

Background: The human equilibrative glucose transporter GLUT1 catalyzes accelerated-exchange transport, but the related isoform GLUT4 does not.
Results: Substitution of GLUT1 transmembrane helix 6 into GLUT4 permits GLUT4 to catalyze accelerated-exchange, whereas GLUT4 helix 6 prevents accelerated-exchange in GLUT1.
Conclusion: Transmembrane helix 6 sequence determines accelerated-exchange behavior.
Significance: Transmembrane helices external to the transport pathway influence transport behavior.
Keywords: Glucose Transport, Membrane Proteins, Membrane Transport, Mutagenesis, Sugar Transport
Abstract
The class 1 equilibrative glucose transporters GLUT1 and GLUT4 are structurally similar but catalyze distinct modes of transport. GLUT1 exhibits trans-acceleration, in which the presence of intracellular sugar stimulates the rate of unidirectional sugar uptake. GLUT4-mediated uptake is unaffected by intracellular sugar. Using homology-scanning mutagenesis in which domains of GLUT1 are substituted with equivalent domains from GLUT4 and vice versa, we show that GLUT1 transmembrane domain 6 is both necessary and sufficient for trans-acceleration. This region is not directly involved in GLUT1 binding of substrate or inhibitors. Rather, transmembrane domain 6 is part of two putative scaffold domains, which coordinate membrane-spanning amphipathic helices that form the sugar translocation pore. We propose that GLUT1 transmembrane domain 6 restrains import when intracellular sugar is absent by slowing transport-associated conformational changes.
Introduction
The GLUT family of glucose transporters catalyzes tissue-specific facilitative monosaccharide transport in mammalian cells (1). GLUT1 mediates sugar uptake in red blood cells, smooth muscle, and across blood-tissue barriers (2–4). GLUT4 is expressed in adipose tissue, skeletal, and cardiac muscle (5), where it is responsible for insulin-stimulated sugar uptake (6, 7).
Although GLUTs 1 and 4 exhibit similar affinities for substrates and antagonists (8, 9), their catalytic behaviors are very different. GLUT4 displays kinetic symmetry (Vmax and Km for net sugar uptake are indistinguishable from the corresponding parameters for net exit (10)), whereas GLUT1 kinetics are asymmetric (Vmax and Km for net sugar uptake are significantly lower than the corresponding parameters for net exit (11)). In addition, GLUT1 displays a behavior termed trans-acceleration,2 whereas GLUT4 does not (10, 12–15). Trans-acceleration (also called accelerated-exchange transport) occurs when unidirectional uptake of sugar is stimulated by the presence of intracellular sugar or, conversely, when unidirectional exit of sugar is stimulated by the presence of extracellular sugar (16). Trans-acceleration may provide a metabolic advantage to the cell because it results in a more rapid equilibration of the cytoplasm with extracellular sugar.3 Trans-acceleration is one of several behaviors that distinguishes carrier-mediated- from channel-mediated facilitative diffusion systems (17, 18), but the physical basis of accelerated-exchange transport is unknown. Comparative analysis of GLUTs 1 and 4 may, therefore, permit definition of the sequence determinants and thereby the physical basis of trans-acceleration.
GLUTs 1 and 4 are structurally similar, containing cytoplasmic N and C termini, 12 transmembrane spanning α-helices (TMs),4 and a large intracellular loop connecting TMs 6 and 7 (19–21). In the absence of GLUT crystal structures, our understanding of GLUT1 tertiary structure derives largely from scanning cysteine mutagenesis (22–25) and modeling studies (26, 27), which align and thread the GLUT1 sequence through the crystal structures of Major Facilitator Superfamily bacterial transporter homologs GlpT (28) and LacY (29). Although these homology-based threaded structures provide quite accurate descriptions of transporter topography and helix packing arrangements, they fail to accurately predict helix and amino acid side chain orientation within the active sites (30).
Some functional domains of GLUT1 have been mapped at low resolution. These include components of the GLUT1 nucleotide binding domain (31–33), substrate binding sites (24, 34), inhibitor binding sites (35, 36), allosteric modulation sites (37–39) and oligomerization domains (40).5 However, detailed structures of these domains and the conformational changes associated with transport are not yet available. In addition, analogous modeling studies have yet to be extended to GLUT4. Thus the available data do not yet provide an explanation for substrate binding and translocation by GLUTs 1 and 4, or why GLUT1 catalyzes trans-acceleration but GLUT4 does not.
This study attempts to investigate the determinants of transporter function using homology-scanning mutagenesis of structurally related but functionally different members of a transporter family. We engineered GLUT1 and GLUT4 chimeras in which we substituted progressively smaller domains of one transporter by the corresponding domains of the other transporter. These chimeras exhibit sequence-dependent trans-acceleration gain- or loss-of-function.
We observe trans-acceleration in GLUT1-transfected HEK cells but not in cells transfected with human GLUT4. Homology-scanning mutagenesis reveals that TM6 of GLUT1 is both necessary and sufficient to confer trans-acceleration to the GLUT4 scaffold. Similarly, the replacement of GLUT1 TM6 with the corresponding region in GLUT4 ablates trans-acceleration in the GLUT1 scaffold.
These results confirm that trans-acceleration is sequence-dependent, requiring a motif within the putative scaffold region of GLUT1, rather than in the translocation pore-forming region of the protein. The implications of our findings are discussed in the context of the prevailing models for GLUT-mediated sugar transport.
EXPERIMENTAL PROCEDURES
Materials
[3H]2-Deoxy-d-glucose was purchased from MP Biomedical. HEK-293 cells were purchased from ATCC. DMEM, DPBS, penicillin/streptomycin, Lipofectamine 2000, DH5α-Subcloning cells, PCDNA 3.1(+) mammalian expression vector, BisTris gels, and MES buffer were obtained from Invitrogen. All restriction enzymes and associated buffers were obtained from New England Biolabs. All primers were purchased from Integrated DNA Technologies. Herculase polymerase, XL1-Blue Competent cells, and QuikChange Multisite-directed Mutagenesis kits were obtained from Stratagene. RNeasy, Qiashredder, One-Step RT-PCR, MinElute Gel Purification, PCR Purification, and HiSpeed Maxi kits were from Qiagen. iScript One-Step PCR kit with SYBR Green was purchased from Bio-Rad. PVDF membranes were obtained from ThermoFisher. 10% Bovine serum albumin was from American Bioanalytical. SuperSignal Pico West, NeutrAvidin Gel, micro-BCA kits, spin columns, and EZ-Link Sulfo-NHS-SS-Biotin were from Pierce. Protease inhibitor mixture tablets were from Roche Applied Science. Other reagents were purchased from Sigma.
Solutions
Cell lysis buffer consisted of DPBS, 0.5% Triton X-100 plus protease inhibitiors with EDTA. TBS contained 20 mm Tris base, 135 mm NaCl, pH 7.6. Biotin lysis and column wash buffer comprised TBS, 0.5% Triton X-100, and protease inhibitors with EDTA. The sample buffer consisted of 0.5 m Tris-Cl, pH 6.8, 40% (v/v) glycerol, 8% SDS, bromphenol blue, and 150 mm DTT; biotinylation sample buffer did not contain bromphenol blue. DPBS-Mg contained 5 mm MgCl2. 2-DG uptake solution contained 100 μm 2-DG with 2.5 μCi/ml of [3H]2-DG in DPBS-Mg. Stop solution consisted of 10 μm cytochalasin B and 100 μm phloretin in DPBS-Mg. Triton extraction buffer contained 0.5% Triton X-100 and 50 μm EDTA in DPBS-Mg. 2-DG/3-MG uptake solution consisted of 100 μm 2-DG with 2.5 μCi/ml of [3H]2-DG in DPBS-Mg with 40 mm 3-MG.
Antibodies
A custom-made (New England Peptide) affinity-purified rabbit polyclonal antibody raised against a peptide corresponding to GLUT1 C-terminal residues 480–492 was used at 1:10,000 dilution as described previously (33). A rabbit polyclonal anti-GLUT4 C-terminal antibody (Sigma G4048) was used at 1:3,000 dilution. A rabbit polyclonal anti-Myc antibody (Abcam ab9106) was used at 1:5,000 dilution. Horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (Jackson ImmunoResearch) was used at 1:50,000 dilution. A rabbit polyclonal anti-Na,K-ATPase antibody (Cell Signaling Technologies 3010) was used at 1:500 dilution.
Tissue Culture
HEK-293 cell culture was as described previously (42). All experiments were performed with confluent cells. Plates were subcultured into 12-well plates at a ratio of 1:2–1:4 2–4 days prior to transfection. Passages 4–19 were used for all experiments.
