

Background: Deubiquitylases (DUBs) oppose the action of E3-ligases and influence key signalling pathways.
Results: USP15 stabilizes the E3 ligase BRAP/IMP, regulates CRAF expression, and is a positive regulator of MEK.
Conclusion: USP15 is a positive regulator of the MAPK pathway while stabilizing the E3 ligase BRAP/IMP.
Significance: Evidence is provided for novel modes of MAPK pathway regulation by DUBs.
Keywords: Deubiquitination, E3 Ubiquitin Ligase, MAP Kinases (MAPKs), Raf, Ubiquitin, BRAP, CRAF, DUB, IMP, USP15
Abstract
The opposing regulators of ubiquitylation status, E3 ligases and deubiquitylases, are often found to be associated in complexes. Here we report on a novel interaction between the E3 ligase BRAP (also referred to as IMP), a negative regulator of the MAPK scaffold protein KSR, and two closely related deubiquitylases, USP15 and USP4. We map the interaction to the N-terminal DUSP-UBL domain of USP15 and the coiled coil region of BRAP. USP15 as well as USP4 oppose the autoubiquitylation of BRAP, whereas BRAP promotes the ubiquitylation of USP15. Importantly, USP15 but not USP4 depletion destabilizes BRAP by promoting its proteasomal degradation, and BRAP-protein levels can be rescued by reintroducing catalytically active but not inactive mutant USP15. Unexpectedly, USP15 depletion results in a decrease in amplitude of MAPK signaling in response to EGF and PDGF. We provide evidence for a model in which the dominant effect of prolonged USP15 depletion upon signal amplitude is due to a decrease in CRAF levels while allowing for the possibility that USP15 may also function to dampen MAPK signaling through direct stabilization of a negative regulator, the E3 ligase BRAP.
Introduction
Ubiquitylation is a reversible post-translational modification that regulates the stability of substrate proteins, protein interactions, and enzymatic activity. The NFκB signaling cascade has provided a paradigm for how reversible ubiquitylation can contribute to signal transduction cascades (1). Much less is known about the impact of ubiquitin on the RAS-MAP6 kinase pathway, which regulates cell growth and differentiation (2). Ubiquitylation of H-RAS by Rabex-5 has been reported to promote its association with endosomes and impede activation of ERK1/2 (3, 4), whereas MEKK1 E3 ligase activity has been proposed to ubiquitylate ERK, leading to its degradation (5). The RAS effector, Impedes Mitogenic Propagation (IMP) (6), hereafter referred to by its Entrez gene symbol BRAP (BRCA1-associated protein), is a RING finger-type ubiquitin E3 ligase that undergoes auto-ubiquitylation. BRAP is proposed to regulate sustained MAPK signaling, at least in part through limiting KSR-1-dependent BRAF-CRAF-MEK complex formation (6–8). Binding of activated RAS leads to autoubiquitylation of BRAP, which relieves its suppression of MEK/ERK signaling (6).
One feature of E3 ligases is that they are often found in association with deubiquitylating enzymes (DUBs) (9–11), bringing to mind the juxtaposition of signal transduction and termination components that has been noted for certain kinases and phosphatases (12). In principal these DUB-E3 pairs can provide a means by which the stability of either partner can be controlled. Alternatively they may coordinately regulate the stability of a third partner as exemplified by the association of MDM2 with USP7 (HAUSP), both regulators of p53 (13). A most extreme case of E3-DUB coupling may be found in the NFκB pathway regulator A20, which has been reported to encode both E3 ligase and DUB activity within a single polypeptide chain (14, 15).
The human genome encodes around 79 DUBs predicted to be catalytically active (16, 17) and >300 members of the RING family of E3 ligases (18). We have undertaken a directed yeast two-hybrid screen combining 55 DUBs with a representative collection of 133 RING E3 ligases (19). Two closely related DUBs, USP4 and USP15, were identified as the sole interactors of BRAP. These proteins together with USP11 share a common domain structure (Fig. 1A) featuring an N-terminal combination of DUSP and UBL domains together with a further UBL domain inserted within the linker region of the catalytic domain (16, 20, 21). We have characterized the interplay between these proteins with respect to ubiquitylation status and show that BRAP protein levels are controlled by USP15 deubiquitylase activity. Furthermore we are able to demonstrate that USP15 also contributes to the regulation of sustained MAP kinase signaling after acute stimulation by modulating the expression level of the upstream kinase CRAF.
