

Background: RIG-I is an essential innate immune receptor that detects viral RNAs in infected cells.
Results: RIG-I uses distinct subdomains to recognize specific characteristics of viral RNAs.
Conclusion: The 5′-triphosphate is critical for high affinity RIG-I/RNA interaction.
Significance: Characterizing the RIG-I/RNA interface is essential for understanding early stages of immune response against RNA viruses.
Keywords: ATPases, Interferon, RNA Helicase, RNA-Protein Interaction, Viral Immunology, RNA Triphosphate
Abstract
RIG-I is a cytoplasmic surveillance protein that contributes to the earliest stages of the vertebrate innate immune response. The protein specifically recognizes 5′-triphosphorylated RNA structures that are released into the cell by viruses, such as influenza and hepatitis C. To understand the energetic basis for viral RNA recognition by RIG-I, we studied the binding of RIG-I domain variants to a family of dsRNA ligands. Thermodynamic analysis revealed that the isolated RIG-I domains each make important contributions to affinity and that they interact using different strategies. Covalent linkage between the domains enhances RNA ligand specificity while reducing overall binding affinity, thereby providing a mechanism for discriminating virus from host RNA.
Introduction
The innate immune system first responds to invading viruses by activating surveillance proteins that sensitively detect the presence of viral gene products. RIG-I (retinoic acid-inducible gene I)-like receptors (RLRs)2 are a family of surveillance proteins that specifically recognize invading viral RNA molecules and then signal a subsequent anti-viral host defense. The RLR proteins are localized in the cytoplasm, and they include several family members, such as RIG-I, MDA-5 (melanoma differentiation-associated gene 5) (1, 2), and LGP2 (laboratory of genetics and physiology 2) (3–5). Although RIG-I and MDA-5 have similar activation pathways, they detect distinct RNA viruses. RIG-I detects viral RNA from paramyxoviruses, influenza virus, Japanese encephalitis virus (6), and hepatitis C virus (7), whereas MDA-5 senses viral RNAs from picornaviruses (6). Upon interaction with viral RNA containing double-stranded character and/or a triphosphate moiety at the 5′ terminus (5′-ppp), RIG-I initiates a signaling cascade by interacting with the adaptor protein MAVS (mitochondrial anti-viral signaling protein; also known as IPS-1, CARDIF, or VISA). The formation of this complex induces expression of interferon regulatory factors and NF-κB to produce type I interferons and cytokines (8). The importance of RIG-I to the antiviral interferon response has been demonstrated by RIG-I knock-out mice, which are more susceptible to infection by RNA viruses (6). Therefore, therapeutics that target RIG-I activity may ultimately be useful for modulating the immune response, potentially providing new strategies for preventing viral infection and treating autoimmune disorders.
The RLR proteins belong to a phylogenetically conserved group of enzymes that are classified as superfamily 2 RNA-dependent ATPases (SF2 proteins). These enzymes are involved in diverse cellular processes that range from nucleic acid metabolism to signaling (9, 10). SF2 proteins contain conserved motifs within their mechanical core (often referred to as the “helicase” core) that are essential for binding nucleic acids and nucleoside triphosphates (NTPs). ATP binding and hydrolysis induces protein conformational changes that power nucleic acid remodeling events, such as duplex unwinding, translocation, strand annealing, protein displacement, and RNA transport activity (9, 10). Like other SF2 proteins, RLRs contain a conserved mechanical core that consists of two RecA-like domains (helicase domain 1 (HEL1) and helicase domain 2 (HEL2)). Additional modular domains are appended to this central core, thereby enabling the RLRs to function specifically during the immune response. Each RLR contains a C-terminal regulatory domain (CTD), which, in the case of RIG-I, contributes to recognition of 5′-ppp double-stranded RNA (dsRNA) (11, 12). At the N terminus, RIG-I and MDA-5 contain two caspase activation and recruitment domains (CARDs) that, upon RNA binding, interact directly with MAVS to initiate signal transduction. Crystallographic studies on a human RIG-I variant lacking the CARDs (RIG-I (ΔCARDs)) and domain variants of duck RIG-I (dRIG-I) have revealed specialized protein domains that have evolved specifically within the RLR subfamily. An insertion domain within the helicase core (HEL2i) confers specificity for duplex RNA and binds both strands in the protein-RNA complex (13, 14). Remarkably, the same domain interacts with the CARDs when RIG-I is not bound to RNA (14). A second innovation is the V-shaped α-helical structure, referred to as the pincer domain, which covalently links HEL2 to the CTD (13, 14). The pincer domain is required for communication between these domains, specifically coupling ATP hydrolysis and RNA binding (13).
RIG-I recognizes specific features on its viral RNA targets, thereby differentiating pathogenic RNAs from host RNAs (11, 12). However, the energetic determinants for recognition remain unclear. The isolated CTD has been studied most intensively, and biophysical studies have shown that it preferentially binds RNA molecules containing a 5′-triphosphate, forming high affinity complexes with 12–14-base pair RNA duplexes that lack single-stranded overhangs at the termini (referred to as blunt-ended) (11, 12, 15). This allows the protein to distinguish viral transcripts from host RNAs that are terminated by 5′-cap structures. A blunt-ended 5′-ppp dsRNA is required for maximal binding affinity (11, 12), although weak binding can be observed for single-stranded transcripts. In contrast to these results with the isolated CTD, a recent study on a longer RIG-I variant (lacking only the CARDs) indicated that the protein bound 5′-ppp dsRNA and 5′-OH dsRNA with similar affinity (16), making it difficult to understand how RIG-I targets viral transcripts specifically. Given these conflicting results and the importance of RIG-I to antiviral host defense, it is essential to identify the molecular determinants for RIG-I recognition of viral RNAs and to understand the role of each subdomain in this important process. To address these issues, we have determined the binding affinities of full-length human RIG-I and its isolated domain variants (Fig. 1B) to various dsRNA substrates, enabling us to dissect the molecular and energetic determinants for recognition of viral RNA and to determine the role of each protein domain in the detection of viral RNA. The work complements recent crystallographic investigations that reveal a network of contacts between RNA and RIG-I functional domains (13, 14, 16).
FIGURE 1.