Mutagenesis
GLUT1- or GLUT4-encoding cDNA was inserted into the EcoRV-NotI restriction sites of PCDNA 3.1(+). Myc-tagged constructs were made using overlapping primers to insert the Myc tag in exofacial loop 1, between GLUT1 residues 55 and 56 or GLUT4 residues 72 and 73. All TM domain chimeras were engineered by designing overlapping primers for each region of interest, amplifying each fragment via PCR using Herculase polymerase, purifying each fragment with the MinElute Gel Purification kit, joining fragments by PCR, and repeating until a full-length insert was obtained. The insert was purified by the PCR purification kit, digested with restriction enzymes, purified by the MinElute Gel Purification kit, and inserted into PCDNA 3.1(+) with the same restriction sites. All final constructs were subcloned into either XL1-Blue competent cells or DH5α subcloning cells, purified using a HiSpeed Maxi kit, and verified by sequencing analysis (Davis Sequencing, Davis, CA). All point mutations and amino acid substitutions were engineered using QuikChange Multisite-directed Mutagenesis kits and verified by sequencing.
Quantitative and End Point Reverse Transcriptase-PCR
Total RNA was isolated from HEK cells using the RNeasy kit and Qiashredder. End point RT-PCR was performed as per the One-Step RT-PCR kit instructions using GLUT-specific primers. RT-PCR products were resolved on a 1.5% agarose gel and visualized by ethidium bromide staining. Expression levels of detected GLUTs were measured by quantitative RT-PCR using the iScript One-Step PCR kit with SYBR Green. Samples were run in duplicate on an MJ Research PTC-200 Peltier Thermal Cycler with a Chromo4 real time-PCR detector running Opticon Monitor 3 software (Bio-Rad). Results were analyzed by using the ΔΔCt method (43) and normalized to a GAPDH control.
Transient Transfection
Cells (70–90% confluence) were transfected with 2 μg of DNA per well (12 well plates) or 5 μg of DNA per well (6 well plates), unless otherwise specified. Transfections were performed 36–48 h prior to analysis of sugar uptake or protein expression.
Western Blotting
Cells were pelleted, washed with DPBS, lysed in cell lysis buffer, and protein concentration was assessed using a micro-BCA kit. Lysates were normalized for total protein concentration and resolved by SDS-PAGE on a 10% BisTris gel in MES buffer. Gels were transferred onto PVDF membranes, blocked with 10% bovine serum albumin in TBS-T, probed with primary antibody overnight at 4 °C, probed with secondary antibody for 1 h at room temperature, and developed using SuperSignal Pico West Chemiluminescent substrate. Blots were imaged on a FujiFilm LAS-3000 and relative band densities were quantitated using ImageJ software.
Biotinylation
6-Well plates of HEK cells were washed twice with ice-cold DPBS and incubated on ice with ice-cold DPBS containing 5 mm EZ-Link Sulfo-NHS-SS-Biotin for 30 min with gentle rocking. Reactions were quenched by adjusting each well to 12.5 mm Trizma (Tris base). Cells were harvested, re-suspended in biotin lysis buffer, and lysates were bound to Neutravidin Gel in spin columns according to kit instructions. Protein concentration was determined spectrophotometrically, and normalized loads were analyzed by Western blot as described above.
2-Deoxy-d-glucose Sugar Uptake
2-Deoxy-d-glucose (2-DG) is an analog of the natural GLUT1 substrate, d-glucose. At physiologic temperature, cytoplasmic 2-DG is phosphorylated by hexokinase to form 2-deoxy-d-glucose 6-phosphate (2-DG-6-P), which is neither metabolized further nor is a GLUT1 substrate (44, 45). Imported [3H]2-DG is therefore trapped within the cell as [3H]2-DG-6-P.
2-DG uptake was measured as described previously (42). Briefly, 36–48 h post-transfection, 12-well plates of confluent HEK-293 cells were serum- and glucose-starved for 2 h at 37 °C in FBS- and penicillin/streptomycin-free DMEM lacking glucose. Cells were washed with 0.5 ml of DPBS-Mg at 37 °C, then exposed to 0.5 ml of [3H]2-DG uptake solution for 0 to 30 min at 37 °C. Uptake was stopped by addition of 1 ml of ice-cold stop solution. Cells were washed twice with ice-cold stop solution and extracted with Triton extraction buffer. Total protein concentration was analyzed in duplicate. Each sample was counted in duplicate by liquid scintillation spectrometry. Each condition was performed in triplicate on at least 3 separate assay occasions.
Zero-trans and Hetero-exchange Transport Measurements
Zero-trans sugar uptake describes uptake of sugar into cells lacking intracellular sugar. Hetero-exchange sugar uptake describes uptake of sugar into cells preloaded with a different, but transportable sugar (46). Zero-trans uptake of [3H]2-DG by sugar-depleted cells was measured as described above with one modification, the transported sugar 3-O-methylglucose (3-MG; 40 mm) was included in the uptake medium. Extracellular 3-MG competitively inhibits [3H]2-DG uptake and thus permits more accurate measurement of transport over a 5-min interval at physiologic temperature (37 °C). Because 3-MG is not a hexokinase substrate (47), any 3-MG that enters the cells during transport or by pre-loading the cells does not compete with intracellular 2-DG for interaction with hexokinase. In hetero-exchange uptake, the concentration of 3-MG inside and outside of the cell is identical (40 mm). This is accomplished by adding 40 mm 3-MG to glucose-free DMEM during serum starvation and DPBS-Mg used in pre-uptake washes.
Data Analysis
All data analysis was performed using GraphPad Prism (La Jolla, CA, version 5.0). For sugar uptake experiments, background counts were subtracted from all samples and uptake, v, was normalized to [total protein]/well. [3H]2-DG uptake (DPM/μg) was then converted to mole/μg of protein/min by using the measured specific activity of the uptake solution. Average uptake in mock-transfected controls was subtracted from the uptake of the corresponding transfected samples. For experiments where [3-MG]i was varied, sugar uptake was fitted to the following approximation,
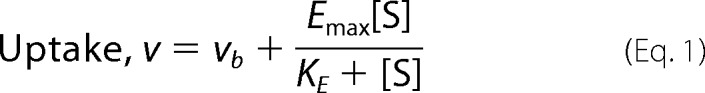 |
by nonlinear regression analysis, where vb is sugar uptake in the absence of 3-MGi, Emax is the stimulation of sugar uptake produced by saturating 3-MGi, [S] is the concentration of 3-MGi and KE is that concentration of 3-MGi, which stimulates sugar uptake by 50%.
In experiments evaluating whether trans-acceleration is observed, the ratio of hetero-exchange (HE) uptake:zero-trans uptake (ZT) was computed for each construct. As we shall show, this normalizes for experimental variation in transporter cell surface expression. When this ratio is significantly greater than 1, trans-acceleration is present. The results of paired experiments were analyzed using Student's t test.
RESULTS
GLUT1 and GLUT4 Chimeras
To identify GLUT1 domain(s) required for trans-acceleration, we swapped specific transmembrane regions of GLUT1 with an equivalent GLUT4 sequence. This allows us to map the involvement of large regions of the transporter in trans-acceleration and thereby narrow our focus to smaller subdomains (Fig. 1). Chimera nomenclature divides the GLUTs into 4 sets of three contiguous TMs (1–3, 4–6, 7–9, 10–12). A chimera comprising the first half of GLUT1 plus the second half of GLUT4 is termed “1144,” GLUT1 TMs 1–3 and 4–6 plus GLUT4 TMs 7–9 and 10–12. If loop 6 linking TMs 6 and 7 is the focus, this is indicated in parentheses. Thus, 44(1)11 is GLUT4 TMs 1–6, GLUT1 loop 6, and GLUT1 TMs 7–12. 1411 is GLUT1 TMs 1–3 plus GLUT4 TMs 4–6 plus GLUT1 loop 6 and TMs 7–12 (Fig. 1B). Mutations involving only 1 or 2 TMs list the scaffold GLUT with substitutions from the other GLUT in parentheses, e.g. GLUT4(5,6 G1) is GLUT4 containing GLUT1 TMs 5–6.
FIGURE 1.

GLUT1/GLUT4 chimeras. Schematic of GLUT topology; trans-membrane domains (1–12) are numbered in blocks of 3. Intracellular loop 6 (L6) connecting TMs 6–7, and the carboxyl terminus (Ct) are cytoplasmic. The location of the inserted exofacial c-Myc epitope (EQKLISEEDL) is shown in red (GLUT1, between residues 55 and 56 and in GLUT4, between residues 72 and 73). Chimeric junctions between GLUT1 and GLUT4 quarter chimeras and in chimeras containing specific substitutions of TMs 4, 5, and 6 are shown schematically. Their specific sequence is described in Table 1.