FIGURE 1.

Interactions between three closely related DUBs and a subset of RING E3 ligases. A, shown is a schematic representation of USP15, -4, and -11 domain structures. % indicates identity at amino acid level. B, shown is a summary of interactions between USP4, USP15, and USP11 (baits) and RING E3 ligases (preys) observed in a directed DUB:RING E3 ligase Y2H mating screen. Yeast diploids were scored after 5 days for growth using HIS3 and ADE2 reporters. The relative level of colony growth (+, ++, +++) is indicated by line thickness. Note that USP15 shares interactors with both USP4 and USP11. BRAP exclusively interacts with USP4 and USP15 among 55 DUBs tested in this screen. C, shown is a summary of the fragment-based mapping of the interaction between USP15 and BRAP described in Table 1.
EXPERIMENTAL PROCEDURES
DNA Constructs
USP and BRAP plasmids were generated using the GatewayTM system (Invitrogen). pDONR223 entry constructs were generated using PCR to amplify open reading frames with primer extensions containing the GatewayTM recombination sequences (“Gateway” primers) and subsequent use of PCR products in BP clonase reactions as per the manufacturer's instructions. A USP4 (NM_003363.3) template was kindly donated by Rohan Baker, Australia. USP11 (NM_004651), USP15 (NM_006313), and BRAP (NM_006768) ORFs were amplified from brain (USPs), and liver (BRAP) Marathon-Ready cDNA libraries (Clontech, Mountain View, CA) using a nested PCR-based protocol. All PCR reactions were performed with KOD HotStart DNA polymerase (Novagen, Nottingham) or PfuUltra HotStart DNA polymerase (Stratagene), and all pDONR223 construct inserts were verified by sequencing (Dundee Sequencing Services, Dundee, UK) and subcloned into Gateway compatible pEGFP-C2 (Clontech), pcDNA3-myc-C1, pGBDU-C1, and pACT2-C1 (Clontech) by incubation with LR clonase. Primer details and full details on sequences used in the Y2H screen are available on request. USP4, -15, -11, and BRAP fragments for interaction mapping were generated by PCR using pDONR223 constructs as templates. Fragment ORFs were fully sequenced before subcloning into Gateway compatible pGBDU-C1 and pACT2-C1 vectors. The following fragments are discussed in this study: full-length, USP4 (1–963), USP15 (1–952), USP11 (1–963); catalytic domain, USP4 (300–963), USP15 (223–952); DUSP-UBL USP4 (1–227), DUSP-UBL USP15 (1–223), DUSP-UBL USP11 (1–218), UBL USP4 (125–227), UBL-USP15 (123–223), UBL-USP11 (148–249); DUSP USP4 (1–125), DUSP USP15 (1–121); BRAP, full-length (1–592); BRAP ΔNT (141–592); BRAP coiled coil (393–592). Catalytically inactive mutants of USP4 and USP15, siRNA-resistant forms of USP15, and the putative E2 binding-deficient mutant of BRAP were generated using QuikChangeTM site-directed mutagenesis (Stratagene) using the following forward and respective complementary primers (mutated bases are followed by an asterisk, mutated codons are underlined): USP4 (C311S), 918cctgggaaacacca*gcttcatgaactccgc; USP15 (C269S), 788gtaacttgggaaatacga*gtttcatgaactcagc; USP15 (siRES), 1850gcatacatgaagaaggg*a*g*c*ccaagtgaaatgg; BRAP (W295A), 867ccagtgtctacagcgcgc*ggacgataccacgtgt.
Yeast Two-hybrid Assay
All bait (pGBDU) constructs were transformed into the PJ69–4A MATa strain, whereas prey constructs (pACT2) were expressed in the complementary mating type-switched strain PJ69–4A MATα. Targeted yeast two-hybrid matrix experiments were performed as described previously (22). Selective growth of diploid yeast was assessed on (−) His/plates containing 2.5 mm 3-aminotriazole (Sigma) and (−) Ade/plates. The screen was repeated twice, and only reproducible growth phenotypes were scored as positive. In each case, colony growth was recorded after 5 days of incubation at 30 °C.
