cytometry. Horizontal lines indicate the mean ± S.D. ns, not significant.
Histone Methylation and CpG DNA Methylation Are Involved in Notch-1 Gene Regulation
Given the observed differences in Notch-1 expression between active SLE patients and inactive patients as well as healthy controls, we next investigated the underlying molecular mechanisms that mediate decreased Notch-1 expression in active SLE patients. We have previously demonstrated the prominent role of CREMα and histone and CpG DNA methylation in T cell-dependent target gene regulation (5–7, 32). Thus, we hypothesized that Notch-1 gene expression may be regulated by an aberrant methylation status of the Notch-1 gene promoter, which is highly conserved throughout evolution and comprises several putative CREs. To this end, we performed ChIP experiments in total T cells obtained from three age-, gender-, and ethnicity-matched pairs of active SLE patients (all SLEDAI > 3) and healthy controls with an antibody that specifically detects trimethylated Lys-27 of histone H3 (H3K27me3). Immunoprecipitated DNA was PCR-amplified, covering a region within the Notch-1 gene promoter that spans a CRE half-site, −991 to −988 bp upstream of the start codon (TGAC) (see Fig. 5A). Histone H3K27 trimethylation was significantly higher in SLE patients compared with healthy controls (Fig. 4, A and B). Furthermore, CpG DNA methylation was examined using methylated DNA immunoprecipitation assays in a cohort of 15 healthy individuals and 18 SLE patients (with 12 of them classified as active patients). Notably, all (active and inactive) SLE patients displayed an elevated CpG DNA methylation status compared with healthy individuals (Fig. 4C). Taken together, our findings argue for a concordant histone and CpG DNA hypermethylation at the Notch-1 gene promoter in T cells from active SLE patients.
FIGURE 5.
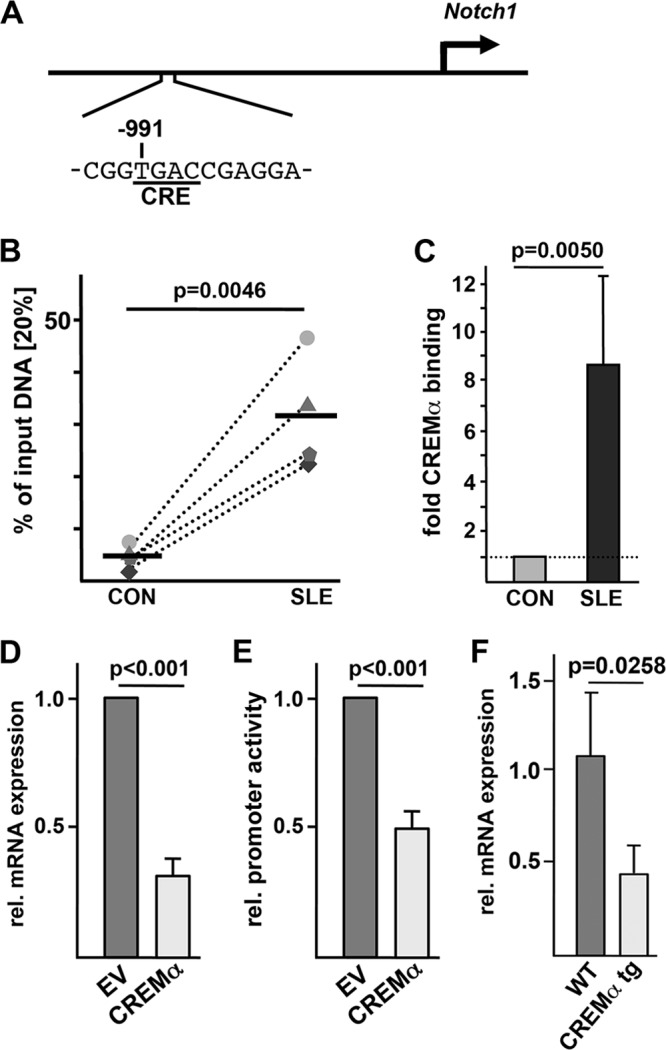
CREMα binds to and trans-represses the Notch-1 promoter. A, schematic of the human Notch-1 gene promoter indicating the CRE of interest located 991 bp upstream of the start codon. B, ChIP was performed using total T cells from four matched pairs of SLE patients and healthy controls (CON) and anti-CREMα antibody. Immunoprecipitated DNA was analyzed by real-time qRT-PCR using the same primers as used for the data in Fig. 4. Ratios between anti-CREMα antibody-immunoprecipitated and input DNAs are shown. Dotted lines associate data from the matched control/SLE pairs. Horizontal lines represent mean values. C, the percentage of anti-CREMα antibody-immunoprecipitated DNA in T cells from a control individual was set to 1.0, and the relative CREMα binding in the corresponding SLE patient was calculated. Values are given as mean ± S.D. D, pcDNA3 empty vector (EV) or CREMα expression plasmid was transfected into human Jurkat T cells, and 5 h after transfection, mRNA was analyzed for Notch-1 and 18 S rRNA expression by real-time qRT-PCR. E, a 2.1-bp spanning Notch-1 promoter sequence (within luciferase vector pGL3) was transfected into Jurkat T cells in the presence or absence of a CREMα expression plasmid, and 5 h after transfection, the relative (rel.) luciferase activity was determined. F, spleens from FVB WT and CREMα transgenic (tg) mice were collected, and CD4+ T cells were isolated by MACS® sorting. Murine Notch-1 and 18 S rRNA expression was analyzed by real-time qRT-PCR in these cells.
FIGURE 4.