Trans-acceleration in HEK Cells
HEK-293 cells were selected for heterologous expression of GLUT1, GLUT4, and GLUT1-GLUT4 chimeras because of their very low endogenous expression of human GLUTs 1 and 4, as determined by quantitative PCR (Fig. 2A). Net uptake of 100 μm 2-DG from medium containing 40 mm 3-MG increases linearly with time (0–10 min) in GLUT1-transfected cells (Fig. 2B). The experiments reported in this study employ a 5-min uptake interval, which provides an ample range of linearity to detect a 2–3-fold increase in 2-DG uptake during hetero-exchange transport catalyzed by transfected GLUTs. To characterize differences in sugar uptake in response to intracellular sugar, [3H]2-DG uptake from medium containing 40 mm 3-MG was measured in GLUT1- or GLUT4-transfected HEK cells pre-loaded with 0 to 40 mm 3-MG (Fig. 3A). GLUT1-transfected HEK cells show a dose-dependent stimulation of 2-DG uptake with increasing intracellular 3-MG, whereas GLUT4-transfected HEK cells do not. This confirms that human GLUT1 displays trans-acceleration at 37 °C, whereas human GLUT4 does not. GLUT1-mediated 2-DG uptake increases in a saturable manner with [3-MG]i, showing a maximal stimulation (Emax) of 1.72 ± 0.02-fold with 50% stimulation (KE) at 25.6 ± 1.5 mm 3-MGi. 40 mm 3-MGi was used in all further hetero-exchange experiments.
FIGURE 2.
Characterization of HEK cell GLUT expression and transport. A, analysis of HEK cell endogenous GLUT message expression by quantitative PCR analysis. HEK cells were screened for the presence of all GLUTs by end point RT-PCR. Quantitative RT-PCR was then used to compare message expression levels. Expression relative to GLUT12 message (ordinate) is plotted versus GLUT identity (abscissa). The significance of the difference between GLUT12 and other GLUTs (***, p ≤ 0.005) was computed by an unpaired, two-tailed Student's t test. B, time course of [3H]2-DG uptake in control and GLUT1-transfected HEK cells. Ordinate, 2-DG uptake in disintegrations/min per μg of total cell protein; abscissa, time in minutes. 2-DG uptake over time were plotted for mock-transfected (●) and GLUT1-transfected (○) cells. Lines drawn through the points were computed by linear regression. GLUT1-mediated hetero-exchange is indicated by the triangle, and the gray rectangle indicates the range of uptake observed during stimulated hetero-exchange transport at 5 min (see Table 1).
FIGURE 3.

Sugar uptake in HEK cells expressing GLUT1, GLUT4, and Myc-tagged GLUTs. A, dose-response of 100 μm 2-DG uptake (ordinate) from medium containing 40 mm 3-MG into HEK cells pre-loaded with 0–40 mm 3-MG (abcissa) and transfected with GLUT1 (○) or GLUT4 (●). Basal uptake by mock-transfected cells was subtracted at each 3-MG concentration. Data points represent the mean ± S.E. for three separate assays. The curve drawn through the GLUT1 data was computed by nonlinear regression, assuming that uptake is described by Equation 1. The resulting analysis yields parameters of vb = 87 ± 3 fmol/μg/min; Exmax = 63 ± 2 fmol/μg/min; Ke = 25.8 ± 1.5 mm. The line drawn through the GLUT4 data was computed by linear regression. B, representative Western blot of whole cell lysates from HEK cells transfected with GLUT1myc or GLUT4myc constructs. Lysates were resolved by SDS-PAGE, transferred to PVDF membranes, and probed with α-Myc Ab. C, zero-trans 2-DG (100 μm) uptake in HEK cells transfected with GLUT1myc, GLUT4myc, and GLUT4myc mutants engineered to increase surface expression (GLUT4Myc F5A, GLUT4Myc L489A/L490A, and GLUT4Myc-3x (GLUT4Myc F5A/L489A/L490A)). Uptake is normalized to zero-trans uptake by GLUT1myc-transfected cells. D, uptake of 100 μm 2-DG from medium containing 40 mm 3-MG in HEK cells transfected with GLUT1myc, GLUT4myc, and GLUT4myc-3x under zero-trans (ZT, empty bars) and hetero-exchange (HE, gray bars) conditions. For each construct, ZT is normalized to 100% to show whether stimulation is present under hetero-exchange conditions. For all transport experiments, results are shown as mean ± S.E. for three separate assays. The significance of the difference between ZT and hetero-exchange conditions was computed by an unpaired, two-tailed Student's t test analysis yielding ***, p ≤ 0.001.
Modification of GLUT4 to Increase Surface Expression
The pre-loading experiment (Fig. 3A) indicates that zero-trans 2-DG uptake in GLUT4myc-transfected HEK cells is significantly slower than uptake in GLUT1myc-transfected cells. Total protein expression levels appear similar by Western blot (Fig. 3B), suggesting either that significantly less GLUT4Myc is expressed at the cell surface or that GLUT4Myc has lower intrinsic activity (kcat) than GLUT1Myc. We show below that cell surface protein biotinylation analysis indicates that GLUT4Myc surface expression is less than half of GLUT1Myc.
GLUT4Myc surface expression was improved by engineering 3 GLUT4 mutations known to affect surface expression in a variety of cell types. GLUT4 N and C termini contain internalization (48, 49) and surface targeting (50, 51) motifs. GLUT4Myc mutations F5A and L489A/L490A were tested individually and together in assays of 100 μm 2-DG uptake under zero-trans conditions (Fig. 3C). In these experiments, 2-DG uptake by GLUT4Myc is only one-quarter of that catalyzed by GLUT1Myc. 2-DG uptake by GLUT4Myc F5A and GLUT4Myc L489A/L490A approaches 80% of GLUT1myc-mediated uptake. The triple mutant (GLUT4Myc F5A/L489A/L490A) catalyzes a level of 2-DG uptake indistinguishable from that of GLUT1Myc. This mutant (GLUT4Myc-3x) was used as the GLUT4 scaffold in all further mutational analysis. GLUT1Myc typically displays a 1.8 ± 0.15-fold stimulation of sugar uptake under hetero-exchange conditions (Table 1). However, both GLUT4Myc and GLUT4Myc-3x display no trans-acceleration (Fig. 3D). This confirms that the loop 1 exofacial Myc tag and the cell surface expression mutations introduced into GLUT4 do not significantly perturb wild-type exchange-transport behavior.
TABLE 1.
Zero-trans and hetero-exchange 2-DG uptake by GLUT1Myc-GLUT4Myc chimeras
ZT and HE uptakes were measured for GLUT1Myc in every assay. This table reports the GLUT1Myc data as a global mean ± S.E. for a minimum of n = 30 assays. The range observed in these assays for zero-trans uptake was 39.2 ± 5.36 to 185 ± 18.8 fmol/μg/min. The range observed for HE:ZT was 1.48 ± 0.11 to 2.3 ± 0.31.
| Chimeraa | Residuesb | ZT uptakec | Fold-stimulation during hetero-exchange (HE) HE:ZTd | Trans-acceleratione |
|---|---|---|---|---|
| fmol/μg/min | p value | |||
| GLUT1Myc | 1–492 | 117.2 ± 14.9 | 1.80 ± 0.15 | Y |
| p ≤ 0.001 | ||||
| GLUT4Myc | 1–509 | 42.0 ± 6.8 | 0.96 ± 0.08 | N |
| GLUT4Myc-3x | 1–509 | 77.4 ± 7.4 | 1.04 ± 0.45 | N |
| 44(1)11 | G4 1–223 | 35.4 ± 4.6 | 0.94 ± 0.17 | N |
| G1 208–492 | ||||
| 11(1)44 | G1 1–266 | 66.4 ± 7.4 | 1.90 ± 0.21 | Y |
| G4 283–509 | p ≤ 0.001 | |||
| 44(4)11 | G4 1–282 | 40.3 ± 9.0 | 0.96 ± 0.18 | N |
| G1 267–492 | ||||
| 11(4)44 | G1 1–207 | 35.3 ± 7.3 | 2.00 ± 0.31 | Y |
| G4 224–509 | p ≤ 0.01 | |||
| 1444 | G1 1–119 | 103.0 ± 7.0 | 1.10 ± 0.11 | N |
| G4 136–509 | ||||
| 4111 | G4 1–135 | 47.3 ± 5.9 | 1.70 + 0.21 | Y |
| G1 120–492 | p ≤ 0.01 | |||
| 1411 | G1 1–119; 208–492 | 146.1 ± 10.2 | 1.00 ± 0.13 | N |
| G4 136–223 | ||||
| 4144 | G4 1–135; 224–509 | 61.6 ± 8.1 | 1.80 ± 0.09 | Y |
| G1 120–207 | p ≤ 0.00001 | |||
| GLUT4Myc (4,5 G1) | G4 1–135; 203–509 | 60.1 ± 4.5 | 0.65 ± 0.15 | N |
| G1 120–186 | ||||
| GLUT4Myc (5,6 G1) | G4 1–166; 224–509 | 38.1 ± 3.9 | 1.90 ± 0.14 | Y |
| G1 151–207 | p ≤ 0.00001 | |||
| GLUT4Myc (5, G1) | G4 1–166; 203–509 | 53.2 ± 5.7 | 0.57 ± 0.09 | N |
| G1 151–186 | ||||
| GLUT4Myc (6, G1) | G4 1–203; 224–509 | 25.8 ± 6.2 | 1.80 ± 0.23 | Y |
| G1 187–207 | p ≤ 0.01 | |||
| GLUT1Myc (6, G4) | G1 1–186; 208–492 | 168.3 ± 12.5 | 1.10 ± 0.11 | N |
| G4 203–223 | ||||
| GLUT1Myc SIIFI 191–195 GLTVL | G1 1–190; 196–492 | 162.4 ± 7.9 | 1.30 ± 0.05 | Y |
| G4 208–212 | p ≤ 0.001 | |||
| GLUT1Myc CIV 202–204 LVL | G1 1–201; 205–492 | 64.1 ± 7.2 | 2.20 ± 0.18 | Y |
| G4 218–220 | p ≤ 0.00001 | |||
| GLUT4Myc GLTVL 208–212 SIIFI | G4 1–207; 213–509 | 55.8 ± 4.9 | 0.59 ± 0.09 | N |
| G1 191–195 | ||||
| GLUT4Myc LVL 218–220 CIV | G4 1–217; 221–509 | 92.1 ± 5.1 | 0.97 ± 0.05 | N |
| G1 202–204 |
a The chimeras employed in this study were constructed using two backbones: GLUT1Myc (wt GLUT1 residues 1–492 with a c-Myc epitope (EQKLISEEDL) inserted between residues 55 and 56) and GLUT4Myc-3x (wt GLUT4 residues 1–509 in which Phe-5, Leu-489, and Leu-490 is each mutagenized to Ala, and where a c-Myc epitope (EQKLISEEDL) inserted between residues 72 and 73). All residue numbering ignores the inserted c-Myc sequence. Chimera nomenclature is described under “Results” and in the legend to Fig. 1.