Antibodies
Primary antibodies used were as follows: mouse monoclonal anti-myc (4A6) (Millipore), polyclonal affinity-purified sheep anti-GFP (gift from Ian Prior, Liverpool, UK), mouse anti-FLAG (M2) (Sigma), rabbit anti-USP4 (A300–830A) and rabbit anti-USP15 (A300–923A), rabbit anti-BRAP (A302–681A and A302–682A) (Bethyl Laboratories), rabbit anti-CRAF (C20) (Santa Cruz), and rabbit anti-BRAP (Fig. 5 only (23)). Rabbit anti-phospho Ser-217/Ser-221 MEK1/2 (9121 and 9154), rabbit anti-MEK1/2 (9122), mouse anti-phospho Thr-202/Tyr-204 ERK1/2 (9106), mouse anti-BRAF (9434), rabbit anti-CRAF (9422), and rabbit anti-ERK1/2 (9102) were from Cell Signaling Technologies (Danvers, MA), and mouse USP15 was from Abcam. Secondary IR680- and IR800-coupled donkey anti-mouse, anti-rabbit, and anti-sheep were from Rockland and Licor. Blots were analyzed using the Licor Odyssey system, and 16-bit images were analyzed using ImageJ or Odyssey analysis software (24).
FIGURE 5.

USP15 is a positive regulator of MAP kinase signaling. USP15, but not USP4 depletion inhibits MEK and MAPK activation. A, HeLa cells were treated >72 h with 40 nm siRNA oligos against BRAP, USP4, USP15, USP4, and USP15 and KSR1. Cells were starved overnight in serum-free medium and stimulated with EGF (2 ng/ml, 30 min). MEK and MAPK (ERK) activation was assessed with phospho-specific antibodies (pMEK, pERK). CTRL, control. B and C, three individual siRNA oligos (#1, #2, #17) were used to deplete USP15 in HeLa cells, which were processed as in A. Shown are the relative phospho-MEK to total MEK levels in cells stimulated for 30 min with EGF, averaged over two (USP4, USP15-17) or three (all others) independent experiments carried out in duplicate (error bars, S.E., two-tailed paired t test compared with control. B, USP15-1: p < 0.05, USP15-2 p < 0.05. C, USP15-1: p < 0.05, USP15-2: p < 0.01).
Cell Culture, Transfection, and RNA Interference Experiments
HeLa, U2OS and WM266-4 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 1% non-essential amino acids. Seeding densities per well of a 6-well plate for 72 h siRNA experiments were as follows: HeLa, 0.12 × 106; U2OS, 0.125 × 106; WM266-4, 0.12 × 106. For siRNA experiments, HeLa cells were treated for 72 h with either BRAP, USP4, or KSR1 ON-Target PLUS oligo pools (Dharmacon, Lafayette, CO), or USP15 siGENOME (#1, #2) and ON-Target PLUS oligos (#17) at 45 nm concentration using Oligofectamine (Invitrogen) in the absence of serum. Control samples were treated with Oligofectamine alone. WM266-4 cells were treated with siRNA at 45 nm for 72 h using Lipofectamine 2000. U2OS cells were treated for 72 h with siRNA at 20 nm using Lipofectamine RNAiMax (Invitrogen). Fetal bovine serum (10%) was added in each case 4 h post-transfection. For rescue experiments, HEK293T cells were first treated with siRNA and the following day transfected with either GFP-USP15siRES, GFP-USP15-C269S-siRES, or myc-CRAF for another 48 h.