Histone methylation and CpG DNA methylation of the Notch-1 promoter are increased in T cells from SLE patients. A, histone H3K27 methylation was analyzed in total T cells from three age-, gender-, and ethnicity-matched control (CON)/SLE pairs by ChIP assays. Dotted lines associate matched samples. A region of interest within the human Notch-1 promoter (harboring a putative CRE) was amplified by qPCR, and the proportion of immunoprecipitated DNA was calculated as relative to the non-immunoprecipitated input DNA in each sample. Subsequently, the ratio of relative expression was calculated between each SLE patient and the corresponding control individual. Horizontal lines represent mean values. B, the dotted line represents the H3K27 methylation status in control T cells, for each of which was set to 1.0. Changes in the methylation status in the matched SLE patient are given in the bar diagram (mean ± S.D.). C, total T cells from 15 healthy controls (light gray bar), 6 inactive SLE patients (dark gray bar), and 12 active SLE patients (black bar) were subjected to methylated DNA immunoprecipitation. The percentage of methylated DNA is given as mean ± S.D.
CREMα Affects Notch-1 Gene Transcription
The transcription factor CREMα has been demonstrated to be crucially involved in the trans-regulation and epigenetic “remodeling” of several gene loci that contribute to SLE pathogenesis (4, 5, 7). As the human Notch-1 promoter defines a putative CRE half-site within the region that we identified to be sensitive to methylation (Fig. 5A), we investigated whether CREMα indeed may bind to this element. Thus, we performed ChIP experiments in total T cells from four matched SLE/control pairs using a polyclonal anti-CREMα antibody. CREMα binding to the CRE (−991/−988) was significantly increased in T cells from SLE patients (all of them being active patients) compared with that in healthy control individuals (Fig. 5, B and C). To test for the functional relevance of CREMα binding to the Notch-1 promoter, we transiently overexpressed CREMα in a human T cell line (Jurkat). This approach was chosen because T cells from SLE patients display increased CREMα levels. Five hours after transfection, mRNA was collected and analyzed for Notch-1 transcript numbers by real-time qRT-PCR. An increased presence of CREMα in T cells led to significantly decreased Notch-1 mRNA expression (Fig. 5D). Most likely, this effect is mediated by direct trans-repression of the Notch-1 gene promoter through CREMα as evidenced by luciferase experiments in human T cells. A reporter construct harboring 2.1 bp of the human Notch-1 gene promoter showed significantly decreased activity when CREMα was co-introduced into these cells (Fig. 5E). Taken together, these findings support the hypothesis that CREMα physically binds to the Notch-1 promoter and acts as a potent repressor of Notch-1 gene transcription. Next, we analyzed splenic T cells from FVB mice that were transgenically engineered to express increased CREMα levels in their T lymphocytes (33). These mice have recently been shown to produce increased amounts of proinflammatory IL-17A and to be more prone to develop signs of autoimmunity that resemble the SLE phenotype. We examined Notch-1 mRNA expression in CD4+ T cells from these mice. Interestingly, we observed decreased Notch-1 mRNA levels in the CREMα-overexpressing mice compared with wild-type control mice (Fig. 5F). Hence, these mice display yet another feature of human SLE T cells, i.e. decreased Notch-1 expression.