b The sequence composition of chimeras is described as fusions of GLUT1Myc (G1) and GLUT4Myc-3x (G4) sequence in which G1 and G4 sequence numbering ignores the inserted c-Myc epitope.
c ZT of 100 μm 2-DG (fmol/μg of protein/min) from medium containing 40 mm 3-MG was measured in transfected HEK cells depleted of intracellular sugar. Values are reported as mean ± S.E. for a minimum of n = 3 assays and are background-corrected for 2-DG uptake measured in non-transfected cells (41 ± 4 fmol/μg of protein/min).
d Stimulation of 2-DG uptake observed under hetero-exchange conditions (extra- and intracellular [3-MG] = 40 mm) was determined as the ratio of hetero-exchange (HE) 2-DG uptake to ZT uptake (fmol/μg/min). Values are reported as mean ± S.E. for a minimum of n = 3 assays.
e Trans-acceleration is absent (N) when HE:ZT is not significantly greater than 1. Trans-acceleration is present (Y) when HE:ZT is significantly greater than 1. Significance was determined using an unpaired, two-tailed Student's t test.
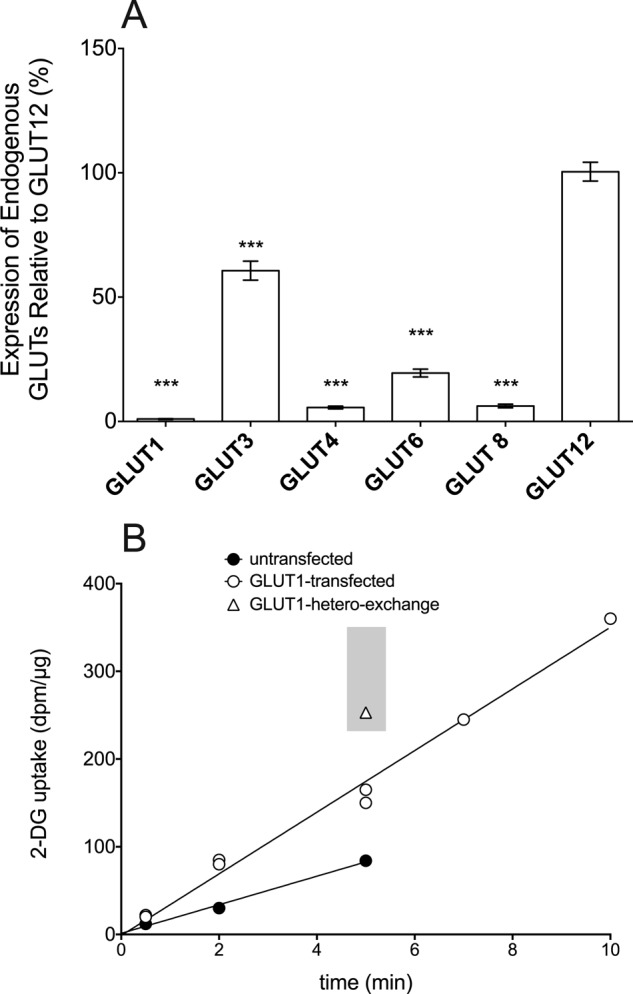
Transport Rates Are Proportional to Cell Surface GLUT Expression
The absolute rate of GLUT1-mediated zero-trans (ZT) and hetero-exchange 2-DG uptake is proportional to the amount of transporter at the cell surface. However, transport behavior (GLUT1-mediated trans-acceleration) is unaffected by expression level. To illustrate this, HEK cells were transfected with a range of [GLUT1myc DNA], and the relationship between cell surface GLUT1Myc expression and GLUT1Myc-mediated zero-trans and hetero-exchange transported was investigated (Fig. 4). Surface expression was quantitated by biotinylation of cell surface protein at 4 °C followed by membrane solubilization, streptavidin affinity purification of labeled proteins, and quantitation of their GLUT1Myc content by immunoblot analysis using α-Myc antibody. Although the relationship between cell surface expression and GLUT1Myc-dependent zero-trans or hetero-exchange 2-DG uptake is linear (Fig. 4), the nearly 2-fold increase in hetero-exchange over zero-trans uptake rates remains constant at every level of cell surface GLUT1Myc observed. We show below that whereas GLUT4 and its engineered variants achieve differing cell surface expression levels, their inability to catalyze trans-acceleration is independent of expression level (see Figs. 3D and 6).
FIGURE 4.
The effect of GLUT1Myc cell surface expression on transport rates and hetero-exchange transport. A, HEK cells were transfected with varying [GLUT1myc DNA]. Two days later, cell surface proteins were biotinylated, solubilized, and affinity purified on streptavidin beads, and GLUT1Myc was detected by immunoblot analysis using either α-Myc Ab or α-Ct Ab. As a loading control, the α-subunit of the Na,K-ATPase was detected using α-Na,K-ATPase Ab. The mobility of molecular weight standards is indicated. The amount of GLUT1myc DNA added at transfection is shown above the blots. B, data obtained in the above experiment and from two similar experiments were analyzed by densitometry, background corrected, normalized to loading controls, and averaged. Ordinate, relative cell surface [GLUT1]; abscissa, μg of DNA added at transfection. The curve is a section of a single rectangular hyperbola characterized by K0.5 = 0.47 ± 0.13 μg of DNA; maximum expression = 1.45 ± 0.12 with expression normalized to unity at 1 μg of DNA. C, rate of GLUT1Myc-catalyzed zero-trans (●) and hetero-exchange (○) 2-DG uptake as a function of cell surface [GLUT1Myc] as detected by cell surface biotinylation. Cells were transfected with GLUT1myc DNA as described in the legend to Fig. 4A, and measurements of ZT and HE 2-DG uptake or cell surface [GLUT1Myc] were made in triplicate on 3 separate occasions. Uptake measured in mock-transfected cells was subtracted. Results are shown as mean ± S.E. The lines drawn through the points were computed by the method of least squares and have the following parameters: ZT, slope = 43.7 ± 2.8 fmol/μg/min/unit biotinylation, y intercept = 2.6 ± 5.1 fmol/μg/min, R2 = 0.98; hetero-exchange, slope = 82.8 ± 8.5 fmol/μg/min/unit biotinylation, y intercept = 8.8 ± 15.3 fmol/μg/min, R2 = 0.96.
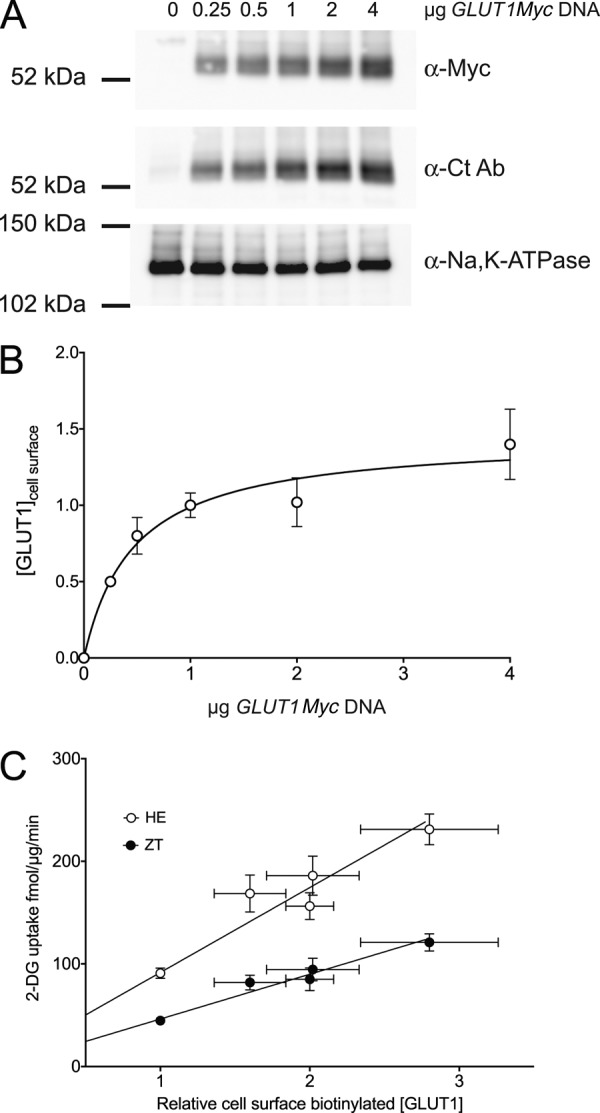
FIGURE 6.