Growth Factor Stimulation and Lysis of Cells
Cells were serum-starved for 12–16 h and stimulated with EGF (1–2 ng/ml, HeLa) or platelet-derived growth factor (PDGF; 10 ng/μl, U2OS), washed with ice-cold PBS, and incubated for 10 min on ice in Nonidet P-40 lysis buffer (0.5% Nonidet P-40, 25 mm Tris/HCl, pH 7.5, 100 mm NaCl, 50 mm NaF) or RIPA lysis buffer (10 mm Tris-HCl pH7.5, 150 mm NaCl, 1% w/v Triton X-100 or Nonidet P-40, 0.1% w/v SDS, 1% sodium deoxycholate) supplemented with mammalian protease inhibitors and phosphatase inhibitor mixture II (Sigma) or PhosSTOP tablets (Roche Applied Science). For Fig. 4, C and D and 8, B and C, 10 mm N-ethylmaleimide was added to the lysis buffer. Lysates were precleared by centrifugation at 21,000 × g, protein concentrations were determined using a BCA protein assay (Pierce), and equal amounts of protein were analyzed by SDS-PAGE and Western blotting.
FIGURE 4.

siRNA-mediated depletion of USP15, but not USP4, destabilizes endogenous BRAP by promoting its ubiquitylation and proteasomal degradation. A, HeLa cells were treated with siRNA as indicated for 72 h before lysis. Lysates were probed with anti-USP15, BRAP, and actin antibodies. Quantitation shows the mean of three biological replicates (error bars. S.E.; two-tailed paired t test compared with control; BRAP, p < 0.001; USP15-1, p < 0.005; USP15-17, p < 0.025). IB, immunoblot. B, HEK293T cells were either stimulated or not for 30 min with EGF. Lysates were subjected to immunoprecipitation (IP) with anti-BRAP and protein A-agarose beads or control beads, and samples were probed with mouse anti-USP15 and rabbit anti-BRAP antibodies. C, BRAP was immunoprecipitated from HeLa cells treated with either control reagents or USP15 siRNA (72 h) and lysed in RIPA buffer including 10 mm N-ethylmaleimide. Cell lysates and immunoprecipitation samples were probed with anti-ubiquitin, BRAP, USP15 antibodies (Ab). USP15 depletion promotes accumulation of a high molecular weight ubiquitylated species (*) of BRAP. D, proteasome inhibition rescues BRAP in USP15 depleted cells. HeLa cells were treated as in B for 66 h before incubation with 0.5 μm epoxomicin or DMSO as a control for a further 8 h. Cells were lysed as in B, and lysates were probed with anti-BRAP. E, HeLa cells were treated with control reagents or USP15 siRNA (#17) for 66 h before cycloheximide (CHX, 100 μg/ml) treatment. Cells were lysed at indicated time points, and lysates were probed as shown. * indicates high molecular weight BRAP species indicative of ubiquitylation. (n = 3, error bars: S.E.). F, HEK293T cells were treated with control reagents or USP15 siRNA (#1) for 24 h before transfection with GFP or siRNA resistant forms of GFP-USP15 and GFP-USP15-C269S (CS) and further incubation for 48 h. Lysates were probed with anti-BRAP and USP15 (light and darker exposures).
Immunoprecipitation
Precleared lysates were incubated with anti-GFP, anti-BRAP, or anti-CRAF and protein A- or G-agarose for 4 h at 4 °C. Beads were washed 3 times with YP-IP buffer (0.1% Nonidet P-40, 25 mm Tris/HCl, pH 7.5, 150 mm NaCl) and once with 10 mm Tris/HCl, pH 7.5, and resuspended in 1.5× SDS-PAGE sample buffer. Proteins were resolved on 4–12% NuPAGE gels (Invitrogen) and analyzed by Western blotting.
For endogenous co-immunoprecipitation experiments, HEK293T cells were treated for 30 min with 20 ng/ml EGF or left untreated. Cells were lysed with Triton X-100 lysis buffer (0.3% Triton X-100, 25 mm Tris, pH 7.5, 100 mm NaCl, 50 mm NaF, supplemented with protease and phosphatase inhibitors), lysates were cleared by centrifugation, and 2 mgs of lysates were incubated for 2 h with protein A-agarose and anti-BRAP antibody (Bethyl). Beads were washed, and samples were processed as indicated above.
Epoxomicin and Cycloheximide Treatment
HeLa cells were treated with control reagents or siRNA for 66 h before incubation with 0.5 μm epoxomycin (for 8 h, DMSO treatment as control) or 100 μg/ml cycloheximide (for 2–10 h). Cells were lysed with RIPA (epoxomicin) or Nonidet P-40 (cycloheximide) lysis buffer, and protein concentration was measured with the BCA assay kit. Equal amounts of lysates were subjected to Western blot analysis.