RIG-I (ΔCARDs) dsRNA binding interface and construct design. A, RNA-binding domains involved in the protein-RNA interaction as observed in a previous crystal structure of RIG-I (ΔCARDs) in complex with 10-mer dsRNA. Blue, HD; red, CTD; green, dsRNA. B, schematic diagram of RIG-I constructs. Yellow, CARDs; blue, HD; light blue, HD insertion; blue with black border, Pincer; red, CTD.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
The human RIG-I domain variants tested include RIG-I (ΔCARDs) (amino acids 229–925) (13); the helicase domain (HD; amino acids 229–793); and the CTD (amino acids 792–925). Full-length RIG-I and variants were cloned into the pET SUMO expression vector (Invitrogen), and the first amino acid in the coding sequence of each construct is a Ser to ensure proper cleavage of the N-terminal SUMO (small ubiquitin-like modifier) tag. The expression plasmid was transformed into Rosetta II cells. The Escherichia coli culture was grown to an optical density of 0.6, inoculated with 0.2 mm isopropyl 1-thio-β-d-galactopyranoside, and grown overnight at 16 °C. The cells were harvested and subsequently lysed with cold buffer consisting of 25 mm HEPES buffer, 10 mm imidazole, 10% glycerol, 5 mm β-ME, and 500 mm NaCl, pH 7.4. The lysed cells were clarified by centrifugation, and the supernatant was bound to nickel-nitrilotriacetic acid-agarose (Qiagen). The protein was eluted with 25 mm HEPES buffer, 160 mm imidazole, 10% glycerol, 5 mm β-ME, and 500 mm NaCl, pH 7.4. The recovered protein was digested using SUMO protease and further purified by a His trap column (GE Healthcare), heparin column (GE Healthcare), and gel filtration chromatography (GE Healthcare). The proteins were concentrated and stored in 25 mm HEPES, 10% glycerol, 5 mm β-ME, and 500 mm NaCl, pH 7.4.
RNA Preparations
RNA oligonucleotides were synthesized on an automated MerMade 6 synthesizer (BioAutomation) using phosphoramidite chemistry (phosphoramidites purchased from Glen Research). The oligonucleotides were deprotected and gel-purified by methods described previously (17). The “top strand” of the 5′-OH RNA duplex (5′-OH 14-mer dsRNA) has the sequence 5′-ACCUCCCACCCCCG. In the case of the triphosphorylated top strand, the 5′-ppp is at the 5′-end of this RNA sequence. The complementary strand of the RNA duplex has the sequence 5′-CGGGGGUGGGAGGU. The complementary strand was labeled at the 5′-end with [γ-32P]ATP (PerkinElmer Life Sciences) and T4 polynucleotide kinase (New England BioLabs). The labeled strand was precipitated, and a 2-fold excess of the top strand was added in buffer containing 10 mm MOPS, pH 6.5, 1 mm EDTA, and 150 mm NaCl. The strands were heated to 95 °C and slow cooled to 4 °C for a period of 1 h. The annealed duplexes were then passed through a G-25 column to remove free [γ-32P]ATP and then gel-purified on a 15% native polyacrylamide gel. The same procedure was carried out for the 5′-ppp 14 dsRNA, which has the same sequence.
In Vitro Transcription
To incorporate the 5′-ppp, we used T7 in vitro transcription. We carried out transcription under a promoter initiated by ATP (18). We did not transcribe the “top strand” directly (because short oligonucleotides do not transcribe with high efficiency) but instead incorporated it at the 5′ terminus of a long RNA (74 nt). This long RNA transcript was gel-purified, eluted, and subsequently precipitated. Because the long RNA transcript contained a guanosine at the 14th position, we selectively and quantitatively cleaved the transcript at nucleotide 14 with RNase T1, thereby generating a perfect 5′-ppp 14-mer top strand. To carry out this procedure, 7 μg of long RNA was cleaved for 2 h at room temperature with T1. To ensure that no residual RNase T1 carried over to subsequent steps, proteinase K was added to the completed cleavage reaction and incubated at 50 °C for 1 h. PMSF was added to this reaction in order to ensure that proteinase K was subsequently inhibited. The reaction was then phenol/chloroform-extracted, precipitated, and purified on a 15% denaturing gel. The faster migrating 5′-ppp top strand was cut out of the gel, eluted, and precipitated. Because the 5′-ppp top strand had been cut by RNase T1, it contained a 2′3′ cyclic phosphate that was removed using a described dephosphorylation method (protocol 2 (19)). The synthetic 5′-OH top strand, 5′-ppp top strand with 2′3′ cyclic phosphate, and the 5′-ppp top strand with 3′-OH were run on a 20% denaturing gel to test for purity, size, and triphosphorylation state.
Electrophoretic Mobility Shift Assay (EMSA)
Binding reactions included 5′-ppp 14 dsRNA or 5′-OH 14 dsRNA at concentrations that were at least 5-fold lower than the determined binding affinity (as indicated), varying concentrations of protein, and binding buffer (25 mm Hepes, pH 7.4, 150 mm NaCl, 1.5 mm DTT, and 0.1 mg/ml BSA). We tested the return to equilibrium, relaxation time (τ), and let our binding reactions incubate at room temperature for 3τ to ensure that the system had reached equilibrium (20). In the case of RIG-I (ΔCARDs), which had a Kd value of 41 pm, the equilibration time was 8 h. In order to determine whether this incubation time was sufficient for the system to reach equilibrium even at the lowest protein concentrations, we also incubated reactions at the two lowest protein concentrations for 16 and 24 h. We found that the amount of the RNA-protein complex formed at the lowest protein concentration after 8, 16, and 24 h of incubation with the RNA was nearly identical within experimental error. The same experiment was conducted with the second lowest protein concentration as well. This indicates that the system has approached equilibrium at 8 h for all protein concentrations, including the lowest protein concentrations. The binding reactions were loaded on 6% non-denaturing polyacrylamide gels (Invitrogen) run in 0.5× TBE at a constant 100 V for 1 h at 4 °C. The gels were dried and exposed to phosphorimaging screens for 2–6 days, visualized with a Storm 820 PhosphorImager, and quantified with ImageQuant software (GE Healthcare). The fraction bound was quantified at each protein concentration by the following formula: (protein-dsRNA complex)/(protein-dsRNA complex + free dsRNA). The analyzed data were fit to the quadratic equation,
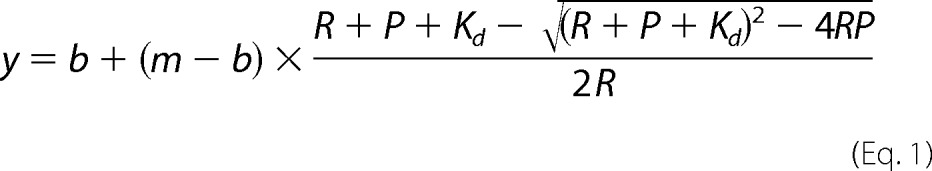 |
using GraphPad Prism version 5.0 software by inserting values for the minimum fraction bound (b), the maximum fraction bound (m), the RNA concentration (R), and the protein concentration (P), in order to determine the equilibrium binding constant (Kd). The experiments were carried out with the dsRNA in trace concentrations (at least 5-fold lower than the Kd value). The error bars represent the S.D. value of at least three individual experiments.