Catalytic activity of cell surface GLUT1, GLUT4, and TM6 mutants. A, cell surface expression of GLUT1Myc, GLUT4Myc, GLUT4Myc-3x, GLUT1 (6, G4), and GLUT4 (6, G1) quantitated by cell surface biotinylation. The streptavidin pulldowns were probed using antibodies for α-Myc to detect transfected transporter and α-Na,K-ATPase (α-subunit) as a loading control. Average densities of bands detected by α-Myc were corrected for density observed in mock-transfected cells, normalized to GLUT1Myc density, and used to calculate the results shown in B. B, zero-trans 100 μm 2-DG/40 mm 3-MG uptake (mol/μg/min, ordinate) of GLUT1Myc, GLUT1 (6, G4)), GLUT4Myc, GLUT4Myc-3x, and GLUT4 (6, G1) adjusted for cell-surface GLUT expression. Adjusted rates were obtained by scaling the average zero-trans rate of each construct by its surface expression relative to GLUT1Myc. Results are shown as mean ± S.E. for three separate assays. An unpaired, two-tailed Student's t test analysis yields the following p values: *, p ≤ 0.07; ****, p ≤ 0.0001.
Trans-acceleration (or lack thereof) is therefore independent of the amount of transporter expressed at the cell surface and is an intrinsic property of the transport protein. This is not unexpected. Endothelial cell GLUT1-mediated zero-trans and accelerated exchange 3-MG uptake (the latter being twice as fast as zero-trans uptake) are both doubled when endothelial cell surface [GLUT1] is doubled by acute metabolic stress (21, 52). Rat erythrocytes express 1,000-fold less GLUT1 than do human erythrocytes, but both cells display accelerated-exchange sugar transport (53, 54). Rat adipocyte GLUT4-mediated zero-trans and equilibrium exchange 3-MG uptake are both increased ∼12-fold by insulin-induced GLUT4 recruitment to the cell surface, but the characteristic GLUT4 kinetic behavior (lack of trans-acceleration) is unchanged (10). Provided that heterologous expression of the transporter is sufficient to measure its function over background, parental transport, the kinetic behavior of the GLUTs (trans-acceleration or lack of trans-acceleration) is independent of cell surface expression levels. The measurement of some kinetic constants, such as kcat, does require specific knowledge of cell surface expression (see below).
Analysis of Half- and Quarter-protein Domain Chimeras for Trans-acceleration
Zero-trans and hetero-exchange 2-DG transport were measured in HEK cells transfected with either GLUT1myc or the half-protein domain chimeras containing GLUT1 loop 6 (11(1)44 and 44(1)11). GLUT1 TMs 7–12 are not important for trans-acceleration (Table 1). Although 2-DG uptake by GLUT1Myc and 11(1)44 is increased under hetero-exchange conditions, transport catalyzed by 44(1)11 is not (Table 1). To ascertain whether loop 6 sequence is critical, we tested an analogous set of half-domain chimeras containing the GLUT4 sequence of loop 6 (Table 1). 2-DG uptake by 11(4)44 or 11(1)44 is significantly increased under HE conditions but transport by 44(4)11 or 44(1)11 is not. Cytoplasmic loop 6, therefore, does not contain isoform-specific sequence that is essential for trans-acceleration. These results allowed us to focus on GLUT1 TMs 1–6 for further analysis.
2-DG uptake by 4111 shows trans-acceleration, whereas uptake by 1444 does not (Table 1). These data show that the isoform-specific sequence in TMs 1–3 is not essential for trans-acceleration. In contrast, TMs 4–6 appear essential for trans-acceleration (Table 1). GLUT1Myc-mediated 2-DG accelerated exchange is ablated when GLUT4 TMs 4–6 are substituted into GLUT1 (1411). Conversely, swapping TMs 4–6 of GLUT1 into a GLUT4Myc-3x scaffold (4144) produces a gain-of-function chimera characterized by robust trans-acceleration (Table 1).
Analysis of TMs 4–6
We next examined paired TM substitutions in TMs 4–6. We tested for gain-of-function in GLUT4Myc-3x containing either GLUT1 TMs 4–5 (GLUT4 (4,5 G1)) or TMs 5–6 (GLUT4 (5,6 G1); Table 1). Our results show that GLUT4 (4,5 G1) does not show exchange stimulation. However, GLUT4 (5,6 G1) displays trans-acceleration gain-of-function. This result indicates that TMs 5–6 are required for trans-acceleration. Because TM5 is also present in the TMs 4–5 chimera, these data suggest either that TM6 alone is required for trans-acceleration, or TM6 in combination with TM5 is required. Indeed, when we substitute GLUT1 TM6 into GLUT4Myc-3x (GLUT4 (6, G1)), we observe a trans-acceleration gain-of-function (Table 1). Conversely, GLUT1 (6, G4) displays a trans-acceleration loss-of-function, indicating that GLUT4 TM6 cannot substitute for GLUT1 TM6. GLUT4 (5, G1) does not show trans-acceleration, indicating that GLUT1 TM5 alone is insufficient to produce trans-acceleration in GLUT4 (Table 1). Taken together, these data confirm that GLUT1 TM6 is both necessary and sufficient for trans-acceleration.
Analysis of Transmembrane Domain 6-Amino Acid Substitutions
Sequence alignment of GLUT1 and GLUT4 TM6 reveals two regions of sequence disparity (Fig. 5). Region A comprises GLUT1 SIIFI(191–195), corresponding to GLUT4 GLTVL(208–212). Region B is GLUT1 CIV(202–204), corresponding to GLUT4 LVL(218–220). We chose to exchange amino acids in Regions A or B between GLUT1 and GLUT4. We observe that neither Region A nor B of the GLUT1 sequence confers trans-acceleration when individually substituted into GLUT4 (Table 1). Similarly, substitution of either Region A or B of GLUT4 into GLUT1 does not produce a loss-of-function (Table 1). These results suggest that all or some of the 8 disparate amino acids within TM6 are required for trans-acceleration.
FIGURE 5.

Sequence alignment and conservation of TM6 in GLUTs 1 and 4. A, sequence alignment of the GLUT1 TM6 subdomains critical for trans-acceleration (cyan background) in 18 mammals (amino acids 191–195PALLQ201–203). B, WebLogo plot of this alignment. Dolphin GLUT1 also displays trans-acceleration (41), but its sequence is not yet known. C, sequence alignment of the equivalent trans-acceleration subdomains (cyan background) of GLUT4 TM6 in 11 mammals (amino acids 208–212PALLQ218–220). D, WebLogo plot of this alignment.
Analysis of kcat/Km for Wild-type and TM6 GLUT1 and GLUT4 Mutants
Despite the use of GLUT4Myc-3x to increase GLUT4 surface expression, there remains consistently lower levels of 2-DG transport among the GLUT4-based chimeras (Table 1). This may be related in part to protein stability, as we observe similar amounts of message for transfected constructs but different protein expression levels (data not shown). Due to these differences, we measured relative surface GLUT expression by cell-surface biotinylation and used this value to scale zero-trans uptake rates for constructs of interest.
Streptavidin pulldowns of biotinylated cell surface proteins confirm the presence of transfected Myc-tagged GLUTs (Fig. 6A). The identity of each Myc-tagged transporter was verified by either anti-GLUT1 or anti-GLUT4 antibodies (data not shown). Quantitation reveals that GLUT4Myc surface expression is 42 ± 2% relative to GLUT1Myc expression, whereas surface expression of GLUT4Myc-3x is only slightly improved (55 ± 18%). GLUT1 (6, G4) shows comparable surface expression to GLUT1Myc (94 ± 29%), whereas GLUT4 (6, G1) achieves only 17 ± 3% of the GLUT1Myc level. Scaling the measured zero-trans uptake rate by relative surface expression allows us to compare differences in catalytic activity (Fig. 6B).
