

Background: mTORC1 directly phosphorylates p70S6K1 and the 4E-BPs.
Results: Although necessary, eIF4E·eIF4G complex formation was insufficient for sustaining global rates of protein synthesis.
Conclusion: p70S6K1-mediated phosphorylation of eIF4B and PDCD4 also was required to optimally stimulate protein synthesis.
Significance: Both eIF4E·eIF4G complex formation and activation of p70S6K1 individually stimulate global rates of protein synthesis.
Keywords: eIF4E, mTOR, mTOR Complex (mTORC), Translation Control, Translation Initiation Factors, Translation Regulation, PDCD4, eIF4A, eIF4B, p70S6K1
Abstract
Modulation of mRNA binding to the 40 S ribosomal subunit during translation initiation controls not only global rates of protein synthesis but also regulates the pattern of protein expression by allowing for selective inclusion, or exclusion, of mRNAs encoding particular proteins from polysomes. The mRNA binding step is modulated by signaling through a protein kinase known as the mechanistic target of rapamycin complex 1 (mTORC1). mTORC1 directly phosphorylates the translational repressors eIF4E binding proteins (4E-BP) 1 and 2, releasing them from the mRNA cap binding protein eIF4E, thereby promoting assembly of the eIF4E·eIF4G complex. mTORC1 also phosphorylates the 70-kDa ribosomal protein S6 kinase 1 (p70S6K1), which subsequently phosphorylates eIF4B, and programmed cell death 4 (PDCD4), which sequesters eIF4A from the eIF4E·eIF4G complex, resulting in repressed translation of mRNAs with highly structured 5′-untranslated regions. In the present study, we compared the role of the 4E-BPs in the regulation of global rates of protein synthesis to that of eIF4B and PDCD4. We found that maintenance of eIF4E interaction with eIF4G was not by itself sufficient to sustain global rates of protein synthesis in the absence of mTORC1 signaling to p70S6K1; phosphorylation of both eIF4B and PDCD4 was additionally required. We also found that the interaction of eIF4E with eIF4G was maintained in the liver of fasted rats as well as in serum-deprived mouse embryo fibroblasts lacking both 4E-BP1 and 4E-BP2, suggesting that the interaction of eIF4G with eIF4E is controlled primarily through the 4E-BPs.
Introduction
Global rates of protein synthesis in the liver are suppressed following an overnight fast but recover rapidly upon feeding (1). The feeding-induced stimulation of hepatic protein synthesis is in large part due to activation of signaling through the serine/threonine protein kinase known as the mechanistic target of rapamycin (mTOR;2 previously known as the mammalian target of rapamycin) (2, 3). The polypeptide mTOR functions in two distinct multiprotein complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) (3). Both complexes contain mTOR, mLST8, DEPTOR, and the Tti1/Tel2 complex; however, mTORC1 contains the raptor (regulatory-associated protein of mTOR) and PRAS40 (proline-rich Akt substrate 40 kDa), whereas mTORC2 contains rictor (raptor-independent companion of mTOR), mSin1, and protor1/2 (3). mTORC1 regulates cell growth and proliferation by integrating signals from nutrients and mitogens to acutely stimulate protein synthesis through multiple mechanisms, including phosphorylation and activation of several downstream proteins involved in the mRNA binding step in translation initiation. During this step, the 43 S pre-initiation complex, consisting of the 40 S ribosomal subunit, the eIF2·GTP·initiator methionyl-tRNA complex, and eIF3, is recruited to the m7GTP cap at the 5′-end of mRNA and scans for an AUG start codon that is in the proper context (4). The role of mTORC1 in regulating global rates of protein synthesis is critical, as Torin1 and PP242, potent inhibitors of mTOR, significantly impair protein synthesis in both proliferating wild-type and rictor−/− cells (5, 6).
The best characterized substrates for mTORC1 are the ribosomal protein S6 kinases S6K1 and S6K2 and the eukaryotic initiation factor eIF4E-binding proteins (4E-BPs). The 4E-BPs bind to the mRNA m7GTP cap binding protein eIF4E and prevent it from interacting with the scaffolding protein eIF4G. Phosphorylation of the 4E-BPs by mTORC1 releases them from eIF4E, allowing eIF4E to interact with eIF4G. The interaction promotes the binding of the mRNA to the 43 S preinitiation complex through the association of the mRNA·eIF4E·eIF4G complex with eIF3 that is part of the preinitiation complex. Thus, in part, the binding of the mRNA to the 43 S preinitiation complex is controlled through mTORC1-mediated phosphorylation of the 4E-BPs, although other mechanisms, e.g. phosphorylation of eIF4E and/or eIF4G, have also been described (7–9). Once the 40 S ribosome is located at the m7GTP cap at the 5′-end of the mRNA, the next step involves scanning of the 40 S ribosomal subunit along the 5′-untranslated region (UTR) until an AUG start codon in the proper context is located. During scanning, secondary structure is unwound by the RNA helicase, eIF4A. The helicase activity of eIF4A is stimulated by eIF4B, allowing the unwinding of longer, more stable secondary structures. The interaction of both eIF4A and eIF4B with eIF4G is regulated by the S6 kinases. Thus, the 70-kDa ribosomal protein S6 kinase p70S6K1 phosphorylates PDCD4 (programmed cell death protein 4), releasing it from eIF4A, allowing eIF4A to interact with the C terminus of eIF4G (10). p70S6K1 also phosphorylates eIF4B, promoting its interaction with eIF4G and eIF4A (11).
The vast majority of studies assessing mTORC1-mediated stimulation of mRNA translation have focused on phosphorylation of the 4E-BPs and the interaction of eIF4E with eIF4G as a mechanism for regulating the mRNA binding step in initiation. However, a recent study (12) suggests that the decrease in protein synthesis associated with mTORC1 inhibition cannot be attributed solely to a reduction in the interaction of eIF4E with eIF4G and that other processes not identified in the study are additionally required. In the present study, we confirmed the results of the previous study (12) to show that the interaction of eIF4E with eIF4G was not sufficient to stimulate mRNA translation in either the liver of fasted mice or in serum-deprived mouse embryo fibroblasts (MEF) in culture. We extend the earlier study to show that activation of p70S6K1 and subsequent phosphorylation of eIF4B and PDCD4 act in concert with eIF4E·eIF4G complex assembly in mTORC1-mediated stimulation of mRNA translation. We also demonstrated that the primary mechanism for regulating the interaction of eIF4E with eIF4G involves 4E-BP1 and -BP2. Overall, the results are consistent with a model in which multiple inputs downstream of mTORC1 are required to optimally stimulate protein synthesis.