Determining the Binding Free Energy
The apparent binding energy of the individual domains, the HD (ΔGHD0*) and the CTD (ΔGCTD0*), and the linked domains RIG-I (ΔCARDs) (ΔGΔCARDs0*) and full-length RIG-I (ΔGΔfull-length0*) were determined from experimentally obtained binding affinities.
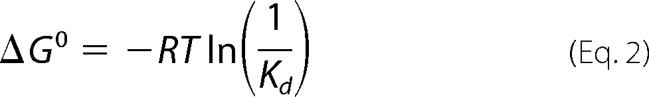 |
The sum of the individual domains was labeled (Σ ΔGHD0* and ΔGCTD0*). To determine the difference between the sum of the domains and the covalently linked domains, the following equation (22) was used for full-length RIG-I,
and also for RIG-I (ΔCARDs),
Fluorescence Polarization (FP) Assay
FP direct binding and competition experiments were performed in Corning 384-well low flange black flat bottom polystyrene non-binding microplates at 25 °C. For both experiments, the reaction conditions were 25 mm HEPES, pH 7.2, 1 mm DTT, and 150 mm NaCl. Reactions were incubated for 1 h at room temperature, and then the polarization values were determined on an Analyst AD (Molecular Devices). The G factor was determined to be 0.98 for the fluorescein standard. For direct binding experiments, RNA concentrations were held at 2 or 5 nm, and protein concentrations were varied from low nanomolar to low micromolar. The fraction of protein bound was determined for each data set, and the Kd value of the complex was determined by fitting the data to Equation 1. Obviously, this method is not suitable for determining Kd values less than nanomolar affinity.
For competition experiments, the RNA and protein concentrations were held constant, and the unlabeled RNA concentration was varied over a range of concentrations in order to displace the bound fluoresceinated RNA. In these experiments, the fluorophore-labeled RNA used to form the protein-RNA complex and the unlabeled RNA used to conduct the competition were identical. The data were fit to the following equation,
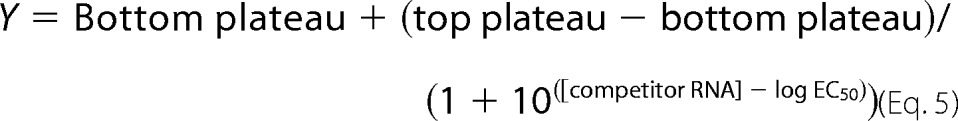 |
using GraphPad Prism version 5.0 software.
RESULTS
5′-ppp RNA Duplex Substantially Increases Full-length RIG-I Binding Affinity
To assess the effect of the 5′-ppp on RNA recognition by RIG-I, recombinant full-length human RIG-I binding was analyzed using an EMSA (Fig. 2). This assay was used throughout the study because it is a sensitive method for detection of protein-RNA interactions with high binding affinities. In addition to EMSAs, FP competition and direct binding experiments were conducted in order to provide orthogonal, independent information on binding affinity (data not shown). The data obtained using all of these methods are in good agreement.
FIGURE 2.

Binding analysis of full-length RIG-I to 14 dsRNA substrates. A, EMSA of full-length RIG-I binding to 5′-ppp 14 dsRNA. Full-length RIG-I concentrations for lanes 1–7 are 9, 18, 35, 70, 140, 280, and 561 pm, respectively; concentrations for lanes 8–15 are 1, 2, 5, 9, 18, 36, 72, and 0 nm, respectively. The dsRNA concentration is 10 pm. The binding curve of full-length RIG-I to 5′-ppp 14 dsRNA is as follows: Kd = 0.159 ± 0.020 nm; R2 = 0.99. B, EMSA of full-length RIG-I binding to 5′-OH 14 dsRNA. Full-length RIG-I concentrations for lanes 1–12 are 0.5, 1, 2, 5, 9, 18, 36, 72, 140, 290, 580, and 0 nm, respectively. The dsRNA concentration is 1 nm. The binding curve of full-length RIG-I to 5′-OH 14 dsRNA is as follows: Kd = 20 ± 3 nm; R2 = 0.99. Insets, data for the six to seven lowest protein concentrations of the respective plots.
RNA binding studies on the full-length RIG-I protein were conducted under conditions comparable with those previously used for studying the isolated CTD (25 mm Hepes, pH 7.4, 150 mm NaCl, 1.5 mm DTT, and 0.1 mg/ml BSA (11)). EMSA experiments show that RIG-I forms a single shifted band upon binding a 14-mer duplex RNA (14 dsRNA; Fig. 2, A and B), which is consistent with the 1:1 binding stoichiometry reported previously for this RNA duplex length (13). Multimerization of RIG-I is evident on longer duplexes because formation of a second higher migrating band was observed upon binding of RIG-I to 16 base pairs or longer (data not shown). RIG-I displays high affinity for the 5′-OH 14-mer duplex (Kd = 20 ± 3 nm; Fig. 2B). This binding increases significantly upon interaction with 5′-triphosphorylated duplex (5′-ppp 14 dsRNA, Kd = 159 ± 20 pm; Fig. 2A), resulting in a 126-fold enhancement in affinity. This dramatic increase in binding affinity confirms that the 5′-triphosphate is a critical recognition determinant for the RIG-I protein.
As an independent quantitative test of RIG-I binding affinity, FP was used to assess RIG-I binding to RNA ligands. RIG-I was observed to bind fluorophore-labeled 5′-OH dsRNA with a Kd value similar to that determined by EMSA (Kd = 30 ± 4 nm; data not shown). This method could not be used to assess full-length RIG-I binding to 5′-ppp dsRNA because the low dsRNA concentration needed to carry out ultra-high affinity experiments is below the detection limit of the instrument. In such cases, radioanalytic detection methods are the only viable option.