Adjusted rates of zero-trans uptake by GLUT1Myc, GLUT4Myc, and GLUT4Myc-3x are similar (Fig. 6B). However, the trans-acceleration loss-of-function GLUT1 chimera GLUT1 (6, G4) has an adjusted rate that is 1.5-fold greater than GLUT1Myc. In contrast, the gain-of-function mutant, GLUT4 (6, G1), has an adjusted zero-trans rate that is lower than both wt GLUT4Myc and GLUT4Myc-3x.
DISCUSSION
Using homology-scanning mutagenesis, we demonstrate that GLUT1 TM6 is both necessary and sufficient to confer a trans- acceleration gain-of-function to the GLUT4 scaffold. Conversely, substituting GLUT4 TM6 into the GLUT1 scaffold ablates trans-acceleration. These results establish that trans-acceleration is intrinsic to GLUT1 sequence, and is not due to modulating co-factors or other cellular contexts. Although GLUT1 and GLUT4 TM6 differ by a total of 8 amino acids in two subregions, homology substitution of either region alone does not materially affect the trans-acceleration profile of each transporter. This suggests that these subdomains work in concert to effect GLUT1 trans-acceleration of sugar transport.
The canonical explanation of trans-acceleration centers on two kinetic models for carrier-mediated transport: the simple carrier and the fixed-site carrier. The simple carrier (Fig. 7A) is proposed to alternate between exofacial and endofacial orientations (18, 37, 56, 57). During sugar uptake, an external sugar binds to the exofacial orientation, which then undergoes a conformational change to the endofacial state, from which the sugar dissociates into cytoplasm. For an additional round of sugar uptake to occur, the endofacial orientation of the carrier must now reorient to the exofacial state. Conformational changes (exofacial to endofacial and vice versa) are termed translocation when a sugar is bound, and relaxation when no sugar is bound (16). Trans-acceleration of sugar uptake occurs when translocation (endofacial to exofacial) is faster than relaxation. The absence of trans-acceleration is observed when translocation proceeds at the same rate as relaxation. Trans-inhibition would be observed if translocation were slower than relaxation.
FIGURE 7.

Models for carrier-mediated sugar transport. A, the simple carrier presents 3 conformations at any instant: e2, exposing an exofacial sugar binding site; e(), an intermediate state in which neither surface of the carrier exposes a sugar binding sites and where a central cavity is occluded from extra- and intracellular water; and e1, exposing an endofacial sugar binding site. These 3 states inter-convert reversibly as e2 ⇌ e() ⇌ e1. When sugar is complexed to e2, e(), or e1, the inter-conversions are termed “translocation.” When no sugar is bound, the inter-conversions are termed “relaxation.” In trans-acceleration, translocation proceeds more rapidly than relaxation. B, the fixed site carrier simultaneously presents two sugar binding sites and a central cavity. The exofacial and endofacial sugar binding sites are called e2 and e1, respectively. The central cavity is large enough to permit 2 sugar molecules to pass in opposite directions. Dissociation of sugar from e2 to the cavity and subsequent association with e1 is called exchange. When e1 and e2 are occupied by sugar, exchange of bound sugar with cavity sugar is “geminate exchange.” Trans-acceleration occurs when geminate exchange is faster than exchange. C, a two-site variant of the simple carrier in which the carrier comprises 2 (or more) subunits in which subunits are functionally coupled in an antiparallel fashion. If one subunit presents an e2 site, the adjacent subunit must present an e1 site and vice versa. Transacceleration is observed when both subunits are occupied by transported sugar because the coupled conformational change may proceed at the full translocational rate. If only one subunit contains a bound sugar, the unoccupied subunit impedes translocation via the occupied subunit because the unoccupied subunit undergoes relaxation, which (as with the simple carrier) is slower than translocation.
The fixed-site carrier model (Fig. 7B) proposes that the carrier exposes endofacial and exofacial sugar binding sites simultaneously (11, 58–60). Transport proceeds concurrently in both directions, implying that sugars initially bound at exo- or endofacial sites exchange into a central cavity, whence they associate with the trans-binding site prior to release into the cytoplasm or interstitium, respectively. Simple exchange describes the release of a bound exo- or endofacial sugar into the central cavity when the trans-site is unoccupied by sugar. Geminate exchange describes the release of a bound exo- or endofacial sugar into the central cavity when the opposite site is occupied by sugar (61). Trans-acceleration is observed when geminate exchange is faster than simple exchange (61).
A hybrid model (Fig. 7C) has also been proposed, in which the transporter comprises 4 simple carriers arranged in a coupled, anti-parallel configuration. At any instant, two subunits (carriers) present exofacial orientations and two subunits present endofacial orientations (62). If one exofacial subunit undergoes a reorientation to the endofacial state, the adjacent endofacial subunit must undergo a reorientation to the exofacial state. If translocation is faster than relaxation, it is easy to see how intracellular sugar could stimulate sugar uptake.
The current study suggests that the GLUT4 TM6 sequence allows equal rates of simple carrier relaxation and translocation or equal rates of fixed-site carrier exchange and geminate-exchange. In contrast, the GLUT1 TM6 sequence inhibits simple carrier relaxation but not translocation or inhibits fixed-site carrier exchange but not geminate-exchange, thereby allowing intracellular sugar to stimulate unidirectional sugar uptake. Whichever kinetic model is correct, the following generalization is consistent with experimental evidence. In carriers containing the GLUT1 TM6 sequence, an empty endofacial sugar-binding site is inhibitory to the rate of uptake. In carriers containing the GLUT4 TM6 sequence, this inhibition is removed and the rate of uptake is unaffected by the presence of intracellular sugar.
This hypothesis is further supported by the observed differences in kcat/Km ratios for GLUT1, GLUT4, and the TM6 chimeras. Vmax/Km for enzyme-catalyzed reactions is normally obtained by measuring the rate constant, k, for the reaction at limiting substrate concentrations, which is converted to kcat/Km by dividing k by [enzyme]. Vmax/Km is obtained from measurements of 2-DG uptake and then normalized to cell surface GLUT expression to give kcat/Km. Although it is possible that TM6 mutants could alter the affinity (≈1/Km) of GLUT1 and GLUT4 for substrate, this seems unlikely because TM6 is a putative scaffold TM quite distant to the hypothesized GLUT1 substrate-binding cavity (25, 26). Moreover, Km(app) for GLUT1- and GLUT4-mediated sugar uptake is similar for both 2-DG (9–10 mm (63)) and 3-MG (∼6 mm (10)). We therefore hypothesize that the observed changes in kcat/Km (Fig. 6) largely reflect changes in kcat.
If TM6 affects the relative rates of simple carrier relaxation and translocation, or of fixed-site carrier exchange and geminate-exchange, we predict that the “inhibitory” sequence of GLUT1 TM6 would reduce GLUT4-catalyzed zero-trans uptake. Indeed, we observe that kcat/Km for GLUT4 (6, G1) is ∼70% lower than that for either wt GLUT4Myc or the surface expression mutant GLUT4Myc-3x (Fig. 6). In contrast, substituting GLUT4 TM6 sequence into GLUT1 should increase zero-trans kcat/Km relative to that of wt GLUT1, and this is observed.
GLUT1 TM6 trans-acceleration subdomains are highly conserved (Phe-194 and Cys-202 are 100% conserved among 18 mammalian species; Ser-191 and Ile-193 are 94% conserved; see Fig. 5). A homology-modeled GLUT1 three-dimensional structure (26) juxtaposes putative scaffold TMs 6 and 3 with the translocation pore-forming TM1 (Fig. 7, A and B). A study by Liu et al. (64), aimed at identifying sequences important for ATP-modulation of GLUT1, showed that a point mutation in TM3 (G111A) abolishes trans-acceleration of GLUT1 expressed in Xenopus laevis oocytes. We did not observe this effect in TMs 1–3 chimeras because this glycine is conserved between GLUT1 and GLUT4, and is therefore present in both chimeras. Although our data suggest that Gly-111 alone is not sufficient for trans-acceleration, it does not rule out the possibility that Gly-111 makes critical contacts with TM6. The sequence of the membrane-spanning region of TM1 is invariant between GLUTs 1 and 4, with the exception of GLUT1 Thr-30 (Fig. 8C). This position is conserved in GLUTs 1 and 3 (those carriers showing trans-acceleration), but not in GLUTs 2 and 4 (carriers lacking trans-acceleration). However, a potential role of Thr-30 in trans-acceleration is eliminated by the observation that the 4111 chimera contains substitution T30I, yet still displays trans-acceleration. It is tempting to speculate that GLUT1 TM6 residues 191–195 and 202–204 interact with partners in TM1 and/or TM3 (Fig. 8C) to stabilize endo- and exofacial orientations of the substrate-deficient carrier, thereby restraining conformational changes between exo- and endofacial states (e.g. relaxation). We hypothesize that when sugar binds to exofacial or endofacial sites, these interactions are weakened, TM arrangements are destabilized, and conformational change is accelerated.
FIGURE 8.