RNA Extraction and Real-time PCR
Cultured HeLa cells were treated with control reagent (Oligofectamine) or one of three oligos targeting USP15 (#1, #2, and #17) at 45 nm for 24 h. Cells were harvested, and total RNA was extracted using RNeasy columns (Qiagen). cDNA was reverse-transcribed from 1 μg of RNA with RevertAid H-minus M-MuLV reverse transcriptase (Fermentas) using either an oligo(dT) primer (Promega, Fig. 9A) or random hexanucleotide primers (Fig. 9C). Quantitative real-time RT-PCR was performed in triplicate using SYBR Green Supermix and a real-time PCR detection system (Bio-Rad). Primer sequences used were as follows to amplify mRNA (actin (forward 5′-GATCATTGCTCCTCCTGAGC, reverse 5′-CGTCATACTCCTGCTTGCTG); USP15 (forward 5′-CAGACAGCACCATTCAGGATGC, reverse 5′-AAAATTGGATGCACCTGGGGAC); CRAF (forward 5′-CATCAGACAACTCTTATTGTTTCC, reverse 5′-TGTGCTGAGAACTAACAGGCA); CRAFe/e, (forward 5′- CGAATCAGCCTCACCTTCAGC, reverse 5′-CTGTCCACGAGGCCTAATTTTGT)) or to amplify pre-mRNA (CRAF e/i (forward 5′-AACAATCTGAGCCCAACAGG, reverse 5′-TTACTGAACCCTAATTGGCAG)). Samples underwent 2-step amplification, and melt curves were analyzed after 40 cycles. For assessment of pre-mRNA, a DNA removal step was included in the protocol, and primers were validated against RT− reactions. The Ct values for test genes were normalized to beta-Actin and relative expression represented as 2−[ΔΔCt].
FIGURE 9.
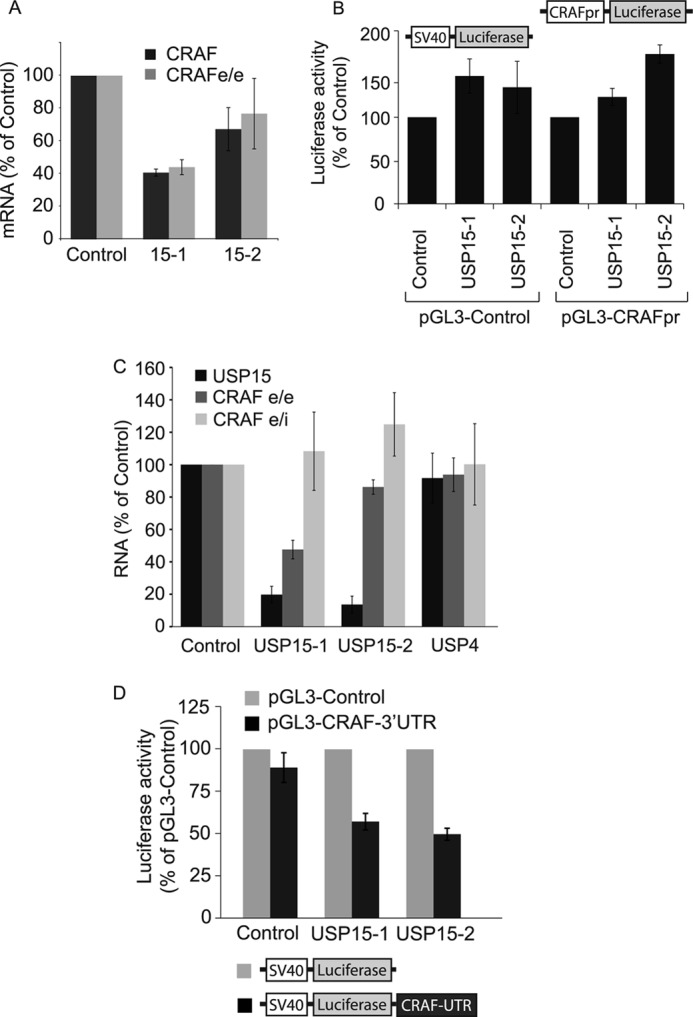
USP15 regulates CRAF at a post-transcriptional level. A, CRAF mRNA from HeLa cells treated for 24 h with two individual oligos targeting USP15 was assessed by RT-PCR using two primer sets (CRAF and CRAFe/e) designed to amplify different regions of the CRAF transcript. Values from four biological replicates were normalized to actin mRNA (error bars, S.D. two-tailed t test compared with control; CRAF-USP15-1, p < 0.00025; USP15-2, p < 0.05; CRAFe/e-USP15-1, p < 0.0005). B, USP15 depletion does not decrease the transcriptional activity of a minimal CRAF promoter. HeLa cells were cotransfected with two individual siRNAs targeting USP15 (USP15-1, USP15-2) or a control siRNA (Control) together with a plasmid expressing firefly luciferase under the control of a SV40 promoter (pGL3-Control) or a minimal CRAF promoter (pGL3-CRAFpr) and the Renilla luciferase reporter construct, phRL-tk. Cells were lysed 48 h post-transfection and analyzed using the Promega dual-luciferase reporter assay system. Results from six biological replicates are shown normalized to phRL-tk reporter activity (n = 6, error bars, S.D.). C, USP15 depletion does not markedly alter the amount of CRAF pre-mRNA. Randomly primed cDNA, derived from RNA extracted from HeLa cells treated for 24 h with