EXPERIMENTAL PROCEDURES
Materials
Protease Inhibitor Mixture was purchased from Sigma and ECL Western blotting detection reagent from Pierce. Preparation of the 4E-BP1 and eIF4E antibodies has been described previously (13, 14). Anti-S6K1, and goat anti-rabbit IgG horseradish peroxidase-conjugated antibodies were purchased from Bethyl Laboratories. Anti-GAPDH antibody was purchased from Santa Cruz Biotechnology, whereas all other antibodies were purchased from Cell Signaling Technology. Protein content was measured by DC protein assay (Bio-Rad).
Animals
Eif4ebp1;Eif4ebp2 mice bearing disruptions in the genes encoding both 4E-BP1 and 4E-BP2 were a kind gift from Dr. Nahum Sonenberg (McGill University). Male mice weighing ∼30 g were maintained on a 12:12-h light:dark cycle with food (Harlan Teklad), and water was provided ad libitum. Mice were fasted for 10 h, and some of the mice were then allowed free access to food for 45 min where indicated. All procedures involving the mice were approved by the Pennsylvania State University College of Medicine Institutional Animal Care and Use Committee.
Processing of Liver Samples
For the analysis of protein phosphorylation state, a portion (∼0.3 g) of liver was homogenized in 7 volumes of lysis buffer (20 mm HEPES, 2 mm EGTA, 50 mm NaF, 100 mm KCl, 0.2 mm EDTA, 50 mm β-glycerophosphate, 1 mm DTT, 1 mm benzamidine, 50 mm sodium vanadate, 1 mm microcystin, and 10 μl/ml Sigma protease inhibitor mixture) using a Polytron homogenizer. The homogenate was centrifuged at 1,000 × g for 3 min at 4 °C, and the resulting supernatant fraction was subjected to SDS-PAGE and Western blot analysis as described previously (15). Phosphorylation of eIF4E, eIF4G, eIF4B, S6K1, and PDCD4 was measured in the supernatant fraction using phospho-specific antibodies.
Cell Culture and Transfections
Cultures of wild-type and Eif4ebp1;Eif4ebp2 double knock-out (DKO) MEF, a kind gift from Dr. Nahum Sonenberg (McGill University), were maintained in Dulbeco's modified Eagle's medium lacking sodium pyruvate and containing high glucose (Invitrogen) supplemented with 10% fetal bovine serum (Atlas) and 1% penicillin/streptomycin (Invitrogen). Transfections were performed using Xtremegene HP (Roche Applied Science) with a 3:1 ratio of reagent to DNA (μl:μg) according to the manufacturer's instructions. Cells were deprived of serum for 3 h and treated with IGF (insulin-like growth factor) 1 (10 ng/ml) as indicated. Cells were harvested in cell lysis buffer supplemented with 1% Triton X-100. To evaluate the rate of protein synthesis, cells were treated as described above. After 5 min of treatment with IGF1, cells were radiolabeled with 50 μCi of 35S-labeled EasyTag Express Protein Labeling Mix (PerkinElmer Life Sciences) for 15 min at 30 °C. Labeled cells were washed 3× with PBS and harvested in ice-cold lysis buffer supplemented with 1% Triton X-100 and 0.5% deoxycholate. Lysates were rocked at 4 °C for 30 min and centrifuged at 1,000 × g for 5 min. Global rates of protein synthesis were calculated by the incorporation of [35S]methionine and [35S]cysteine into TCA-precipitable protein as described previously (16). HA-tagged constitutively active S6K1 expression plasmid was a kind gift from Dr. John Blenis (Harvard Medical School) and has been described previously (17). Constitutively active HA-tagged eIF4B was generated through mutation of Ser-422 to Asp using a previously described plasmid (18) that was also provided by Dr. Blenis. Site-directed mutagenesis was performed using QuikChange Lightning (Agilent Technologies) and the following primers to mutate Ser-422 to Asp: 5′-GGAACGGTCGAGGACAGGAGATGAGTCATCACAAACTG-3′ and 5′-CAGTTTGTGATGACTCATCTCCTGTCCTCGACCGTTCC-3′. The product was then cloned into pCDNA3.1 (Invitrogen) with an N-terminal HA-tag using HindIII and NotI and the following primers: 5′-AGCTAAGCTTACCATGTACCCATACGATGTTCCAGATTACGCTATGGCGGCCTCAGCAAA-3′ and 5′-GAAGATTATGCCGATTAAGCGGCCGCAGCT-3′. The plasmid was then sequenced to confirm the mutation. For siRNA studies, the following complementary RNAs were transfected using DharmaFECT (Thermo Scientific) according to the manufacturer's suggested protocol to knock down PDCD4 expression: 5′-CCTATATCGATAGTTACAAAGGAAC-3′ and 5′-GTTCCTTTGTAACTATCGATATAGGTA-3′.
Immunoprecipitations
Immunoprecipitations were performed by incubating 1,000 × g supernatant fractions of liver homogenates or cell lysates with monoclonal anti-human eIF4E or eIF4G antibody. Homogenate or lysate fractions containing ∼1 mg of protein were incubated with 15 μg of antibody for 1 h at 4 °C. For each immunoprecipitation reaction, 200 μg of anti-mouse IgG beads (Qiagen) previously blocked with lysis buffer containing 1% BSA was added to each sample, and the suspension was rocked at 4 °C for 1 h. For eIF4E immunoprecipitation, beads were washed 3× with 1 ml of cold lysis buffer. For eIF4G, immunoprecipitation beads were washed 3× with PBS. After washing, beads were suspended in 1× SDS sample buffer, and boiled for 5 min. Supernatant fractions were subjected to Western blot analysis using antibodies to 4E-BP1, 4E-BP2, eIF4E, eIF4G, or PDCD4, and the results were normalized for the amount of the target protein in the immunoprecipitate.
Statistical Analysis
The data are expressed as means ± S.E. One-way analysis of variance and Student's t test were used to compare differences among groups. p < 0.05 was considered statistically significant.