Reduced Notch-1 Expression Is Linked to Increased IL-17A Levels
One of the most recently discovered T helper subsets (denoted Th17 cells) is characterized by abundant production of IL-17 cytokines. We and others have demonstrated the prominent role of IL-17 and Th17 cells in SLE pathogenesis. CREMα has been shown to act as a strong inducer of IL17A gene transcription and synthesis in human and murine T cells (5, 33). As Notch-1 expression is down-regulated in T cells from SLE patients, we wondered whether this might be of functional relevance for IL-17A production. To this end, we mimicked the “SLE phenotype” by silencing endogenous Notch-1 levels in human Jurkat T cells using siRNAs. mRNA was collected from these cells and analyzed for IL17A expression by real-time qRT-PCR. Indeed, reduced Notch-1 expression, as observed in T cells from active SLE patients, was associated with increased IL17A transcript numbers (Fig. 6A). Vice versa, we overexpressed a constitutively active Notch-1 construct that spans only the intracellular receptor domains in this T cell line. This approach yielded robustly decreased IL17A mRNA expression (Fig. 6B).
FIGURE 6.

Notch-1 levels control IL-17A synthesis. A, siRNA was used to down-regulate endogenous Notch-1 levels in human Jurkat T cells. (Irrelevant siRNA transfection was used in the control (CON) assays.) mRNA from these cells was analyzed for relative (rel.) IL17A expression by real-time qRT-PCR. B, human Jurkat T cells were transfected with a constitutively active Notch-1 construct (i.e. the intracellular receptor domains (Notch1ICD)) or pcDNA3 empty vector. Cells were harvested 5 h after transfection, and relative IL17A expression was determined by real-time qRT-PCR.
DISCUSSION
In this study, we have presented evidence that T cells from active SLE patients display significantly decreased basal levels of the transmembrane receptor Notch-1. This is in line with the previous report by Sodsai et al. (34) that SLE T cells fail to up-regulate Notch-1 expression after T cell receptor-mediated cell activation. However, the authors did not examine basal Notch-1 expression levels. Thus, our findings constitute the first description of Notch-1 expression in unstimulated T cells in a large cohort of active and non-active SLE patients and healthy control individuals. Furthermore, it has not been shown before that decreased Notch-1 transcript numbers are transduced into reduced Notch-1 protein levels.
Aberrant gene expression in immune cells from SLE patients has largely been attributed to specific epigenetic modifications, including histone and CpG DNA methylation, as well as aberrant transcriptional activities (32, 35). DNA hypomethylation is a well recognized key determinant in SLE pathogenesis, and several gene loci have been identified that are hypomethylated in SLE T cells, contributing to increased gene expression, e.g. IL4, IL6, IL10, IL13, IL17A, IFNγ, and protein phosphatase-2A (5, 36–39). Recently, it has become clear that there are also genes that may be hypermethylated in T cells from SLE patients, including IL2 (6, 7). We have now provided evidence that Notch-1 marks another gene with a markedly hypermethylated promoter region in T cells from SLE patients. This involves concordant histone H3 and CpG DNA hypermethylation. The epigenetic pattern under pathophysiological conditions is governed by specialized enzymes that are recruited to promoter regions and/or additional cis-regulatory regions. These enzymes alter histone tail or DNA modifications and comprise histone deacetylases and DNA methyltransferases (32). Usually, histone methylation and CpG DNA methylation follow the same pattern, i.e. either hypo- or hypermethylation (40). The mechanisms by which these modifications are induced during the development of autoimmune diseases remain poorly understood. We have previously reported that CREMα, which is the dominant CREM isoform in T cells from SLE patients, may regulate gene expression not only by virtue of its transcription factor capacities but also because it may directly interact with HDAC1 and DNMT3a and thus affect epigenetic modifications (7, 41). CREMα differentially induces or represses cytokine expression in SLE T cells.