Removal of the CARDs Increases RIG-I Binding Affinity to Duplex RNA
In order to understand how the CARDs influence dsRNA recognition and binding, we compared RNA affinities of full-length RIG-I with a construct that lacks the CARDs (RIG-I (ΔCARDs) (13)). Because the CARDs are not an RNA-binding domain, it was unclear how they might influence stability of the RNA complex. We observed that RIG-I (ΔCARDs) binds the 5′-OH 14 dsRNA with relatively high affinity (Kd = 3.6 ± 0.9 nm; Fig. 3B) and single site stoichiometry, consistent with previous structural and functional studies of this construct (Fig. 1A) (13). Interestingly, affinity is increased upon removal of the CARDs because RIG-I (ΔCARDs) binds 5′-OH dsRNA 5.5-fold more tightly than full-length RIG-I (Table 1). Results from FP, direct binding, and competition experiments are in reasonable agreement with the Kd value determined by the EMSA (data not shown). We then examined whether the CARDs influence affinity for the triphosphate. We observed that RIG-I (ΔCARDs) bound 5′-ppp 14 dsRNA with very high affinity (Kd = 41 ± 2 pm; Fig. 3A), which is only 3.8-fold higher affinity than observed for full-length RIG-I to the same triphosphorylated RNA (Fig. 2A). These data indicate that the CARDs do not affect triphosphate recognition, but they do obstruct interactions with the RNA duplex, as suggested by available crystal structures.
FIGURE 3.

Binding analysis of RIG-I (ΔCARDs) to 14 dsRNA and ssRNA substrates. A, binding curve of RIG-I (ΔCARDs) to 5′-ppp 14 dsRNA (Kd = 0.041 ± 0.002 nm; R2 = 0.98). RIG-I (ΔCARDs) concentrations used in experiments are 3, 5, 10, 20, 40, 85, 170, 340, 680, 1400, 2700, 5500, 11,000, and 22,000 pm. The dsRNA concentration in the reactions is 10 pm. B, binding curve of RIG-I (ΔCARDs) to 5′-OH 14 dsRNA (Kd = 3.6 ± 0.9 nm; R2 = 0.98) determined by EMSA data. RIG-I (ΔCARDs) concentrations used in experiments are 0.070, 0.140, 0.280, 0.560, 1, 2, 5, 9, 18, 36, 72, 140, and 290, and 580 nm. The dsRNA concentration in the reactions is 0.5 pm. Insets, data for the five to six lowest protein concentrations of the respective plots. To evaluate whether the radioactive label on the 5′ terminus of the complementary strand influences RIG-I binding affinity to the substrates, a radioactive binding competition assay with unlabeled 5′-OH dsRNA was conducted and found to be almost identical (KI = 13 nm; data not shown). C, binding curve of RIG-I (ΔCARDs_238aa) to 12-mer ssRNA (Kd = 1.3 ± 0.2 μm; R2 = 0.95) determined by FP. The protein concentrations used in the experiments are 5, 10, 20, 40, 80, 160, 320,480, 720, 1080, 1620, 2430, 3650, and 5470 nm. Note that the data in C were obtained with a slightly shorter RIG-I construct than described under “Experimental Procedures.” This construct contains the same RNA-binding domains as RIG-I (ΔCARDs) and binds dsRNA with similar affinity (17 ± 9 nm; data not shown).
TABLE 1.
Equilibrium dissociation binding constants of RIG-I constructs with 14 dsRNA
| Full-length RIG-I Kd | RIG-I (ΔCARDs) Kd | HD Kd | CTD Kd | |
|---|---|---|---|---|
| nm | nm | nm | nm | |
| 5′-OH 14 dsRNA | 20 ± 3 | 3.6 ± 0.9 | 1830 ± 250 | 5900 ± 750 |
| 5′-ppp 14 dsRNA | 0.159 ± 0.020 | 0.041 ± 0.002 | 760 ± 20 | 0.500 ± 0.060 |
Because RIG-I is an SF2 protein that contains the same minimal RNA-binding domains as single-stranded RNA-binding proteins (such as hepatitis C virus NS3, which also has HEL1 and HEL2), it was important to establish the relative affinity of the RIG-I constructs to single-stranded RNA (ssRNA) in order to provide a basis for comparison with other members of the family. We observe that RIG-I (ΔCARDs) binds ssRNA with weak affinity compared with dsRNA. FP data of RIG-I (ΔCARDs) binding to a 3′-fluorophore-labeled ssRNA was fit to a 1:1 binding model (Kd = 1.3 ± 0.2 μm; Fig. 3C). To test the validity of these results and to evaluate whether ssRNA binding was affected by the presence of the fluorophore at the 3′ terminus, FP competition experiments were conducted, resulting in similar values for ssRNA affinity (KI = 1.9 ± 0.9 μm; data not shown). Collectively, these results indicate that RIG-I has evolved to bind dsRNA with exceptionally high affinity and to discriminate against the interaction with ssRNA.
Affinity of the Isolated HD and CTD Domains to 5′-ppp and 5′-OH RNA Duplexes
RIG-I contains two subsections (the CTD and HD) that contribute to recognition of viral RNA. Previous structural and biochemical studies have shown that the CTD is important for 5′-ppp recognition and high affinity binding (11, 12). The HD contains a conserved “helicase” core that binds RNA and stimulates ATP hydrolysis (9, 10), along with an insertion domain that facilitates recognition of duplex RNA. Here, we dissected the relative contributions of the isolated domains to recognition of RNA. The HD construct used in our studies contains the “helicase” core (consisting of HEL1, HEL2, and HEL2i) and the pincer domain. However, the HD construct lacks the proline-rich sequence (residues 796–804) that covalently links HEL2 and the CTD and that facilitates interdomain communication within the full-length protein.
Despite its decoupling from the CTD, the isolated HD retains affinity for dsRNA ligands, as suggested by crystallographic studies on the duck RIG-I helicase complex, which lacks the CTD (14). HD bound to the 5′-OH 14 dsRNA as a 1:1 complex with weak affinity (Kd = 1.8 ± 0.3 μm; Fig. 4) but displayed a slightly enhanced affinity in the presence of a 5′-triphosphate (Kd = 760 ± 20 nm; Fig. 4). The enhancement in binding observed in the presence of a 5′-ppp may represent a specific binding mode, or it may be due to nonspecific electrostatic contacts between the HD and the electronegative 5′-ppp moiety on the RNA duplex.
FIGURE 4.

Binding analysis of HD to 14 dsRNA substrates. Shown is the binding curve of HD to 5′-ppp 14 dsRNA (circles; Kd = 760 ± 20 nm; R2 = 0.97) and 5′-OH 14 dsRNA (triangles; Kd = 1830 ± 250 nm; R2 = 0.96) determined by EMSAs. HD concentrations for experiments with 5′-ppp 14 dsRNA are 20, 40, 77, 150, 310, 620, 1230, 2450, 4900, and 9800 nm. The HD concentrations for experiments with 5′-OH 14 dsRNA are 10, 20, 40, 80, 150, 310, 610, 1230, 2450, 4900, 9800 nm. The dsRNA concentration in the reactions is 1 nm.