RESULTS
Fasting Fails to Reduce the Interaction of eIF4E with eIF4G in the Liver of DKO Mice
Three mechanisms have been proposed to regulate the interaction of eIF4E with eIF4G: the reversible interaction of eIF4E with the eIF4E binding proteins (7), phosphorylation of eIF4E on Ser-209 (8), and phosphorylation of eIF4G on multiple residues including Ser-1108 (9). To examine the relative contribution of each of these mechanisms to regulation of the eIF4E·eIF4G interaction, mice containing disruptions in the genes encoding 4E-BP1 and 4E-BP2 were fasted overnight and then refed in the morning to activate mTORC1 and promote changes in phosphorylation of the 4E-BPs, eIF4E and eIF4G. eIF4E was then immunoprecipitated from liver supernatants and the presence of eIF4G was assessed in the immunoprecipitates. As shown in Fig. 1A, the amount of eIF4G bound to eIF4E was low in the livers of fasted, wild-type mice, and re-feeding resulted in a 2-fold elevation in eIF4G bound to eIF4E. To confirm the feeding-induced elevation in the interaction of the two proteins, eIF4G was instead immunoprecipitated from liver supernatant fractions and assessed for eIF4E present in the immunoprecipitate. In agreement with the results of the eIF4E immunoprecipitation, refeeding significantly elevated the interaction of eIF4E with eIF4G (Fig. 1B). The enhanced interaction of the two proteins was associated with a reduction in the interaction of 4E-BP1 (Fig. 1C) and 4E-BP2 (Fig. 1D) with eIF4E as well as enhanced phosphorylation of 4E-BP1 (Fig. 1E) and eIF4G (Fig. 1F), concomitant with a reduction in phosphorylation of eIF4E on Ser-209 (Fig. 1G). Interestingly, the amount of eIF4G in the eIF4E immunoprecipitate was the same in livers from fasted DKO mice as in fed wild-type or DKO mice, but significantly greater compared with fasted wild-type mice (Fig. 1A). Similarly, the amount of eIF4E in the eIF4G immunoprecipitate was the same in fasted DKO mice as in fasted wild-type or refed DKO mice but greater than in fasted wild-type mice (Fig. 1B). In contrast, the magnitude of the feeding-induced changes in eIF4E and eIF4G phosphorylation was similar in wild-type and DKO mice. Thus, in mouse liver, the interaction of eIF4E with eIF4G is regulated primarily through the interaction of eIF4E with 4E-BP1 and 4E-BP2.
FIGURE 1.

Effects of fasting and refeeding on eIF4E and eIF4G phosphorylation and interaction in the liver of wild-type and 4E-BP1/2 DKO mice. Wild-type and 4E-BP1/2 DKO mice were fasted for 12 h and refed for 45 min as indicated. The interaction of eIF4G with eIF4E was evaluated in liver supernatant fractions by immunoprecipitating eIF4E and measuring the amount of eIF4G in the immunoprecipitate (A) as well as by immunoprecipitating eIF4G and measuring the amount of eIF4E in the immunoprecipitate by Western blot analysis (B). The interaction of 4E-BP1 (C) and 4E-BP2 (D) with eIF4E was evaluated by immunoprecipitating eIF4E and measuring the amount of each protein in the immunoprecipitate. E, phosphorylation of 4E-BP1 was assessed in liver supernatant fractions as the proportion of the protein present in the γ-form relative to the total amount of 4E-BP1 in all forms (α+β+γ). Phosphorylation of eIF4G on Ser-1108 (F) and eIF4E on Ser-209 (G) was assessed by Western blot analysis with phosphospecific antibodies. Representative blots are shown. Values are means + S.E. (n = 5–8). Statistical significance is denoted by the presence of different letters above the bars on the graphs. Bars with different letters are statistically different, p < 0.05.
Feeding Enhances mTORC1 Signaling Downstream of S6K1 in the Liver of Wild-type and DKO Mice
In addition to promoting phosphorylation of 4E-BPs, activation of mTORC1 also leads to enhanced phosphorylation of p70S6K1. In the present study, hyperphosphorylation of p70S6K1 was enhanced in the liver of both wild-type and DKO mice in response to refeeding (Fig. 2A). The enhanced phosphorylation of p70S6K1 in the liver of DKO compared with wild-type mice was likely due to the absence of 4E-BP1/2, as mTORC1 substrates have been previously observed to compete for phosphorylation (17, 19). Upon activation, p70S6K1 phosphorylates multiple downstream targets, including eIF4B on Ser-422 (18), PDCD4 on Ser-67 (18, 20), and eukaryotic elongation factor 2 (eEF2) kinase on Ser-336 (21). In response to refeeding, phosphorylation of eIF4B on Ser-422 was enhanced by 176% in the liver of wild-type mice (Fig. 2B), and a similar response was observed in the liver of DKO mice. Furthermore, the feeding-induced stimulation of PDCD4 phosphorylation on Ser-67 was elevated to a similar extent in the liver of wild-type (69%) and DKO mice (54%) (Fig. 2C) compared with fasted mice, whereas PDCD4 expression was reduced by 31 and 33%, respectively (Fig. 2D). The feeding-induced reduction in PDCD4 expression was mirrored by a fall in the amount of the protein detected in eIF4G immunoprecipitates (Fig. 2E). Phosphorylation of eEF2 kinase on Ser-336 by p70S6K1 results in inhibition of eEF2 kinase activity and consequently repressed phosphorylation of eEF2 on Thr-56 (21). However, in the present study, phosphorylation of eEF2 on Thr-56 did not change in the liver of either wild-type or DKO mice upon feeding (Fig. 2F).
FIGURE 2.

Feeding-induced activation of S6K1 promotes eIF4B phosphorylation and reduces the interaction of PDCD4 with eIF4G in the liver of wild-type and 4E-BP1/2 DKO mice. Wild-type and 4E-BP1/2 DKO mice were fasted for 12 h and refed for 45 min as indicated. Phosphorylation of S6K1 on Thr-389 (A), eIF4B on Ser-411 (B), and PDCD4 on Ser-67 (C) were evaluated in liver supernatant fractions with phosphospecific antibodies by Western blot analysis. D, PDCD4 expression was evaluated in liver supernatant fractions by Western blot analysis. E, interaction of PDCD4 with eIF4G/eIF4A was evaluated by immunoprecipitating eIF4G and measuring co-immunoprecipitated PDCD4 by Western blot analysis. F, phosphorylation of eEF2 on Thr-56 was assessed by Western blot analysis. Values are means + S.E. (n = 5). Statistical significance is denoted by the presence of different letters above the bars on the graphs. Bars with different letters are statistically different, p < 0.05.