control reagent or two individual siRNA oligos targeting USP15 (USP15-1, USP15-2), or USP4, was analyzed by quantitative real-time RT-PCR. Primers were designed to anneal to exon junction sequences (USP15, CRAF e/e) or exon-intron junctions (CRAF e/i) to assess mature and pre-mRNA, respectively. Values from three biological replicates were normalized to actin mRNA. Two-tailed t test; USP15-USP15-1, p < 0.005; USP15-2, p < 0.025; CRAFe/e-USP15-1, p < 0.01; USP15-2, p < 0.05. D, HeLa cells were treated for 48 h with individual siRNAs targeting USP15 (USP15-1, USP15-2) or a control siRNA (Control) before transfection for another 24 h with plasmids expressing firefly luciferase under the control of a minimal SV40 promoter with or without the appendage of the 3′-UTR of CRAF. Cells were cotransfected with phRL-tk. Comparison of the relative luciferase activity from the basic and pGL3-CRAF-3′-UTR plasmids suggests that USP15 depletion has a destabilizing effect on the 3′-UTR-harboring transcript. Data are of three biological replicates. Error bars, S.D., paired two-tailed t test for pGL3-CRAF-UTR compared with pGL3-Control, USP15-1 and USP15-2, p < 0.0001.
Dual Luciferase Reporter Assays
The minimal CRAF promoter firefly luciferase reporter construct (pGL3-humanRaf1PR; pGL3-CRAFpr in Fig. 9B) encompassing the first non-coding exon and about 750 bp of promoter upstream cloned into pGL3basic was a kind gift from Siobhan Corbett, New Brunswick, NJ (25). The CRAF 3′-UTR was PCR-amplified using HeLa cDNA as template and the primers forward (5′-cgtctagaTTGACTTTGCACCTGTCTTCAGG-3′) and reverse (5′-cgtctagaACATAATTGAGGGACCATCAGATAAC-3′) and subsequently cloned into the XbaI site downstream of the firefly luciferase expression cassette of the pGL3-control vector(Promega), and the plasmid was sequence verified. Renilla-luciferase control plasmid (phRL-tk, Promega) was used to standardize transfection efficiency. Luciferase activity was measured at 24 or 48 h post-transfection using the Dual-luciferase Reporter Assay System as described by the manufacturer (Promega).
RESULTS
Yeast Two-hybrid Screen Identifies USP4 and USP15 as BRAP Binding Partners
From our collection of 66 sequence-verified open reading frames for DUBs, which were inserted into yeast two-hybrid expression vectors, 55 showed no evidence of self-activation properties. These were taken forward into a directed screen against a collection of 133 RING proteins that were previously been used in a screen for ubiquitin E2-conjugating enzyme interactions (19). Matrices were constructed for three different reporter assays of varying stringency and only those interactions seen in two independent screens were scored as positive.