In addition to binding RNA, the HD is also the site of ATP binding and hydrolysis in RIG-I. The effect of two ATP analogues (AMP-PNP and ADP) on dsRNA binding by HD was tested in order to determine whether nucleotide binding would influence apparent RNA affinity. The presence of AMP-PNP or ADP results in a 2-fold decrease in the Kd value for HD binding to 5′-OH 14 dsRNA. The same analogues decrease HD affinity for 5′-ppp 14 dsRNA by 3-fold (each analog is present at a concentration of 1 mm, which is more than 10-fold above the Kd; data not shown). Given the small extent of these binding effects, there does not appear to be a major role for nucleotide in mediating RNA affinity of RIG-I, although this is an important issue that merits further study.
Because the CTD contributes to RIG-I recognition of triphosphorylated RNAs (11, 12), it was of interest to evaluate the binding affinity of the isolated CTD to both 5′-ppp and 5′-OH 14 dsRNAs. EMSA experiments show that the CTD binds 5′-ppp 14 dsRNA as a high affinity 1:1 complex (Kd = 500 ± 60 pm; Fig. 5A). A similar analysis indicates that the CTD is capable of binding RNA duplexes that lack a triphosphate weakly (5′-OH 14 dsRNA, Kd = 5.9 ± 0.8 μm; Fig. 5B). In contrast with previous studies on duplexes that contained two triphosphates (one on each strand) (11, 12), we observe monomeric binding of the CTD to 5′-ppp 14 dsRNA, which contains only one 5′-ppp, on one strand of the duplex. Despite these differences in experimental systems, the binding affinities we obtained are consistent with previous ITC data on the binding of CTD to a 5′-OH 12 dsRNA (1.1 μm) (11) and with the observation that the CTD binds 5′-ppp 14 dsRNA with higher binding affinity than the 5′-OH dsRNA (11, 12). Although the CTD is smaller (17 kDa) than the HD (64 kDa), it is able to recognize and bind the 5′-ppp dsRNA with much higher binding affinity. This indicates that the CTD is the primary contributor to viral RNA specificity observed in full-length RIG-I, whereas the HD may have a secondary role in facilitating recognition of RNA duplexes with modest affinity. Importantly, the HD binds 5′-OH dsRNA with slightly higher affinity than the CTD, indicating that it contributes to duplex docking.
FIGURE 5.

Binding analysis of CTD to 14 dsRNA substrates. A, binding curve of CTD with 5′-ppp 14 dsRNA (Kd = 0.500 ± 0.060 nm; R2 = 0.95). B, binding curve of CTD with 5′-OH 14 dsRNA (Kd = 5900 ± 750 nm; R2 = 0.98) determined by EMSAs. The CTD concentrations for experiments with 5′-ppp 14 dsRNA are 0.100, 0.200, 0.400, 0.800, 1.5, 3, 6, 13, 25, 51, 102 nm. The CTD concentrations for experiments with 5′-OH 14 dsRNA are 13, 25, 50, 100, 200, 410, 810, 1630, 3250, 6500, 13,000 nm. The dsRNA concentration in the reactions is 0.1 nm.
Enhancing Target Specificity through Antagonistic Domain Binding
Although full-length RIG-I protein binds triphosphorylated duplex tightly (Table 2, column 5) and with higher affinity than the isolated HD or CTD proteins (Table 2, columns 2 and 3, respectively) the observed affinity (159 pm) is actually much weaker than expected. Indeed, if the free energy of RIG-I binding to RNA were the simple sum of HD and CTD affinities for RNA, one would expect a total interaction energy that is 7.7 kcal/mol greater than what is observed (compare column 5 with column 4); i.e. if binding of the individual domains were purely additive, RIG-I would bind triphosphorylated targets with an affinity in the attomolar range (Kd = 436 am, computed from column 4). The fact that this is not the case indicates that the covalently linked HD and CTD bind RNA in an antagonistic fashion during interaction of RIG-I with RNA. The positive ΔΔG0 value of 7.7 kcal/mol (column 7) suggests that RNA binding by the tandem domains in the full-length protein is accompanied by a conformational transition that is energetically unfavorable. This type of effect is a common strategy that often evolves to enhance target binding specificity by macromolecules.
TABLE 2.
Energetic parameters of the RIG-I dsRNA interaction
| Substrate | ΔGHD* | ΔGCTD* | Σ ΔGHD* and ΔGCTD* | ΔGfull-length* | ΔGΔCARDs* | ΔΔGfull-length − domains | ΔΔGΔCARDs − domains |
|---|---|---|---|---|---|---|---|
| kcal/mol | kcal/mol | kcal/mol | kcal/mol | kcal/mol | kcal/mol | kcal/mol | |
| 5′-OH 14 dsRNA | −7.80 ± 0.08 | −7.11 ± 0.08 | −14.9 ± 0.2 | −10.50 ± 0.08 | −11.5 ± 0.2 | 4.4 ± 0.2 | 3.4 ± 0.4 |
| 5′-ppp 14 dsRNA | −8.31 ± 0.02 | −12.7 ± 0.06 | −21.00 ± 0.08 | −13.30 ± 0.07 | −14.10 ± 0.03 | 7.7 ± 0.2 | 6.9 ± 0.1 |
The presence of the CARDs influences this energetic penalty, although the effect is not large. The ΔΔG0 for 5′-ppp RNA binding by full-length RIG-I is 7.7 kcal/mol, whereas that of RIG-I lacking the CARDs is 6.9 kcal/mol (compare columns 7 and 8). The effect is similar upon binding a non-triphosphorylated duplex because the ΔΔG0 for full-length RIG-I is 4.4 kcal/mol, whereas that of the mutant without CARDs is 3.4 kcal/mol (compare columns 7 and 8). These data suggest that the CARDs may slightly enhance the severity of the energetic penalty incurred upon binding of the tandem domains. This would be consistent with crystallographic studies showing that the HD insertion domain plays two mutually exclusive roles, serving as both a binding site for the CARDs in the absence of RNA and as a duplex interface with bound RNA molecules (13, 14). Thus, detachment of the CARDs and subsequent binding of HD to RNA may incur a small but significant energetic cost.
The magnitude of the energetic penalty is most strongly influenced by the presence of the triphosphate moiety because it is 3–4 kcal/mol more pronounced in the case of triphosphorylated RNA duplex (compare rows 1 and 2 for columns 7 and 8). This suggests that binding of the isolated CTD to a triphosphorylated RNA duplex is exceptionally strong, as previously established in the literature, and that this interaction mode differs from (and may rearrange into) the conformation that forms when the CTD is covalently connected to the HD in the full-length protein. Taken together, the data indicate that binding of contiguous CTD and HD domains is less energetically favorable than the sum of individual CTD and HD binding energies. This effect is particularly pronounced for RNAs containing a 5′-triphosphate, and it is somewhat influenced by the presence of CARDs.