In this study, we have demonstrated that CREMα binds to and trans-represses a CRE site within the Notch-1 promoter and thereby contributes to decreased Notch-1 gene expression. Furthermore, lowering endogenous Notch-1 levels using siRNA is associated with increased amounts of IL-17A, which is a hallmark of SLE T cells. Whether the reduced presence of Notch-1 at the surface of T cells from active SLE patients is associated with additional alterations observed in this autoimmune disease, including aberrant synthesis of other cytokines and/or impaired differentiation and proliferation capacities, remains to be elucidated in future studies.
Our findings provide evidence that (i) the gene and protein expression of Notch-1 receptors is markedly decreased in T cells from active SLE patients, (ii) transcript numbers inversely mirror disease activity in these patients, and (iii) epigenetic and CREMα-induced transcriptional effects are important upstream mechanisms to explain this phenomenon. Thus, Notch-1 constitutes a novel molecule within the growing network of T cell-relevant genes that are tightly controlled by the transcription factor CREMα.
The observed epigenetic patterns and transcriptional CREMα effects at the Notch-1 promoter are very similar to those observed at the IL2 gene locus (i.e. histone H3K27 and CpG DNA hypermethylation, increased CREMα binding, and direct trans-repression of the IL2 promoter through CREMα). This suggests congenerous epigenetic and CREMα mechanisms at multiple gene loci in SLE T cells. CREMα expression in SLE T cells strongly correlates with SLE disease activity at the promoter and mRNA and protein expression levels (9, 12). Thus, we hypothesize that, among the observed regulatory mechanisms that control Notch-1 expression in SLE T cells, transcriptional repression through CREMα is the most decisive one. This idea is also supported by our observation that the overall CpG DNA methylation of the Notch-1 promoter is significantly increased in inactive SLE patients, whereas Notch-1 expression levels do not differ between controls and inactive patients. We conclude that the impaired epigenetic pattern of an increased histone and CpG DNA methylation in SLE T cells (inactive and active) constitutes the “background condition” of the Notch-1 promoter, but it is only the repressive effects exerted by CREMα that really make the significant differences in active patients.
Taken together, the results indicate that the CREMα/Notch-1/IL-17A axis appears to be part of the impaired cytokine network in T cells from SLE patients. Corrections along these lines, e.g. by the development of strategies to increase or reactivate Notch-1 signaling in T cells from active SLE patients and/or target the outlined upstream molecules, might well be of therapeutic importance. It has been shown in murine lupus models that a pan-Notch blockade using γ-secretase inhibitors results in decreased autoantibody production and kidney pathology (24). However, application of these agents is not feasible in humans (27). More specific interventions targeting individual Notch receptors in autoimmune diseases appear to be more promising but demand a systematic expression analysis of the involved immune and resident tissue-specific cells. In this context, this study contributes novel data on reduced Notch-1 expression in T cells from active SLE patients (whereas Notch-2 is not regulated) and the underlying molecular mechanisms. Once more, the transcription factor CREMα arises as a promising target to correct cytokine and disease expression in patients with SLE and other autoimmune diseases.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 AI42269, R01 AI49954, and R01 AI85567 (to G. C. T.). This work was also supported by Deutsche Forschungsgemeinschaft Grant RA1927-1/1 and a START Grant for Young Researchers of the Medical Faculty of RWTH University of Aachen (to T. R.) and Interdisziplinäres Zentrum für Klinische Forschung (IZKF) Aachen Grant E6-6 (to K. T.).
- SLE
- systemic lupus erythematosus
- CREMα
- cAMP-responsive element modulator α
- CRE
- cAMP-responsive element
- qRT-PCR
- quantitative RT-PCR
- SLEDAI
- SLE disease activity index.