Maintenance of eIF4E·eIF4G Complex Assembly Fails to Preserve Protein Synthesis upon Serum Deprivation
To evaluate the role of 4E-BP1/2 in regulating global rates of protein synthesis, the incorporation of 35S-labeled methionine and cysteine into cellular protein was measured in wild-type and 4E-BP1/2 DKO MEF following serum deprivation and upon stimulation of serum-deprived cells with IGF1. Protein synthesis is stimulated by IGF1 through binding to the type 1 IGF receptor, which promotes its interaction with the p85α subunit of the phosphatidylinositol-3-kinase and in turn activates Akt (22). Akt directly phosphorylates at least two proteins responsible for the regulation of mTORC1 signaling: tuberous sclerosis complex 2 and the 40-kDa proline-rich Akt substrate. The IGF1 receptor also activates the mitogen-activated protein kinase ERK1 and ERK2, leading to phosphorylation of tuberous sclerosis complex 2 and raptor (23, 24). Together, activation of the Akt and ERK signaling pathways results in a potent up-regulation of mTORC1 signaling (25). IGF1 enhanced the rate of protein synthesis in wild-type MEF by 76% (Fig. 3A) such that there was no difference in the rate of protein synthesis in wild-type and DKO MEF. When wild-type MEF were deprived of serum, a substantial amount of 4E-BP1 was observed to interact with eIF4E, whereas eIF4G could not be detected (Fig. 3B). Upon stimulation with IGF1, the interaction of 4E-BP1 with eIF4E was reduced, and the interaction with eIF4G was enhanced. Similar to the observations in the liver of fasted DKO mice, eIF4G interacted readily with eIF4E in serum-deprived DKO MEF, and no further enhancement was observed with IGF1 treatment (Fig. 3B), despite elevated eIF4G phosphorylation on Ser-1108 and reduced eIF4E phosphorylation on Ser-209 in both wild-type and DKO MEF (Fig. 3C). Thus, protein synthesis is repressed by serum deprivation in MEF lacking 4E-BP1 and 4E-BP2, even though the interaction of eIF4E with eIF4G is preserved. This finding suggests that the interaction of eIF4G with eIF4E is not by itself sufficient to maintain protein synthesis in serum-deprived MEF and that activation of another step in mRNA translation is required for full maintenance.
FIGURE 3.

IGF-1-induced protein synthesis is not dependent on changes in the interaction of eIF4E with eIF4G in 4E-BP1/2 DKO MEF. Wild-type and 4E-BP1/2 DKO MEF were exposed to serum-free medium (SFM) for 3 h to repress the rate of protein synthesis. Cells were treated with IGF1 for 15 min as indicated. A, the rate of protein synthesis was measured by the incorporation of [35S]methionine/cysteine into protein as described under “Experimental Procedures.” Values are means + S.E. (n = 4). Statistical significance is denoted by the presence of different letters above the bars on the graphs. Bars with different letters are statistically different, p < 0.05. DPM, decompositions per minute. B, the interaction of eIF4G and 4E-BP1 with eIF4E was evaluated by immunoprecipitating eIF4E and measuring the amount of each protein in the immunoprecipitate by Western blot analysis. C, phosphorylation of S6K1, eIF4B, PDCD4, eIF4G, eIF4E, and 4E-BP1 was evaluated in whole cell lysate by Western blot analysis using phosphospecific antibodies. Representative blots are shown, of which four were performed.
One potential explanation for the decreased rate of protein synthesis in serum-starved DKO MEF is repression of signaling pathways downstream of p70S6K1. In this regard, in serum-deprived wild-type and DKO MEF phosphorylation of p70S6K1 on Thr-389 was low (Fig. 3C), and IGF1 treatment robustly stimulated phosphorylation in both cell types. Moreover, in both wild-type and DKO MEF, phosphorylation of the p70S6K1 substrates eIF4B on Ser-422 and PDCD4 on Ser-67 was up-regulated in IGF1-treated compared with serum-deprived cells (Fig. 3C).
Activation of p70S6K1 Enhances Protein Synthesis in DKO MEF
To evaluate the role of p70S6K1 in stimulating protein synthesis, we transiently overexpressed constitutively active p70S6K1 (CA-S6K1) in wild-type and DKO MEF. In both wild-type and DKO MEF, overexpression of CA-S6K1 enhanced the phosphorylation of eIF4B on Ser-422 following serum-deprivation (Fig. 4A). This effect was particularly pronounced in serum-starved wild-type MEF, where CA-S6K1 enhanced eIF4B phosphorylation by 151% (Fig. 4B). Overexpression of CA-S6K1 enhanced phosphorylation of eIF4B by 80% in serum-deprived DKO MEF, such that it was no longer different from eIF4B phosphorylation observed in IGF1-treated wild-type MEF (Fig. 4B). Notably, CA-S6K1 did not enhance the level of eIF4B phosphorylation in either wild-type or DKO MEF treated with IGF1, suggesting that IGF1 might promote eIF4B phosphorylation through activation of p70S6K1. Overexpression of CA-S6K1 also reduced the expression of PDCD4 in both wild-type and DKO MEF (Fig. 4C). In response to IGF1 treatment, phosphorylation of PDCD4 on Ser-67 was enhanced (Figs. 3C and 4A), and there was a trend toward reduced expression in both wild-type (7%) and DKO (16%) MEF (Fig. 4C). Overexpression of CA-S6K1 also reduced PDCD4 expression in both wild-type and DKO MEF (Fig. 4C), with the effect being particularly dramatic in serum-deprived wild-type MEF (80%).
FIGURE 4.

Activation of p70S6K1 enhances the rate of protein synthesis in serum-deprived 4E-BP1/2 DKO MEF. Wild-type and 4E-BP1/2 DKO MEF were transfected with an HA-tagged constitutively active p70S6K1 (CA-S6K1) and exposed to serum-free medium (SFM) for 3 h to repress the rate of protein synthesis. Cells were treated with IGF1 for 15 min as indicated. A, phosphorylation of eIF4B and PDCD4 was evaluated in whole cell lysate by Western blot analysis using phosphospecific antibodies. Expression of eIF4B, PDCD4, HA-tagged CA-S6K1, GAPDH, and 4E-BP1 was assessed by Western blot analysis. Representative blots are shown. B, relative phosphorylation of eIF4B on Ser-422. C, expression of PDCD4. D, the rate of protein synthesis was measured by the incorporation of 35S-labeled methionine and cysteine into protein as described under “Experimental Procedures.” Values are means + S.E. (n = 3). Statistical significance is denoted by the presence of different letters above the bars on the graphs. Bars with different letters are statistically different, p < 0.05. DPM, decompositions per minute.