Role of TM6 in GLUT1-mediated trans-acceleration. Putative glucose transport proteins (GLUT1) homology-modeled structure based on the GlpT homology model (26) and visualized using VMD 1.8.5 (University of Illinois 2006). GLUT1 coordinates were obtained from the RCSB Protein Data Bank (entry 1SUK). A, GLUT1 viewed along the bilayer plane. The limits of the bilayer are indicated by the schematic representations of phospholipids. B, putative helix packing arrangement viewed from the cytoplasmic surface. TMs are numbered and colored as in A. Cytoplasmic loops are indicated by solid lines and exofacial loops by dashed lines. C, putative stacking of TMs 6 and 1. TM6 is shown as a ribbon schematic (cyan) and residues 191–195 and 201–203 as surface representations (yellow), respectively. TM1 is shown as a surface representation (cyan), with residue Thr-30 highlighted (yellow). D, sequence alignment of TM6 from human GLUTs 1, 3, 2, and 4. GLUTs 1 and 3 catalyze transacceleration, whereas GLUTs 2 and 4 do not. Numbering corresponds to GLUT1 sequence; the areas lacking homology are shaded cyan. A putative consensus sequence is indicated.
Although the ability to catalyze trans-acceleration has not been studied in all GLUTs, exchange transport has been measured in all four of the class I glucose transporters (GLUTs 1–4). Human GLUT3 catalyzes trans-acceleration in rat cerebellar granule neurons (65) and in transfected HEK cells,6 whereas rat liver GLUT2 does not exhibit trans-acceleration (55). TM6 sequence comparisons across GLUTs 1–4 (Fig. 8D) show that the same 2 subdomains responsible for trans-acceleration in GLUT1 represent the only variable TM6 sequence among all four transporters. Further homology scanning mutagenesis studies extending TM6 substitutions into GLUT2 and GLUT3 may reveal whether TM6 plays a central role in trans-acceleration in all glucose uniporters.
This work was supported, in whole or in part, by National Institutes of Health Grants DK 36081 and DK 44888.
GLUT1 kinetic asymmetry and transacceleration diminish but persist as temperature is raised from 4 to 37 °C.
A. Carruthers, unpublished data.
K. B. Levine, J. K. DeZutter, and A. Carruthers, unpublished observations.
S. S. Vollers and A. Carruthers, unpublished data.
- TM
- membrane spanning α helix
- 2-DG
- 2-deoxy-d-glucose
- 2-DG-6-P
- 2-deoxy-d-glucose 6-phosphate
- 3-MG
- 3-O-methylglucose
- GLUT
- glucose transport protein
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- MES
- 4-morpholineethanesulfonic acid
- DMEM
- Dulbecco's modified Eagle's medium
- DPBS
- Dulbecco's phosphate-buffered saline
- HE
- hetero-exchange
- ZT
- zero-trans.
REFERENCES
- 1. Joost H. G., Bell G. I., Best J. D., Birnbaum M. J., Charron M. J., Chen Y. T., Doege H., James D. E., Lodish H. F., Moley K. H., Moley J. F., Mueckler M., Rogers S., Schürmann A., Seino S., Thorens B. (2002) Nomenclature of the GLUT/SLC2A family of sugar/polyol transport facilitators. Am. J. Physiol. Endocrinol. Metab. 282, E974–976 [DOI] [PubMed] [Google Scholar]
- 2. Gorga F. R., Lienhard G. E. (1982) Changes in the intrinsic fluorescence of the human erythrocyte monosaccharide transporter upon ligand binding. Biochemistry 21, 1905–1908 [DOI] [PubMed] [Google Scholar]
- 3. Mann G. E., Yudilevich D. L., Sobrevia L. (2003) Regulation of amino acid and glucose transporters in endothelial and smooth muscle cells. Physiol. Rev. 83, 183–252 [DOI] [PubMed] [Google Scholar]
- 4. Harik S. I., Kalaria R. N., Andersson L., Lundahl P., Perry G. (1990) Immunocytochemical localization of the erythroid glucose transporter. Abundance in tissues with barrier functions. J. Neurosci. 10, 3862–3872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thorens B., Mueckler M. (2010) Glucose transporters in the 21st Century. Am. J. Physiol. Endocrinol. Metab. 298, E141–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cushman S. W., Wardzala L. J. (1980) Potential mechanism of insulin action on glucose transport in the isolated rat adipose cell. J. Biol. Chem. 255, 4758–4762 [PubMed] [Google Scholar]
- 7. James D. E., Brown R., Navarro J., Pilch P. F. (1988) Insulin-regulatable tissues express a unique insulin-sensitive glucose transport protein. Nature 333, 183–185 [DOI] [PubMed] [Google Scholar]
- 8. Nishimura H., Pallardo F. V., Seidner G. A., Vannucci S., Simpson I. A., Birnbaum M. J. (1993) Kinetics of GLUT1 and GLUT4 glucose transporters expressed in Xenopus oocytes. J. Biol. Chem. 268, 8514–8520 [PubMed] [Google Scholar]
- 9. Hellwig B., Joost H. G. (1991) Differentiation of erythrocyte-(GLUT1), liver-(GLUT2), and adipocyte-type (GLUT4) glucose transporters by binding of the inhibitory ligands cytochalasin B, forskolin, dipyridamole, and isobutylmethylxanthine. Mol. Pharmacol. 40, 383–389 [PubMed] [Google Scholar]
- 10. Taylor L. P., Holman G. D. (1981) Symmetrical kinetic parameters for 3-O-methyl-d-glucose transport in adipocytes in the presence and in the absence of insulin. Biochim. Biophys. Acta 642, 325–335 [DOI] [PubMed] [Google Scholar]
- 11. Naftalin R. J., Holman G. D. (1977) in Membrane Transport in Red Cells (Ellory J. C., Lew V. L., eds) pp. 257–300, Academic Press, New York [Google Scholar]
- 12. Miller D. M. (1968) The kinetics of selective biological transport. IV. Assessment of three carrier systems using the erythrocyte-monosaccharide transport data. Biophys. J. 8, 1339–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Miller D. M. (1968) The kinetics of selective biological transport. III. Erythrocyte-monosaccharide transport data. Biophys. J. 8, 1329–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baker G. F., Naftalin R. J. (1979) Evidence of multiple operational affinities for d-glucose inside the human erythrocyte membrane. Biochim. Biophys. Acta 550, 474–484 [DOI] [PubMed] [Google Scholar]
- 15. Toyoda N., Flanagan J. E., Kono T. (1987) Reassessment of insulin effects on the Vmax and Km values of hexose transport in isolated rat epididymal adipocytes. J. Biol. Chem. 262, 2737–2745 [PubMed] [Google Scholar]
- 16. Stein W. D. (1986) Transport and Diffusion Across Cell Membranes, Academic Press, New York [Google Scholar]
- 17. Lieb W. R., Stein W. D. (1974) Testing and characterizing the simpler pore. Biochim. Biophys. Acta 373, 165–177 [DOI] [PubMed] [Google Scholar]
- 18. Lieb W. R., Stein W. D. (1974) Testing and characterizing the simple carrier. Biochim. Biophys. Acta 373, 178–196 [DOI] [PubMed] [Google Scholar]
- 19. Mueckler M., Caruso C., Baldwin S. A., Panico M., Blench I., Morris H. R., Allard W. J., Lienhard G. E., Lodish H. F. (1985) Sequence and structure of a human glucose transporter. Science 229, 941–945 [DOI] [PubMed] [Google Scholar]
- 20. Zhao F. Q., Keating A. F. (2007) Functional properties and genomics of glucose transporters. Curr. Genomics 8, 113–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cura A. J., Carruthers A. (2012) Role of monosaccharide transport proteins in carbohydrate assimilation, Distribution, metabolism, and homeostasis. Comp. Physiol. 2, 863–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mueckler M., Makepeace C. (2002) Analysis of transmembrane segment 10 of the Glut1 glucose transporter by cysteine-scanning mutagenesis and substituted cysteine accessibility. J. Biol. Chem. 277, 3498–3503 [DOI] [PubMed] [Google Scholar]
- 23. Mueckler M., Makepeace C. (2005) Cysteine-scanning mutagenesis and substituted cysteine accessibility analysis of transmembrane segment 4 of the Glut1 glucose transporter. J. Biol. Chem. 280, 39562–39568 [DOI] [PubMed] [Google Scholar]
- 24. Mueckler M., Makepeace C. (2009) Model of the exofacial substrate-binding site and helical folding of the human Glut1 glucose transporter based on scanning mutagenesis. Biochemistry 48, 5934–5942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mueckler M., Makepeace C. (2008) Transmembrane segment 6 of the Glut1 glucose transporter is an outer helix and contains amino acid side chains essential of transport activity. J. Biol. Chem. 283, 11550–11555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Salas-Burgos A., Iserovich P., Zuniga F., Vera J. C., Fischbarg J. (2004) Predicting the three-dimensional structure of the human facilitative glucose transporter glut1 by a novel evolutionary homology strategy. Insights on the molecular mechanism of substrate migration, and binding sites for glucose and inhibitory molecules. Biophys. J. 87, 2990–2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Holyoake J., Caulfeild V., Baldwin S. A., Sansom M. S. (2006) Modeling, docking, and simulation of the major facilitator superfamily. Biophys. J. 91, L84–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang Y., Lemieux M. J., Song J., Auer M., Wang D. N. (2003) Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science 301, 616–620 [DOI] [PubMed] [Google Scholar]