A total of 47 RING E3s displayed 163 (primarily weak) interactions shared between 27 DUBs, of which 159 have not been reported in protein interaction databases. Interaction of the RING E3 ligase BRAP was restricted to the two closely related DUBs (57% sequence identity) USP4 and USP15 (Fig. 1, A and B). USP4 interactions were limited to BRAP and TRIM21 (shared with USP15 and the highly promiscuous USP2). USP15 displayed a total of 10 binding partners, most of which are RING proteins with promiscuous DUB interaction profiles, with the exceptions of TRIM17 (unique to USP15) and PHF7 (shared with USP11). Among this set of interactions, we chose to follow up those involving BRAP based on its highly selective interaction profile and reported relevance to the MAPK signaling pathway (8).
We next sought to identify the regions within the respective proteins that are necessary for this interaction by directed yeast two-hybrid assays using truncated forms of each protein. We also assayed USP11 in parallel, as the most closely related member of the USP family that did not interact with BRAP in the original screen (Fig. 1A and B). In this independent analysis with retransformed yeast, only USP15 displayed an interaction with full-length BRAP (Table 1). However, truncations incorporating only the DUSP and UBL domains of all three USPs interact with an N-terminal-deleted form of BRAP (141–592), and in the case of USP4 and USP15, this can be further reduced to interaction with the C-terminal region (393–592) that is predicted to form a coiled-coil structure. Neither the DUSP domain nor the adjacent UBL domain alone can recapitulate this interaction (Table 1). In summary, USP4 and USP15 interact with the coiled-coil domain of BRAP through their N-terminal regions, requiring a combination of DUSP and UBL domains (Fig. 1C).
TABLE 1.
The USP15 and USP4 DUSP-UBL mediates binding to the BRAP C-terminal coiled coil region
Fragments of USP4, USP15, USP11, and BRAP were inserted into bait (USPs) and prey (BRAP) Y2H vectors and tested for interaction by directed Y2H. +, interaction seen with HIS3 reporter only; ++, interaction shown with HIS3 and ADE2 reporters. ND, not determined. Cat. domain, catalytic domain; ΔNT, N-terminal deletion mutant; CC, coiled coil.
| BAIT (USPs) | PREY (BRAP) |
||
|---|---|---|---|
| Full-length | ΔNT (141–592) | CC (393–592) | |
| Full-length USP15 | + | ND | ND |
| USP4 | − | ND | ND |
| USP11 | − | ND | ND |
| DUSP-UBL USP15 | − | ++ | + |
| USP4 | − | ++ | + |
| USP11 | − | ++ | − |
| UBL USP15 | − | − | − |
| USP4 | − | − | − |
| USP11 | − | − | − |
| DUSP USP15 | − | − | − |
| USP4 | − | − | − |
| Cat. domain USP15 | − | − | − |
| USP4 | − | − | − |
Interplay between USPs and BRAP within a Cellular Environment
We turned to expression of epitope-tagged proteins to confirm the interactions of USP4 and USP15 with BRAP in the context of a mammalian cell system. GFP-tagged USPs or GFP-BRAP were co-expressed in HEK293T cells together with myc-tagged BRAP. After immunoprecipitation with GFP antibodies and probing with myc antibodies, GFP-BRAP can be found associated with myc-BRAP, consistent with its oligomerization (Fig. 2). Myc-BRAP also clearly co-immunoprecipitates with GFP-USP4 and GFP-USP15, whereas interaction with USP11 is significantly weaker. Higher molecular weight forms of myc-BRAP are evident both in some cell lysates and immunoprecipitates (see Fig. 2, cell lysates lower panel). Co-expression of GFP-BRAP enhances these higher molecular weight forms of myc-BRAP, consistent with trans-ubiquitylation within BRAP oligomers. Higher molecular weight forms of myc-BRAP are also promoted by co-expression of catalytically inactive mutants of USP4 or USP15 but are abolished by expression of wild-type versions of either enzyme but not USP11 (Fig. 2).
FIGURE 2.