REFERENCES
- 1. Tsokos G. C. (2011) Systemic lupus erythematosus. N. Engl. J. Med. 365, 2110–2121 [DOI] [PubMed] [Google Scholar]
- 2. Grammatikos A. P., Tsokos G. C. (2012) Immunodeficiency and autoimmunity: lessons from systemic lupus erythematosus. Trends Mol. Med. 18, 101–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ahlmann M., Varga G., Sturm K., Lippe R., Benedyk K., Viemann D., Scholzen T., Ehrchen J., Müller F. U., Seidl M., Matus M., Tsokos G. C., Roth J., Tenbrock K. (2009) The cyclic AMP response element modulator α suppresses CD86 expression and APC function. J. Immunol. 182, 4167–4174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ghosh D., Kis-Toth K., Juang Y. T., Tsokos G. C. (2012) CREMα suppresses spleen tyrosine kinase expression in normal but not systemic lupus erythematosus T cells. Arthritis Rheum. 64, 799–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rauen T., Hedrich C. M., Juang Y. T., Tenbrock K., Tsokos G. C. (2011) cAMP-responsive element modulator (CREM) α protein induces interleukin-17A expression and mediates epigenetic alterations at the interleukin-17A gene locus in patients with systemic lupus erythematosus. J. Biol. Chem. 286, 43437–43446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hedrich C. M., Rauen T., Kis-Toth K., Kyttaris V. C., Tsokos G. C. (2012) cAMP-responsive element modulator α (CREMα) suppresses IL-17F protein expression in T lymphocytes from patients with systemic lupus erythematosus (SLE). J. Biol. Chem. 287, 4715–4725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hedrich C. M., Rauen T., Tsokos G. C. (2011) cAMP-responsive element modulator (CREM) α protein signaling mediates epigenetic remodeling of the human interleukin-2 gene: implications in systemic lupus erythematosus. J. Biol. Chem. 286, 43429–43436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kyttaris V. C., Juang Y. T., Tenbrock K., Weinstein A., Tsokos G. C. (2004) Cyclic adenosine 5′-monophosphate response element modulator is responsible for the decreased expression of c-fos and activator protein-1 binding in T cells from patients with systemic lupus erythematosus. J. Immunol. 173, 3557–3563 [DOI] [PubMed] [Google Scholar]
- 9. Solomou E. E., Juang Y. T., Gourley M. F., Kammer G. M., Tsokos G. C. (2001) Molecular basis of deficient IL-2 production in T cells from patients with systemic lupus erythematosus. J. Immunol. 166, 4216–4222 [DOI] [PubMed] [Google Scholar]
- 10. Tenbrock K., Tsokos G. C. (2004) Transcriptional regulation of interleukin 2 in SLE T cells. Int. Rev. Immunol. 23, 333–345 [DOI] [PubMed] [Google Scholar]
- 11. Rauen T., Benedyk K., Juang Y. T., Kerkhoff C., Kyttaris V. C., Roth J., Tsokos G. C., Tenbrock K. (2011) A novel intronic cAMP response element modulator (CREM) promoter is regulated by activator protein-1 (AP-1) and accounts for altered activation-induced CREM expression in T cells from patients with systemic lupus erythematosus. J. Biol. Chem. 286, 32366–32372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Juang Y. T., Rauen T., Wang Y., Ichinose K., Benedyk K., Tenbrock K., Tsokos G. C. (2011) Transcriptional activation of the cAMP-responsive modulator promoter in human T cells is regulated by protein phosphatase 2A-mediated dephosphorylation of SP-1 and reflects disease activity in patients with systemic lupus erythematosus. J. Biol. Chem. 286, 1795–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bour-Jordan H., Esensten J. H., Martinez-Llordella M., Penaranda C., Stumpf M., Bluestone J. A. (2011) Intrinsic and extrinsic control of peripheral T-cell tolerance by costimulatory molecules of the CD28/B7 family. Immunol. Rev. 241, 180–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Veillette A., Latour S., Davidson D. (2002) Negative regulation of immunoreceptor signaling. Annu. Rev. Immunol. 20, 669–707 [DOI] [PubMed] [Google Scholar]