DISCUSSION
Here, we report a thermodynamic analysis of the molecular determinants for viral RNA recognition by each domain of RIG-I. We find that the individual domains play distinct mechanistic roles, yet the interplay among domains makes an important contribution to binding specificity.
The Mechanistic Role of Individual RIG-I Components
The CTD Plays a Dominant Thermodynamic Role in Ligand Recognition
Despite its small size, the CTD provides the greatest thermodynamic contribution to the binding of triphosphorylated RNAs. High affinity binding by the CTD was expected because previous studies on the isolated domain had demonstrated that it binds short, triphosphorylated dsRNA ligands with high affinity, particularly in comparison with non-triphosphorylated duplexes (11, 12). These studies hypothesized that the CTD would enhance the affinity of full-length RIG-I to 5′-ppp dsRNA (11, 12, 15, 23), although it was not expected that the CTD would actually dominate the energetics of RNA recognition by RIG-I, given the large molecular interface between the HD and the RNA duplex (see below) (13). Indeed, a recent study on full-length RIG-I and domain variants suggested that RIG-I did not readily discriminate dsRNA ligands with 5′-ppp and 5′-OH termini (16). In those experiments, the affinity for 5′-ppp dsRNA was only 2.7-fold higher than that of 5′-OH dsRNA (16). However, our results are in accord with early work on the isolated CTD (11, 12) because we observe that intact human RIG-I binds 5′-ppp dsRNA with 126-fold tighter affinity than 5′-OH dsRNA. Similarly, a RIG-I variant lacking the CARDs displays radically enhanced binding affinity for 5′-ppp dsRNA compared with 5′-OH dsRNA.
It is likely that previous reports of comparable 5′-ppp dsRNA and 5′-OH dsRNA affinities for full-length RIG-I are due to the experimental design (16). Those studies employed fluorescence anisotropy experiments in which a hydrophobic fluorophore was placed at the 5′ terminus of the RNA. Based on the crystal structures of the CTD and RIG-I (ΔCARDs) in complex with dsRNA, the duplex terminus, and the 5′-end in particular, is a critical binding interface for the protein (11–16). Potential contacts formed between RIG-I and fluorescein may have artificially enhanced affinity between the protein and the labeled 5′-OH duplex. This effect would have been reduced in the case of RIG-I binding to 5′-ppp dsRNA (where the fluorescein was incorporated on the opposite 5′ terminus) because 5′-ppp binding to the CTD establishes a specific polarity for association with the protein (11, 12). As a result of these two opposing effects, similar binding for the two ligands was observed.
The HD Is Essential for Specific Binding to Duplex RNA
The triphosphate is not the only important determinant for discrimination by RIG-I. This innate immune sensor must discriminate duplex RNA from single-stranded host RNA and from viral DNA, which is detected by another set of surveillance proteins. Recent crystal structures of RIG-I show that the HD enfolds the RNA duplex in a complex network of interactions. Our thermodynamic analysis is consistent with this because the HD binds with reasonable affinity to an RNA duplex (∼2 μm), which is somewhat higher than the affinity of the isolated CTD (∼6 μm). The structures reveal that the CTD and HD form different types of molecular interactions with the RNA (13), perhaps explaining the lower affinity of the HD. Whereas the CTD forms hydrophobic and electrostatic interactions with the duplex terminus, the HD forms strictly polar contacts with the dsRNA backbone, mediated largely by glutamine contacts to RNA 2′-hydroxyl groups. Whereas these interactions are likely to contribute to high specificity, they are less likely than hydrophobic contacts to mediate exceptionally strong binding.
The HD plays a central role in the selectivity for duplex RNA. Here we show that RIG-I sensitively distinguishes duplex RNA from a short single-stranded RNA (14-mer) that lacks secondary structure, displaying a 361-fold difference in affinity for this ligand. This is surprising because RIG-I belongs to the same family of proteins (superfamily 2), and contains the same RNA binding motifs, as many motor proteins with high affinity toward single-stranded RNA. One might expect that RIG-I (and the HD) would bind single-stranded RNA with the same affinity as NS3 (Kd = 9 nm) (24) and that it would bind dsRNA with even higher affinity. The fact that this is not observed and that RIG-I binds ssRNA with 144-fold less affinity than its cousin NS3 indicates that it has developed specialized domains for recognition of the second strand (13, 14). This specificity of RIG-I has important implications within the cell. The abundance of cellular single-stranded RNAs is likely to be low because RNA molecules tend to form secondary and tertiary structures involving both canonical and non-canonical base pairings. Therefore, the low abundance of truly single-stranded RNA in the cell and the very weak binding of RIG-I to single-stranded RNA probably allow RIG-I to actively discriminate against binding to single-stranded ligands. In this way, RIG-I will actively respond to viral RNA while ignoring abundant host mRNA.
Although the triphosphate specificity of RIG-I is primarily attributable to the CTD and dsRNA specificity lies predominantly with the HD, the HD nonetheless prefers triphosphorylated ligands to 5′-OH RNA duplexes (Fig. 4). Although this could be the result of a simple electrostatic enhancement, it is also consistent with aspects of the molecular structure. The crystal structure of dRIG-I, which is strikingly similar to the human variant (14), nonetheless lacks the CTD and allows us to visualize RNA binding by the HD in isolation. In that case, the dRIG-I HD interacts with the 5′-end of the top strand using conserved residues within the RLR “helicase” core (motif IIa and HEL2i) (14). In the context of a triphosphorylated RNA, these motifs may communicate with the CTD to enhance the binding affinity to 5′-ppp dsRNA. These observations, together with the fact that the isolated CTD prefers duplexes to single-stranded RNA (11, 12, 15), suggest that the mechanistic roles of the CTD and HD overlap to some extent and that, over time, the domains have evolved to behave synergistically.
The Autoinhibitory Role of the CARDs
As an innate immune sensor, RIG-I must be tightly regulated at the initial step of RNA binding. Several studies have implicated the CARDs in mechanisms to reduce and/or inhibit nonspecific binding of nucleic acids (23, 25). Indeed, the structures of full-length dRIG-I and ΔCTD dRIG-I (14) in the absence of RNA indicate that the CARDs bind the HEL2i domain and partially block the dsRNA binding site in the HD (14). However, in the structures of RIG-I bound to RNA, the same regions of HEL2i interact directly with dsRNA (13, 14). Based on these studies and the observation that RIG-I (ΔCARDs) bound 5′-OH dsRNA with 5.5-fold higher affinity compared with full-length RIG-I, it is likely that the CARDs modulate dsRNA binding to the HD. Interestingly, RIG-I (ΔCARDs) bound 5′-ppp dsRNA with only a 3.8-fold higher affinity than full-length RIG-I. This suggests a lack of significant interaction between the CARDs and the CTD, as previously reported (14). Thus, the CARDs influence RIG-I binding to duplex RNA through control of HD accessibility. Removal of the CARDs effectively unmasks the dsRNA binding site, exposing the surface of the HEL2i subdomain (14). Thus, in their own way, the CARDs play a role in discriminating between potential RIG-I ligands, serving as a type of flexible gate that may select among high affinity ligands.