In serum-deprived wild-type MEF overexpressing CA-S6K1, the rate of protein synthesis was enhanced by 30% as compared with serum-deprived wild-type MEF transfected with an empty vector; however, overexpression of CA-S6K1 had no effect on the rate of protein synthesis following exposure to IGF1 (Fig. 4B). In serum-deprived DKO MEF, overexpression of CA-S6K1 produced a more dramatic enhancement in the rate of protein synthesis (58%) compared with wild-type cells; yet no further enhancement was achieved following exposure to IGF1. Notably, in DKO MEF overexpressing CA-S6K1, serum-deprivation failed to significantly suppress protein synthesis (Fig. 4D). Thus, constitutive activation of p70S6K1 in combination with loss of 4E-BPs was sufficient to maintain protein synthesis in the absence of serum.
Phosphorylation of PDCD4 and eIF4B Stimulates Protein Synthesis
To specifically evaluate the role of PDCD4 and eIF4B phosphorylation in the stimulation of protein synthesis, we treated DKO MEFs with siRNA directed against PDCD4 mRNA and/or transiently overexpressed constitutively active HA-tagged eIF4B (eIF4B S422D). In serum-deprived DKO MEF treated with siRNA directed against PDCD4, the expression of PDCD4 was reduced (Fig. 5A), and the rate of protein synthesis was stimulated by 34% as compared with serum-deprived DKO MEF transfected with an scrambled RNA (Fig. 5B). Similarly, overexpression of eIF4B S422D in serum-deprived DKO MEF stimulated the rate of protein synthesis by 24%. Notably, overexpression of eIF4B S422D and knockdown of PDCD4 simulated protein synthesis in serum-starved DKO MEF to an extent that it was no longer significantly different than values observed with IGF1 treatment (Fig. 5, compare lanes 4 and 7). Thus, phosphorylation of eIF4B and PDCD4 in combination with loss of 4E-BPs is sufficient to maintain protein synthesis in the absence of serum.
FIGURE 5.
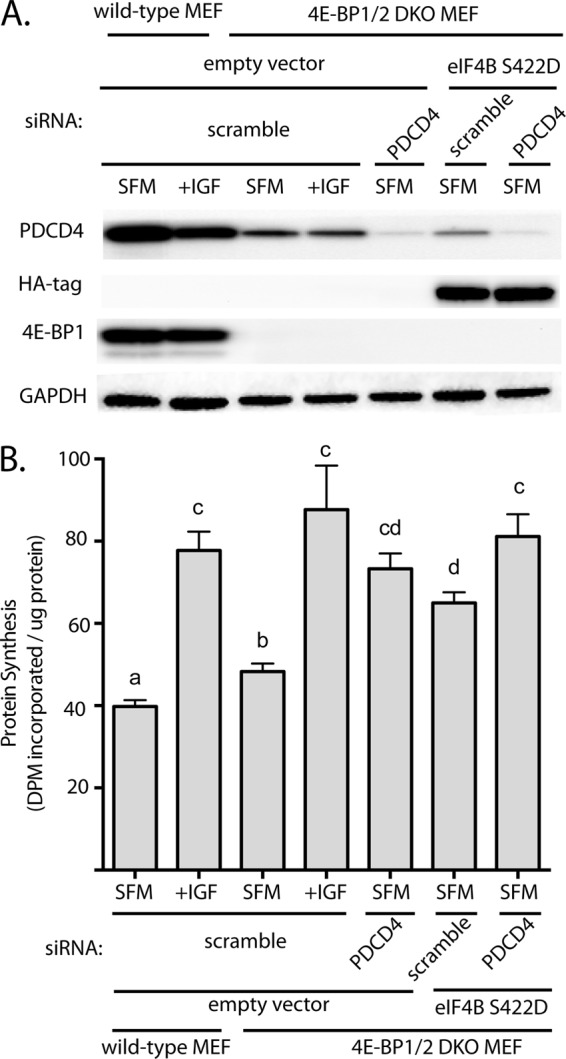
PDCD4 expression and eIF4B phosphorylation regulate the rate of protein synthesis in serum-deprived 4E-BP1/2 DKO MEF. Wild-type and 4E-BP1/2 DKO MEF were transfected with siRNA directed against PDCD4 and/or HA-tagged constitutively active eIF4B (eIF4B S422D) and exposed to serum-free medium (SFM) for 3 h to repress the rate of protein synthesis. Cells were treated with IGF1 for 15 min as indicated. A, expression of eIF4B S422D, PDCD4, 4E-BP1, and GAPDH was evaluated in whole cell lysate by Western blot analysis. Representative blots are shown. B, the rate of protein synthesis was measured by the incorporation of 35S-labeled methionine and cysteine into protein as described under “Experimental Procedures.” Values are means + S.E. (n = 6). Statistical significance is denoted by the presence of different letters above the bars on the graphs. Bars with different letters are statistically different, p < 0.05. DPM, decompositions per minute.
DISCUSSION
Various mechanisms have been proposed to control the interaction of eIF4E with eIF4G. For example, the MAP kinase signal integrating kinases (Mnk1 and Mnk2) bind to the C terminus of eIF4G and phosphorylate eIF4E on Ser-209, and phosphorylation of eIF4E on that residue correlates with enhanced interaction of the protein with capped mRNA and eIF4G (29). However, in the present study, the amount of eIF4G bound to eIF4E was greater in the liver of fasted DKO compared with wild-type mice, even though eIF4E phosphorylation was the same in both conditions. Moreover, eIF4G binding to eIF4E was enhanced in response to refeeding fasted wild-type but not DKO mice even though eIF4E became dephosphorylated in both wild-type and DKO mice after refeeding. Similarly, treating serum-deprived MEF with IGF1 resulted in dephosphorylation of eIF4E in both wild-type and DKO MEF, but eIF4G binding to eIF4E was enhanced only in wild-type cells. Assembly of the eIF4E·eIF4G complex also correlates with enhanced phosphorylation of eIF4G on Ser-1108 (e.g. 26). However, in the present study, eIF4G phosphorylation on Ser-1108 was low in the liver of fasted mice and in serum-deprived MEF, and in response to feeding or IGF1 treatment, respectively, eIF4G phosphorylation was enhanced to a similar extent. However, despite enhanced phosphorylation of eIF4G, the interaction of eIF4E with eIF4G was not elevated. Combined, these observations suggest that 4E-BP1 and 4E-BP2 are the dominant regulators of the interaction of eIF4G with eIF4E in mouse liver and MEF. This conclusion is also consistent with the observation that there is little 4E-BP3 expressed in liver (27).