- 29. Abramson J., Smirnova I., Kasho V., Verner G., Kaback H. R., Iwata S. (2003) Structure and mechanism of the lactose permease of Escherichia coli. Science 301, 610–615 [DOI] [PubMed] [Google Scholar]
- 30. Lemieux M. J. (2007) Eukaryotic major facilitator superfamily transporter modeling based on the prokaryotic GlpT crystal structure (review). Mol. Membr. Biol. 24, 333–341 [DOI] [PubMed] [Google Scholar]
- 31. Carruthers A., Helgerson A. L. (1989) The human erythrocyte sugar transporter is also a nucleotide binding protein. Biochemistry 28, 8337–8346 [DOI] [PubMed] [Google Scholar]
- 32. Levine K. B., Cloherty E. K., Hamill S., Carruthers A. (2002) Molecular determinants of sugar transport regulation by ATP. Biochemistry 41, 12629–12638 [DOI] [PubMed] [Google Scholar]
- 33. Blodgett D. M., De Zutter J. K., Levine K. B., Karim P., Carruthers A. (2007) Structural basis of GLUT1 inhibition by cytoplasmic ATP. J. Gen. Physiol. 130, 157–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Seatter M. J., De la Rue S. A., Porter L. M., Gould G. W. (1998) QLS motif in transmembrane helix VII of the glucose transporter family interacts with the C-1 position of D-glucose and is involved in substrate selection at the exofacial binding site. Biochemistry 37, 1322–1326 [DOI] [PubMed] [Google Scholar]
- 35. Baldwin S. A., Baldwin J. M., Gorga F. R., Lienhard G. E. (1979) Purification of the cytochalasin B binding component of the human erythrocyte monosaccharide transport system. Biochim. Biophys. Acta 552, 183–188 [DOI] [PubMed] [Google Scholar]
- 36. Sogin D. C., Hinkle P. C. (1980) Binding of cytochalasin B to human erythrocyte glucose transport. Biochemistry 19, 5417–5420 [DOI] [PubMed] [Google Scholar]
- 37. Krupka R. M., Devés R. (1981) An experimental test for cyclic versus linear transport models. The mechanism of glucose and choline transport in erythrocytes. J. Biol. Chem. 256, 5410–5416 [PubMed] [Google Scholar]
- 38. Helgerson A. L., Hebert D. N., Naderi S., Carruthers A. (1989) Characterization of two independent modes of action of ATP on human erythrocyte sugar transport. Biochemistry 28, 6410–6417 [DOI] [PubMed] [Google Scholar]
- 39. Cloherty E. K., Levine K. B., Carruthers A. (2001) The red blood cell glucose transporter presents multiple, nucleotide-sensitive sugar exit sites. Biochemistry 40, 15549–15561 [DOI] [PubMed] [Google Scholar]
- 40. Zottola R. J., Cloherty E. K., Coderre P. E., Hansen A., Hebert D. N., Carruthers A. (1995) Glucose transporter function is controlled by transporter oligomeric structure. A single, intramolecular disulfide promotes GLUT1 tetramerization. Biochemistry 34, 9734–9747 [DOI] [PubMed] [Google Scholar]
- 41. Craik J. D., Young J. D., Cheeseman C. I. (1998) GLUT-1 mediation of rapid glucose transport in dolphin (Tursiops truncatus) red blood cells. Am. J. Physiol. 274, R112–119 [DOI] [PubMed] [Google Scholar]
- 42. Levine K. B., Robichaud T. K., Hamill S., Sultzman L. A., Carruthers A. (2005) Properties of the human erythrocyte glucose transport protein are determined by cellular context. Biochemistry 44, 5606–5616 [DOI] [PubMed] [Google Scholar]
- 43. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔCT) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 44. Bachelard H. S. (1972) Deoxyglucose and brain glycolysis. Biochem. J. 127, 83P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bissonnette P., Gagné H., Blais A., Berteloot A. (1996) 2-Deoxyglucose transport and metabolism in Caco-2 cells. Am. J. Physiol. 270, G153–62 [DOI] [PubMed] [Google Scholar]
- 46. Cloherty E. K., Heard K. S., Carruthers A. (1996) Human erythrocyte sugar transport is incompatible with available carrier models. Biochemistry 35, 10411–10421 [DOI] [PubMed] [Google Scholar]
- 47. Jay T. M., Dienel G. A., Cruz N. F., Mori K., Nelson T., Sokoloff L. (1990) Metabolic stability of 3-O-methyl-d-glucose in brain and other tissues. J. Neurochem. 55, 989–1000 [DOI] [PubMed] [Google Scholar]
- 48. Corvera S., Chawla A., Chakrabarti R., Joly M., Buxton J., Czech M. P. (1994) A double leucine within the GLUT4 glucose transporter COOH-terminal domain functions as an endocytosis signal. J. Cell Biol. 126, 979–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Al-Hasani H., Kunamneni R. K., Dawson K., Hinck C. S., Müller-Wieland D., Cushman S. W. (2002) Roles of the N and C termini of GLUT4 in endocytosis. J. Cell Sci. 115, 131–140 [DOI] [PubMed] [Google Scholar]
- 50. Piper R. C., Tai C., Kulesza P., Pang S., Warnock D., Baenziger J., Slot J. W., Geuze H. J., Puri C., James D. E. (1993) GLUT-4 NH2 terminus contains a phenylalanine-based targeting motif that regulates intracellular sequestration. J. Cell Biol. 121, 1221–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Verhey K. J., Birnbaum M. J. (1994) A Leu-Leu sequence is essential for COOH-terminal targeting signal of GLUT4 glucose transporter in fibroblasts. J. Biol. Chem. 269, 2353–2356 [PubMed] [Google Scholar]
- 52. Cura A. J., Carruthers A. (2010) Acute modulation of sugar transport in brain capillary endothelial cell cultures during activation of the metabolic stress pathway. J. Biol. Chem. 285, 15430–15439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Helgerson A. L., Carruthers A. (1989) Analysis of protein-mediated 3-O-methylglucose transport in rat erythrocytes. Rejection of the alternating conformation carrier model for sugar transport. Biochemistry 28, 4580–4594 [DOI] [PubMed] [Google Scholar]
- 54. Naftalin R. J., Rist R. J. (1991) 3-O-Methyl-d-glucose transport in rat red cells. Effects of heavy water. Biochim. Biophys. Acta 1064, 37–48 [DOI] [PubMed] [Google Scholar]
- 55. Craik J. D., Elliott K. R. (1979) Kinetics of 3-O-methyl-d-glucose transport in isolated rat hepatocytes. Biochem. J. 182, 503–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jardetzky O. (1966) Simple allosteric model for membrane pumps. Nature 211, 969–970 [DOI] [PubMed] [Google Scholar]
- 57. Widdas W. F. (1952) Inability of diffusion to account for placental glucose transfer in the sheep and consideration of the kinetics of a possible carrier transfer. J. Physiol. 118, 23–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Baker G. F., Widdas W. F. (1973) The asymmetry of the facilitated transfer system for hexoses in human red cells and the simple kinetics of a two component model. J. Physiol. 231, 143–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Baker P. F., Carruthers A. (1981) 3-O-Methylglucose transport in internally dialysed giant axons of Loligo. J. Physiol. 316, 503–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Naftalin R. J. (1988) Pre-steady-state uptake of d-glucose is inconsistent with a circulating carrier mechanism. Biochim. Biophys. Acta 946, 431–438 [DOI] [PubMed] [Google Scholar]
- 61. Naftalin R. J. (2008) Alternating carrier models of asymmetric glucose transport violate the energy conservation laws. Biophys. J. 95, 4300–4314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hebert D. N., Carruthers A. (1992) Glucose transporter oligomeric structure determines transporter function. Reversible redox-dependent interconversions of tetrameric and dimeric GLUT1. J. Biol. Chem. 267, 23829–23838 [PubMed] [Google Scholar]
- 63. Hansen P., Gulve E., Gao J., Schluter J., Mueckler M., Holloszy J. (1995) Kinetics of 2-deoxyglucose transport in skeletal muscle. Effects of insulin and contractions. Am. J. Physiol. 268, C30–35 [DOI] [PubMed] [Google Scholar]
- 64. Liu Q., Vera J. C., Peng H., Golde D. W. (2001) The predicted atp-binding domains in the hexose transporter glut1 critically affect transporter activity. Biochemistry 40, 7874–7881 [DOI] [PubMed] [Google Scholar]
- 65. Maher F., Davies-Hill T. M., Simpson I. A. (1996) Substrate specificity and kinetic parameters of GLUT3 in rat cerebellar granule neurons. Biochem. J. 315, 827–831 [DOI] [PMC free article] [PubMed] [Google Scholar]