Covalent Connection between CTD and HD Is Required for Highest Affinity Binding
Connectivity of the CTD and HD domains (as in full-length RIG-I and RIG-I (ΔCARDs)) creates an optimized binding site for RNA. For example, RIG-I (ΔCARDs) binds 5′-OH dsRNA (Kd = 3.6 nm) much more tightly than the isolated protein domains. The affinity of RIG-I (ΔCARDs) for the same RNA was ∼500-fold higher than for the HD alone and ∼1600-fold higher than for the isolated CTD. The tandem domains also form an optimal binding site for triphosphorylated RNA, as indicated by the low picomolar binding affinity of RIG-I (ΔCARDs) to 5′-ppp 14 dsRNA (41 pm). By contrast, RIG-I (ΔCARDs) bound 5′-ppp dsRNA ∼18,000-fold more tightly than the HD and 12-fold more tightly than the CTD alone. These results indicate that RIG-I and RIG-I (ΔCARDs) utilize both domains to attain a high degree of structural specificity not only for 5′-OH dsRNA but also for 5′-ppp dsRNA.
Because the RIG-I (ΔCARDs) structure (13) superimposes best with the crystal structure of the CTD bound to a 5′-ppp dsRNA (11, 12), we can surmise that the presence of the 5′-ppp would probably increase the number of existing contacts made by the CTD to the duplex terminus.
Why RIG-I Is Less Than the Sum of Its Parts; the Mechanistic Role of Domain Antagonism
One of the most intriguing findings of this study is that the binding energy of intact RIG-I constructs is significantly less than the sum of CTD and HD binding energies. If RIG-I bound RNA targets with the energy expected from this sum, affinity would be in the attomolar range, and binding would be effectively irreversible. However, when HD and CTD bind RNA in tandem, a substantial amount of binding energy is somehow released into the system. This effect is strikingly pronounced, resulting in a ΔΔG0 of 7–8 kcal/mol for triphosphorylated duplexes to a value of 3–4 kcal/mol for non-triphosphorylated duplexes. What is the molecular basis for this effect, and what advantage might it provide for surveillance and signaling by RIG-I?
The molecular basis for this energetic penalty may be linked to intraprotein conformational changes that occur upon RNA ligand binding. Clues to this effect are provided by crystallographic and small angle x-ray scattering studies on RIG-I constructs (13), which show that the tandem domains become conformationally restricted upon binding 5′-OH dsRNA (13, 26). An overall loss of entropy within the large constructs is likely to account for some of the observed energetic penalty. Induction of strain within the protein may also occur upon RNA binding. For example, the pincer domain is a rigid linker between the HD and CTD, and it sensitively relays information about RNA binding to the ATPase site within RIG-I (13). It is well established that linkers within multidomain proteins and molecular switches can be sites of induced strain, leading to apparent antagonistic behavior among individual protein domains (27). Alternatively, RIG-I binding energy may be released by the rearrangement of contacts at the RNA interface. For example, in the structure of the isolated CTD (11), a loop containing Lys-849 reaches in and binds within the RNA duplex. However, in the structure of the RIG-I (ΔCARDs) (13), this contact has come apart, and the same RNA functional groups interact with the HEL1 domain. Thus, certain contacts by the individual RNA domains are mutually exclusive, and when the same domains are connected within the full-length protein, substantial rearrangement occurs. Not only does this explain the basis for an energetic penalty upon binding of full-length protein, but it suggests that the CTD could bind the 5′ terminus first, subsequently rearranging to accommodate binding of the HD.
The most likely consequence of this effect is a dramatic enhancement in ligand specificity. Antagonistic ligand binding by multidomain macromolecules is critical for specific binding by systems that range from ribozymes to cadherins (28, 29). Simply put, the individual units or domains of these systems bind so strongly that if they were strung together, the macromolecule would be unable to discriminate among important ligand variants. In the case of RIG-I, it is essential that the protein discriminate between viral RNA and a variety of single-stranded, double-stranded, capped, and sometimes 5′-phosphorylated host RNAs. As in other systems, RIG-I accomplishes this by linking two specific ligand binding sites but reducing the buildup of interaction energy through induction of strain or loss of entropy.
CONCLUSION
In conclusion, this work characterizes the molecular determinants for viral RNA recognition by RIG-I and establishes the respective roles for individual protein domains during RIG-I/RNA interaction. Full-length RIG-I forms a particularly high affinity complex with short, triphosphorylated duplexes due to specific 5′-ppp recognition by the CTD. This establishes that the 5′-ppp on dsRNA is a critical moiety important for viral RNA recognition by the full-length RIG-I protein. Thermodynamic analysis of the isolated and covalently linked protein domains reveals that the specialized domains of RIG-I are poised to interact with viral transcripts and that they discriminate against host RNA in a manner that requires expenditure of energy in order to achieve high substrate specificity. The results have important implications for targeting the RIG-I/RNA interface in the development of therapeutics for infection and autoimmune disorders.
Acknowledgments
We thank Dr. Dahai Luo for help in the design of RNA oligonucleotides used in FP experiments and Dr. Megan Fitzgerald for helpful discussions about assays. We thank the laboratory of Alanna Schepartz for use of the plate reader. We also thank Dr. Nora Zingler and Dr. Amanda Solem for initial assistance and technical help with the project. We thank Andrew Kohlway and Dahai Luo for helpful discussions and for initial reading of the manuscript. We also thank Gaby Drews for technical assistance.
Footnotes
- RLR
- RIG-I-like receptor
- HEL1 and -2
- helicase domain 1 and 2, respectively
- CTD
- C-terminal domain
- CARD
- caspase activation and recruitment domain
- dRIG-I
- duck RIG-I
- HD
- helicase domain
- β-ME
- β-mercaptoethanol
- FP
- fluorescence polarization
- AMP-PNP
- 5′-adenylyl-β,γ-imidodiphosphate
- 5′-ppp
- triphosphate moiety at the 5′ terminus
- ΔG*
- apparent binding energy.