Assembly of the eIF4E·eIF4G complex plays a central role in regulating cap-dependent mRNA translation. For example, in rabbit reticulocyte lysate, the small molecule inhibitor, 4EGI-1 causes a dose-dependent reduction in both the amount of eIF4G bound to eIF4E and cap-dependent mRNA translation (27). Similarly, in cardiomyocytes, overexpressing 4E-BP1 attenuates the binding of eIF4G to eIF4E in conjunction with a reduction in rates of global protein synthesis and cap-dependent mRNA translation (12). Interestingly, in both studies, a reduction in the interaction of eIF4E with eIF4G was associated with an enhancement of cap-independent mRNA translation. In the present study, we found that the eIF4E·eIF4G complex was maintained during serum deprivation in MEF lacking both 4E-BP1 and 4E-BP2 but not in wild-type MEF. Interestingly, although global rates of protein synthesis were elevated in serum-deprived DKO compared with wild-type MEF, the rate was significantly lower compared with either cell type after treatment with IGF1. Indeed, the rate of protein synthesis was the same in both wild-type and DKO MEF treated with IGF1. Combined, the results indicate that although assembly of the eIF4E·eIF4G complex is a required step in cap-dependent mRNA translation, it, by itself, is not sufficient to optimally stimulate protein synthesis.
The finding that protein synthesis was reduced in serum-deprived compared with IGF-1-treated DKO MEF despite maintenance of the eIF4E·eIF4G complex suggests that another step(s) in mRNA translation must be rate controlling under such conditions. One possible explanation for this observation is that activation of p70S6K1 and subsequent phosphorylation of its downstream targets is also required to optimally stimulate protein synthesis. Phosphorylation of p70S6K1 by mTORC1 requires the interaction of the N-terminal mTORC1-signaling motif of p70S6K1 with raptor. Phosphorylation of p70S6K1 by mTORC1 occurs on Thr-389, a residue located in a hydrophobic motif that is C-terminal to the kinase domain (17). Phosphorylation of p70S6K1 on Thr-389 creates a docking site for phosphoinositide-dependent kinase 1, which then phosphorylates p70S6K1 on Thr-229 of its activation loop rendering p70S6K1 fully activated (11). p70S6K1 then phosphorylates multiple proteins, including eIF4B, PDCD4, and eEF2 kinase that play important roles in regulating mRNA translation. Notably, phosphorylation of eEF2 kinase by p70S6K1 results in inhibition of eEF2 kinase activity, and because phosphorylation of eEF2 by eEF2 kinase leads to its inactivation, activation of p70S6K1 results in enhanced peptide chain elongation (28). In the present study, we observed no change in the phosphorylation of eEF2 by eEF2 kinase under conditions that activated p70S6K1. This finding supports previous sucrose density gradient analysis of polysome aggregation that has shown inhibition of mTORC1 with either rapamycin (29) or more potent inhibitors such as Torin1 (30) results in disaggregation of polysomes and accumulation of ribosomal subunits not associated with polysomes. Together, these findings suggest that mTORC1 signaling has a greater effect on translation initiation relative to elongation. Therefore, in the present study, we focused on eIF4B and PDCD4 rather than eEF2 kinase as targets of p70S6K1 action.
PDCD4 inhibits eIF4A function through at least two mechanisms. First, PDCD4 inhibits the RNA helicase activity of eIF4A, in part by blocking its RNA-binding domain (31). The inhibition occurs both with eIF4A alone and when it is part of the eIF4F complex. Second, PDCD4 blocks the binding of eIF4A to the C-terminal, but not the middle, eIF4A binding domain of eIF4G (10, 32), an event that is thought to interfere with formation of the active conformation of the eIF4F complex. Consequently, PDCD4 prevents eIF4A from unwinding secondary structure in the 5′-UTR of the mRNA and thereby attenuates scanning by the 40S ribosomal subunit to locate the AUG start codon. This conclusion is supported by the finding that PDCD4 inhibits cap-dependent, but not cap-independent, mRNA translation (10, 33). The RNA helicase activity of eIF4A is stimulated not only through its association with eIF4G, but also upon interaction with eIF4B (34, 35). In part, the stimulation occurs through an eIF4B-induced enhancement of the affinity of eIF4A for both RNA and ATP. Both the binding of PDCD4 to eIF4A and the interaction of eIF4B with eIF4A are regulated by p70S6K1. Thus, phosphorylation of PDCD4 by p70S6K1 results in its release from eIF4A, allowing the helicase to bind to both the middle and C-terminal domains in eIF4G (Fig. 6). In addition, phosphorylation of eIF4B by the kinase promotes its recruitment to the 48 S preinitiation complex which contains eIF4F bound to the 5′-end of the mRNA. Combined, the interaction of eIF4A with eIF4G and eIF4B dramatically enhances its RNA helicase activity and allows for dissolution of secondary structure in the 5′-UTR.
FIGURE 6.
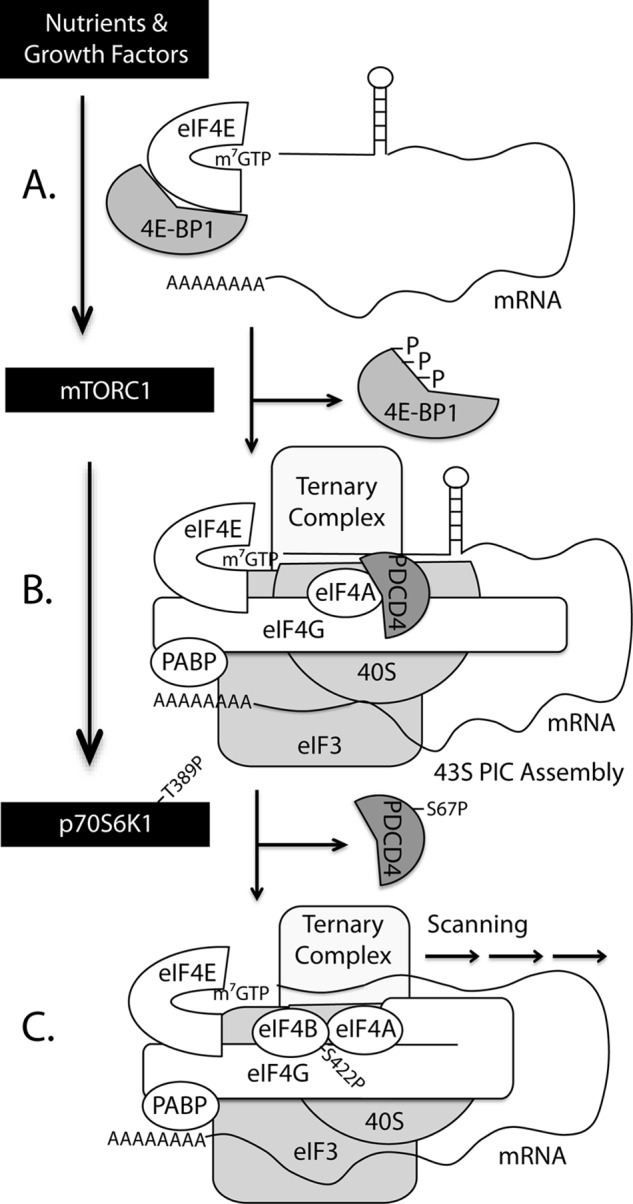
Model proposed for S6K1-mediated enhancement in protein synthesis. A, hypophosphorylated 4E-BP1/2 binds tightly to eIF4E, preventing its interaction with eIF4G and thus inhibiting translation. B, in response to nutrients or growth factors, mTORC1-mediated phosphorylation of 4E-BP1/2 promotes disassociation from eIF4E and allows recruitment of eIF4G to the mRNA 5′-cap, and assembly of the 43 S preinitiation complex. C, upon activation, mTORC1 also phosphorylates and promotes activation of p70S6K1, which in turn phosphorylates PDCD4 and eIF4B. Phosphorylation of PDCD4 on Ser-67 leads to its disassociation from eIF4A and subsequent ubiquitylation and degradation. In the absence of PDCD4, the C-terminal region of eIF4G interacts with eIF4A to promote RNA helicase activity. Phosphorylation of eIF4B on Ser-422 by p70S6K1 promotes its recruitment to the preinitiation complex and enhances the RNA helicase activity of eIF4A. Thus, p70S6K1 regulates protein synthesis by promoting the unwinding of mRNAs with long and structured 5′-untranslated regions to facilitate preinitiation complex scanning.