- 15. Scalapino K. J., Daikh D. I. (2008) CTLA-4: a key regulatory point in the control of autoimmune disease. Immunol. Rev. 223, 143–155 [DOI] [PubMed] [Google Scholar]
- 16. Chatterjee M., Rauen T., Kis-Toth K., Kyttaris V. C., Hedrich C. M., Terhorst C., Tsokos G. C. (2012) Increased expression of SLAM receptors SLAMF3 and SLAMF6 in systemic lupus erythematosus T lymphocytes promotes Th17 differentiation. J. Immunol. 188, 1206–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bray S. J. (2006) Notch signalling: a simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 7, 678–689 [DOI] [PubMed] [Google Scholar]
- 18. D'Souza B., Miyamoto A., Weinmaster G. (2008) The many facets of Notch ligands. Oncogene 27, 5148–5167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rauen T., Raffetseder U., Frye B. C., Djudjaj S., Mühlenberg P. J., Eitner F., Lendahl U., Bernhagen J., Dooley S., Mertens P. R. (2009) YB-1 acts as a ligand for Notch-3 receptors and modulates receptor activation. J. Biol. Chem. 284, 26928–26940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. De Strooper B., Annaert W., Cupers P., Saftig P., Craessaerts K., Mumm J. S., Schroeter E. H., Schrijvers V., Wolfe M. S., Ray W. J., Goate A., Kopan R. (1999) A presenilin-1-dependent γ-secretase-like protease mediates release of Notch intracellular domain. Nature 398, 518–522 [DOI] [PubMed] [Google Scholar]
- 21. Rizzo P., Miao H., D'Souza G., Osipo C., Song L. L., Yun J., Zhao H., Mascarenhas J., Wyatt D., Antico G., Hao L., Yao K., Rajan P., Hicks C., Siziopikou K., Selvaggi S., Bashir A., Bhandari D., Marchese A., Lendahl U., Qin J. Z., Tonetti D. A., Albain K., Nickoloff B. J., Miele L. (2008) Cross-talk between Notch and the estrogen receptor in breast cancer suggests novel therapeutic approaches. Cancer Res. 68, 5226–5235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Iso T., Kedes L., Hamamori Y. (2003) HES and HERP families: multiple effectors of the Notch signaling pathway. J. Cell. Physiol. 194, 237–255 [DOI] [PubMed] [Google Scholar]
- 23. Ehebauer M., Hayward P., Arias A. M. (2006) Notch, a universal arbiter of cell fate decisions. Science 314, 1414–1415 [DOI] [PubMed] [Google Scholar]
- 24. Teachey D. T., Seif A. E., Brown V. I., Bruno M., Bunte R. M., Chang Y. J., Choi J. K., Fish J. D., Hall J., Reid G. S., Ryan T., Sheen C., Zweidler-McKay P., Grupp S. A. (2008) Targeting Notch signaling in autoimmune and lymphoproliferative disease. Blood 111, 705–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jurynczyk M., Jurewicz A., Raine C. S., Selmaj K. (2008) Notch3 inhibition in myelin-reactive T cells down-regulates protein kinase Cθ and attenuates experimental autoimmune encephalomyelitis. J. Immunol. 180, 2634–2640 [DOI] [PubMed] [Google Scholar]
- 26. Riccio O., van Gijn M. E., Bezdek A. C., Pellegrinet L., van Es J. H., Zimber-Strobl U., Strobl L. J., Honjo T., Clevers H., Radtke F. (2008) Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27Kip1 and p57Kip2. EMBO Rep. 9, 377–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu Y., Cain-Hom C., Choy L., Hagenbeek T. J., de Leon G. P., Chen Y., Finkle D., Venook R., Wu X., Ridgway J., Schahin-Reed D., Dow G. J., Shelton A., Stawicki S., Watts R. J., Zhang J., Choy R., Howard P., Kadyk L., Yan M., Zha J., Callahan C. A., Hymowitz S. G., Siebel C. W. (2010) Therapeutic antibody targeting of individual Notch receptors. Nature 464, 1052–1057 [DOI] [PubMed] [Google Scholar]