REFERENCES
- 1. Kang D. C., Gopalkrishnan R. V., Wu Q., Jankowsky E., Pyle A. M., Fisher P. B. (2002) mda-5. An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc. Natl. Acad. Sci. U.S.A. 99, 637–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sarkar D., Desalle R., Fisher P. B. (2008) Evolution of MDA-5/RIG-I-dependent innate immunity. Independent evolution by domain grafting. Proc. Natl. Acad. Sci. U.S.A. 105, 17040–17045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Satoh T., Kato H., Kumagai Y., Yoneyama M., Sato S., Matsushita K., Tsujimura T., Fujita T., Akira S., Takeuchi O. (2010) LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc. Natl. Acad. Sci. U.S.A. 107, 1512–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Murali A., Li X., Ranjith-Kumar C. T., Bhardwaj K., Holzenburg A., Li P., Kao C. C. (2008) Structure and function of LGP2, a DEX(D/H) helicase that regulates the innate immunity response. J. Biol. Chem. 283, 15825–15833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Komuro A., Horvath C. M. (2006) RNA- and virus-independent inhibition of antiviral signaling by RNA helicase LGP2. J. Virol. 80, 12332–12342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kato H., Takeuchi O., Sato S., Yoneyama M., Yamamoto M., Matsui K., Uematsu S., Jung A., Kawai T., Ishii K. J., Yamaguchi O., Otsu K., Tsujimura T., Koh C. S., Reis e Sousa C., Matsuura Y., Fujita T., Akira S. (2006) Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105 [DOI] [PubMed] [Google Scholar]
- 7. Saito T., Owen D. M., Jiang F., Marcotrigiano J., Gale M., Jr. (2008) Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454, 523–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fujita T., Onoguchi K., Onomoto K., Hirai R., Yoneyama M. (2007) Triggering antiviral response by RIG-I-related RNA helicases. Biochimie 89, 754–760 [DOI] [PubMed] [Google Scholar]
- 9. Fairman-Williams M. E., Guenther U. P., Jankowsky E. (2010) SF1 and SF2 helicases. Family matters. Curr. Opin. Struct. Biol. 20, 313–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pyle A. M. (2008) Translocation and unwinding mechanisms of RNA and DNA helicases. Annu. Rev. Biophys. 37, 317–336 [DOI] [PubMed] [Google Scholar]
- 11. Wang Y., Ludwig J., Schuberth C., Goldeck M., Schlee M., Li H., Juranek S., Sheng G., Micura R., Tuschl T., Hartmann G., Patel D. J. (2010) Structural and functional insights into 5′-ppp RNA pattern recognition by the innate immune receptor RIG-I. Nat. Struct. Mol. Biol. 17, 781–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lu C., Xu H., Ranjith-Kumar C. T., Brooks M. T., Hou T. Y., Hu F., Herr A. B., Strong R. K., Kao C. C., Li P. (2010) The structural basis of 5′ triphosphate double-stranded RNA recognition by RIG-I C-terminal domain. Structure 18, 1032–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Luo D., Ding S. C., Vela A., Kohlway A., Lindenbach B. D., Pyle A. M. (2011) Structural insights into RNA recognition by RIG-I. Cell 147, 409–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kowalinski E., Lunardi T., McCarthy A. A., Louber J., Brunel J., Grigorov B., Gerlier D., Cusack S. (2011) Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 147, 423–435 [DOI] [PubMed] [Google Scholar]
- 15. Lu C., Ranjith-Kumar C. T., Hao L., Kao C. C., Li P. (2011) Crystal structure of RIG-I C-terminal domain bound to blunt-ended double-strand RNA without 5′ triphosphate. Nucleic Acids Res. 39, 1565–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jiang F., Ramanathan A., Miller M. T., Tang G. Q., Gale M., Jr., Patel S. S., Marcotrigiano J. (2011) Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature 479, 423–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wincott F., DiRenzo A., Shaffer C., Grimm S., Tracz D., Workman C., Sweedler D., Gonzalez C., Scaringe S., Usman N. (1995) Synthesis, deprotection, analysis and purification of RNA and ribozymes. Nucleic Acids Res. 23, 2677–2684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Coleman T. M., Wang G., Huang F. (2004) Superior 5′ homogeneity of RNA from ATP-initiated transcription under the T7 phi 2.5 promoter. Nucleic Acids Res. 32, e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hartmann R. K. (2005) Handbook of RNA Biochemistry, pp. 33–34, Wiley-VCH, Weinheim, Germany [Google Scholar]
- 20. Fersht A. (1985) Enzyme Structure and Mechanism, 2nd Ed., p. 129, W.H. Freeman, San Francisco CA [Google Scholar]
- 21. Chen C. P., Posy S., Ben-Shaul A., Shapiro L., Honig B. H. (2005) Specificity of cell-cell adhesion by classical cadherins. Critical role for low affinity dimerization through β-strand swapping. Proc. Natl. Acad. Sci. U.S.A. 102, 8531–8536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cantor C. R., Schimmel P. R. (1980) The Behavior of Biological Macromolecules, pp. 874–875, W.H. Freeman, San Francisco [Google Scholar]
- 23. Cui S., Eisenächer K., Kirchhofer A., Brzózka K., Lammens A., Lammens K., Fujita T., Conzelmann K. K., Krug A., Hopfner K. P. (2008) The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol Cell 29, 169–179 [DOI] [PubMed] [Google Scholar]
- 24. Beran R. K., Serebrov V., Pyle A. M. (2007) The serine protease domain of hepatitis C viral NS3 activates RNA helicase activity by promoting the binding of RNA substrate. J. Biol. Chem. 282, 34913–34920 [DOI] [PubMed] [Google Scholar]
- 25. Gee P., Chua P. K., Gevorkyan J., Klumpp K., Najera I., Swinney D. C., Deval J. (2008) Essential role of the N-terminal domain in the regulation of RIG-I ATPase activity. J. Biol. Chem. 283, 9488–9496 [DOI] [PubMed] [Google Scholar]
- 26. Civril F., Bennett M., Moldt M., Deimling T., Witte G., Schiesser S., Carell T., Hopfner K. P. (2011) The RIG-I ATPase domain structure reveals insights into ATP-dependent antiviral signalling. EMBO Rep. 12, 1127–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cutler T. A., Mills B. M., Lubin D. J., Chong L. T., Loh S. N. (2009) Effect of interdomain linker length on an antagonistic folding-unfolding equilibrium between two protein domains. J. Mol. Biol. 386, 854–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Qin P. Z., Pyle A. M. (1999) Antagonistic substrate binding by a group II intron ribozyme. J. Mol. Biol. 291, 15–27 [DOI] [PubMed] [Google Scholar]