In the present study, exogenous expression of a constitutively active variant of p70S6K1 in 4E-BP1/2 DKO but not wild-type MEF prevented the reduction in protein synthesis caused by serum deprivation. Maintenance of protein synthesis was associated with elevated eIF4B phosphorylation and reduced PDCD4 expression. Notably, expression of CA-S6K1 in wild-type MEF slightly, but significantly enhanced protein synthesis in serum-deprived cells compared with cells transfected with an empty vector. Similarly, a small, but significant increase in protein synthesis was observed in serum-deprived DKO compared with wild-type MEF. Together, these observations suggest that assembly of the eIF4E·eIF4G complex and activation of p70S6K1 individually stimulate global rates of protein synthesis, but combined, they have a synergistic effect. Further support for this idea is provided by the observation that knockdown of PDCD4 or exogenous expression of an eIF4B phosphomimetic S422D variant individually increase protein synthesis, and in MEF lacking 4E-BP1 or 4E-BP2 combined repression of PDCD4 expression and expression of eIF4B (S422D) increased protein synthesis to the same extent as IGF1. In summary, the results of the present studies support a model in which optimal rates of mRNA translation require dissociation of eIF4E from 4E-BP1/2, assembly of an active eIF4F complex consisting of eIF4G, eIF4E, and eIF4A associated with both eIF4A binding domains of eIF4G, as well as phosphorylation of eIF4B (Fig. 6).
Acknowledgments
We thank Dr. Nahum Sonenberg for generously providing 4E-BP1/2 DKO mice and 4E-BP1/2 DKO MEF, Dr. John Blenis for providing plasmids containing constitutively active S6K1 and eIF4B, Lydia Kutzler for assistance with animals, and Holly Lacko for assistance with Western bloting.
This work was supported by National Institutes of Health Grants DK13499 (to L. S. J.) and DK088416 (to M. D. D.).
- mTOR
- mechanistic target of rapamycin
- 4E-BP
- eukaryotic initiation factor 4E binding proteins
- MEF
- mouse embryo fibroblast(s)
- DKO
- double knock-out
- eEF2
- eukaryotic elongation factor 2
- IGF
- insulin-like growth factor.
REFERENCES
- 1. Yoshizawa F., Kimball S. R., Vary T. C., Jefferson L. S. (1998) Effect of dietary protein on translation initiation in rat skeletal muscle and liver. Am. J. Physiol. 275, E814–820 [DOI] [PubMed] [Google Scholar]
- 2. Laplante M., Sabatini D. M. (2012) mTOR signaling in growth control and disease. Cell 149, 274–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jacinto E., Loewith R., Schmidt A., Lin S., Rüegg M. A., Hall A., Hall M. N. (2004) Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 6, 1122–1128 [DOI] [PubMed] [Google Scholar]
- 4. Pestova T. V., Kolupaeva V. G., Lomakin I. B., Pilipenko E. V., Shatsky I. N., Agol V. I., Hellen C. U. (2001) Molecular mechanisms of translation initiation in eukaryotes. Proc. Natl. Acad. Sci. U.S.A. 98, 7029–7036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thoreen C. C., Kang S. A., Chang J. W., Liu Q., Zhang J., Gao Y., Reichling L. J., Sim T., Sabatini D. M., Gray N. S. (2009) An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 284, 8023–8032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huo Y., Iadevaia V., Yao Z., Kelly I., Cosulich S., Guichard S., Foster L. J., Proud C. G. (2012) Stable isotope-labelling analysis of the impact of inhibition of the mammalian target of rapamycin on protein synthesis. Biochem. J. 444, 141–151 [DOI] [PubMed] [Google Scholar]
- 7. Pause A., Belsham G. J., Gingras A. C., Donzé O., Lin T. A., Lawrence J. C., Jr., Sonenberg N. (1994) Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature 371, 762–767 [DOI] [PubMed] [Google Scholar]
- 8. Waskiewicz A. J., Johnson J. C., Penn B., Mahalingam M., Kimball S. R., Cooper J. A. (1999) Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Mol. Cell Biol. 19, 1871–1880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Raught B., Gingras A. C., Gygi S. P., Imataka H., Morino S., Gradi A., Aebersold R., Sonenberg N. (2000) Serum-stimulated, rapamycin-sensitive phosphorylation sites in the eukaryotic translation initiation factor 4GI. EMBO J. 19, 434–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang H. S., Jansen A. P., Komar A. A., Zheng X., Merrick W. C., Costes S., Lockett S. J., Sonenberg N., Colburn N. H. (2003) The transformation suppressor Pdcd4 is a novel eukaryotic translation initiation factor 4A binding protein that inhibits translation. Mol. Cell Biol. 23, 26–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Holz M. K., Ballif B. A., Gygi S. P., Blenis J. (2005) mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 123, 569–580 [DOI] [PubMed] [Google Scholar]
- 12. Huang B. P., Wang Y., Wang X., Wang Z., Proud C. G. (2009) Blocking eukaryotic initiation factor 4F complex formation does not inhibit the mTORC1-dependent activation of protein synthesis in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 296, H505–514 [DOI] [PubMed] [Google Scholar]
- 13. Kimball S. R., Horetsky R. L., Jefferson L. S. (1998) Implication of eIF2B rather than eIF4E in the regulation of global protein synthesis by amino acids in L6 myoblasts. J. Biol. Chem. 273, 30945–30953 [DOI] [PubMed] [Google Scholar]
- 14. Kimball S. R., Jurasinski C. V., Lawrence J. C., Jr., Jefferson L. S. (1997) Insulin stimulates protein synthesis in skeletal muscle by enhancing the association of eIF-4E and eIF-4G. Am. J. Physiol. 272, C754–759 [DOI] [PubMed] [Google Scholar]