- 28. Hochberg M. C. (1997) Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 40, 1725. [DOI] [PubMed] [Google Scholar]
- 29. Bailey J., Tyson-Capper A. J., Gilmore K., Robson S. C., Europe-Finner G. N. (2005) Identification of human myometrial target genes of the cAMP pathway: the role of cAMP-response element binding (CREB) and modulator (CREMα and CREMτ2α) proteins. J. Mol. Endocrinol. 34, 1–17 [DOI] [PubMed] [Google Scholar]
- 30. Liu Z., Teng L., Bailey S. K., Frost A. R., Bland K. I., LoBuglio A. F., Ruppert J. M., Lobo-Ruppert S. M. (2009) Epithelial transformation by KLF4 requires Notch1 but not canonical Notch1 signaling. Cancer Biol. Ther. 8, 1840–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sakamoto K., Yamaguchi S., Ando R., Miyawaki A., Kabasawa Y., Takagi M., Li C. L., Perbal B., Katsube K. (2002) The nephroblastoma overexpressed gene (NOV/ccn3) protein associates with Notch1 extracellular domain and inhibits myoblast differentiation via Notch signaling pathway. J. Biol. Chem. 277, 29399–29405 [DOI] [PubMed] [Google Scholar]
- 32. Hedrich C. M., Tsokos G. C. (2011) Epigenetic mechanisms in systemic lupus erythematosus and other autoimmune diseases. Trends Mol. Med. 17, 714–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lippe R., Ohl K., Varga G., Rauen T., Crispin J. C., Juang Y. T., Kuerten S., Tacke F., Wolf M., Roebrock K., Vogl T., Verjans E., Honke N., Ehrchen J., Foell D., Skryabin B., Wagner N., Tsokos G. C., Roth J., Tenbrock K. (2012) CREMα overexpression decreases IL-2 production, induces a TH17 phenotype and accelerates autoimmunity. J. Mol. Cell Biol. 4, 121–123 [DOI] [PubMed] [Google Scholar]
- 34. Sodsai P., Hirankarn N., Avihingsanon Y., Palaga T. (2008) Defects in Notch1 upregulation upon activation of T cells from patients with systemic lupus erythematosus are related to lupus disease activity. Lupus 17, 645–653 [DOI] [PubMed] [Google Scholar]
- 35. Tenbrock K., Juang Y. T., Kyttaris V. C., Tsokos G. C. (2007) Altered signal transduction in SLE T cells. Rheumatology 46, 1525–1530 [DOI] [PubMed] [Google Scholar]
- 36. Mi X. B., Zeng F. Q. (2008) Hypomethylation of interleukin-4 and -6 promoters in T cells from systemic lupus erythematosus patients. Acta Pharmacol. Sin. 29, 105–112 [DOI] [PubMed] [Google Scholar]
- 37. Janson P. C., Marits P., Thörn M., Ohlsson R., Winqvist O. (2008) CpG methylation of the IFNG gene as a mechanism to induce immunosuppression in tumor-infiltrating lymphocytes. J. Immunol. 181, 2878–2886 [DOI] [PubMed] [Google Scholar]
- 38. Sunahori K., Juang Y. T., Tsokos G. C. (2009) Methylation status of CpG islands flanking a cAMP response element motif on the protein phosphatase 2Acα promoter determines CREB binding and activity. J. Immunol. 182, 1500–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhao M., Tang J., Gao F., Wu X., Liang Y., Yin H., Lu Q. (2010) Hypomethylation of IL10 and IL13 promoters in CD4+ T cells of patients with systemic lupus erythematosus. J. Biomed. Biotechnol. 2010, 931018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brenner C., Fuks F. (2007) A methylation rendezvous: reader meets writers. Dev. Cell 12, 843–844 [DOI] [PubMed] [Google Scholar]
- 41. Tenbrock K., Juang Y. T., Leukert N., Roth J., Tsokos G. C. (2006) The transcriptional repressor cAMP response element modulator α interacts with histone deacetylase 1 to repress promoter activity. J. Immunol. 177, 6159–6164 [DOI] [PubMed] [Google Scholar]