- 15. Kimball S. R., Siegfried B. A., Jefferson L. S. (2004) Glucagon represses signaling through the mammalian target of rapamycin in rat liver by activating AMP-activated protein kinase. J. Biol. Chem. 279, 54103–54109 [DOI] [PubMed] [Google Scholar]
- 16. Tuckow A. P., Jefferson S. J., Kimball S. R., Jefferson L. S. (2011) Simvastatin represses protein synthesis in the muscle-derived C(2)C(1)(2) cell line with a concomitant reduction in eukaryotic initiation factor 2B expression. Am. J. Physiol. Endocrinol. Metab. 300, E564–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schalm S. S., Blenis J. (2002) Identification of a conserved motif required for mTOR signaling. Curr. Biol. 12, 632–639 [DOI] [PubMed] [Google Scholar]
- 18. Raught B., Peiretti F., Gingras A. C., Livingstone M., Shahbazian D., Mayeur G. L., Polakiewicz R. D., Sonenberg N., Hershey J. W. (2004) Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO J. 23, 1761–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. von Manteuffel S. R., Dennis P. B., Pullen N., Gingras A. C., Sonenberg N., Thomas G. (1997) The insulin-induced signalling pathway leading to S6 and initiation factor 4E binding protein 1 phosphorylation bifurcates at a rapamycin-sensitive point immediately upstream of p70s6k. Mol. Cell Biol. 17, 5426–5436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dorrello N. V., Peschiaroli A., Guardavaccaro D., Colburn N. H., Sherman N. E., Pagano M. (2006) S6K1- and βTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 314, 467–471 [DOI] [PubMed] [Google Scholar]
- 21. Wang X., Li W., Williams M., Terada N., Alessi D. R., Proud C. G. (2001) Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 20, 4370–4379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Luo J., Field S. J., Lee J. Y., Engelman J. A., Cantley L. C. (2005) The p85 regulatory subunit of phosphoinositide 3-kinase down-regulates IRS-1 signaling via the formation of a sequestration complex. J. Cell Biol. 170, 455–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carriere A., Romeo Y., Acosta-Jaquez H. A., Moreau J., Bonneil E., Thibault P., Fingar D. C., Roux P. P. (2011) ERK1/2 phosphorylate Raptor to promote Ras-dependent activation of mTOR complex 1 (mTORC1). J. Biol. Chem. 286, 567–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miyazaki M., McCarthy J. J., Esser K. A. (2010) Insulin like growth factor-1-induced phosphorylation and altered distribution of tuberous sclerosis complex (TSC)1/TSC2 in C2C12 myotubes. FEBS J. 277, 2180–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Winter J. N., Jefferson L. S., Kimball S. R. (2011) ERK and Akt signaling pathways function through parallel mechanisms to promote mTORC1 signaling. Am. J. Physiol. Cell Physiol. 300, C1172–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bolster D. R., Vary T. C., Kimball S. R., Jefferson L. S. (2004) Leucine regulates translation initiation in rat skeletal muscle via enhanced eIF4G phosphorylation. J. Nutr. 134, 1704–1710 [DOI] [PubMed] [Google Scholar]
- 27. Poulin F., Gingras A. C., Olsen H., Chevalier S., Sonenberg N. (1998) 4E-BP3, a new member of the eukaryotic initiation factor 4E-binding protein family. J. Biol. Chem. 273, 14002–14007 [DOI] [PubMed] [Google Scholar]
- 28. Sans M. D., Xie Q., Williams J. A. (2004) Regulation of translation elongation and phosphorylation of eEF2 in rat pancreatic acini. Biochem. Biophys. Res. Commun. 319, 144–151 [DOI] [PubMed] [Google Scholar]
- 29. Olanich M. E., Moss B. L., Piwnica-Worms D., Townsend R. R., Weber J. D. (2011) Identification of FUSE-binding protein 1 as a regulatory mRNA-binding protein that represses nucleophosmin translation. Oncogene 30, 77–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thoreen C. C., Chantranupong L., Keys H. R., Wang T., Gray N. S., Sabatini D. M. (2012) A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485, 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chang J. H., Cho Y. H., Sohn S. Y., Choi J. M., Kim A., Kim Y. C., Jang S. K., Cho Y. (2009) Crystal structure of the eIF4A-PDCD4 complex. Proc. Natl. Acad. Sci. U.S.A. 106, 3148–3153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Suzuki C., Garces R. G., Edmonds K. A., Hiller S., Hyberts S. G., Marintchev A., Wagner G. (2008) PDCD4 inhibits translation initiation by binding to eIF4A using both its MA3 domains. Proc. Natl. Acad. Sci. U.S.A. 105, 3274–3279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang H. S., Cho M. H., Zakowicz H., Hegamyer G., Sonenberg N., Colburn N. H. (2004) A novel function of the MA-3 domains in transformation and translation suppressor Pdcd4 is essential for its binding to eukaryotic translation initiation factor 4A. Mol. Cell Biol. 24, 3894–3906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nielsen K. H., Behrens M. A., He Y., Oliveira C. L., Jensen L. S., Hoffmann S. V., Pedersen J. S., Andersen G. R. (2011) Synergistic activation of eIF4A by eIF4B and eIF4G. Nucleic Acids Res. 39, 2678–2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Özeş A. R., Feoktistova K., Avanzino B. C., Fraser C. S. (2011) Duplex unwinding and ATPase activities of the DEAD-box helicase eIF4A are coupled by eIF4G and eIF4B. J. Mol. Biol. 412, 674–687 [DOI] [PMC free article] [PubMed] [Google Scholar]