BRAP interacts with both catalytically active and inactive USP4 and 15 but not USP11 in a cellular environment. HEK293T cells coexpressing myc-tagged BRAP and GFP-tagged USP4, -11, -15, GFP-BRAP- or GFP were lysed in RIPA buffer and subjected to immunoprecipitation (IP) with anti-GFP antibodies and probed with anti-myc and anti-GFP. Bound proteins were analyzed alongside 5% of input sample. IB, immunoblot. Arrows indicate higher molecular weight forms of BRAP. The bottom panel shows a higher intensity representation of input lanes. U, USP; CS, catalytically inactive mutant.
To test whether these higher molecular weight forms of myc-BRAP are ubiquitylated, we co-expressed FLAG-ubiquitin together with GFP-USPs and myc-BRAP as described above. We also expressed a mutant version of BRAP that is unable to recruit a cognate E2 enzyme due to a Trp to Ala mutation in its RING finger and, therefore, fails to autoubiquitylate (26, 27). As expected, the FLAG antibody detects bands coinciding with the higher molecular weight forms of myc-BRAP induced by the expression of catalytically inactive USP4 and 15, which are absent in the case of mutant myc-BRAP-WA (Fig. 3A, compare lanes 2 and 4 with 9 and 11). Importantly these bands are also abolished by co-expressing catalytically active USP4 or USP15, suggesting that both USPs are capable of deubiquitylating BRAP in this heterologous expression system (Fig. 3A, lanes 1 and 3, pink arrows).
FIGURE 3.

Co-expression with catalytically inactive DUBs promotes the accumulation of ubiquitylated BRAP. HEK293T cells were transfected with FLAG-ubiquitin, GFP, or GFP-tagged active or inactive (CS) mutants of USP4 and -15 together with empty vector (vector) or myc-tagged BRAP or a mutant BRAP that is unable to recruit a ubiquitin conjugating E2 (WA). Cells were processed as described in Fig. 2, and immunoprecipitates (IP) were probed with anti-FLAG and anti-myc antibodies (A) or anti-GFP antibodies (B) to assess the ubiquitylation status of both BRAP (A) and USP4 and 15 (B). A, pink arrows indicate higher molecular weight bands reactive to anti-myc (BRAP) and anti-FLAG (Ub) antibodies in cells co-expressing wild-type BRAP (wtBRAP) and catalytically inactive USP4 (U4 CS) and USP15 (U15 CS). B, catalytically inactive USP15 and to a lesser extent also USP4 present with higher molecular weight bands that can be detected with FLAG-ubiquitin (blue arrows; see also Fig. 3). U4, USP4; U15, USP15; CS, catalytically inactive mutants; WA, E2-binding deficient mutant. Lane 7, Mr marker. IB, immunoblot.
Intriguingly, GFP-USP15 and USP4 catalytically inactive mutants also display some prominent higher molecular weight species (Fig. 2) that coincide with FLAG-ubiquitin positive bands in Fig. 3 (Fig. 3B, blue arrows). Ubiquitylation of USP15 is clearly increased by co-expression of wild-type but not an E2 binding-deficient mutant of myc-BRAP (Fig. 3B, compare lanes 5 with 4 and 12 with 11). In contrast, ubiquitylation of GFP-USP4 is not differentially affected by myc-BRAP or its mutant counterpart (Fig. 3B, compare lanes 2 and 9). In summary, our data show that USP4 and -15 can both interact with BRAP and thereby attenuate its autoubiquitylation. In turn, BRAP can ubiquitylate USP15, which is opposed by its intrinsic catalytic activity.
USP15 but Not USP4 Depletion in Cells Destabilizes BRAP
We next examined the effects of siRNA mediated knock-down of endogenous proteins (USP4, USP15, and BRAP). Only USP15, but not USP4 depletion, results in a significant reduction in BRAP levels (Fig. 4A). This reinforces the notion that although both USP4 and USP15 can interact with BRAP under certain experimental conditions, the USP15:BRAP rather than USP4:BRAP interaction may be of most relevance within a physiological setting. Indeed, we were able to detect a constitutive interaction between endogenous USP15 and BRAP in (both stimulated and unstimulated) HEK293T cells (Fig. 4B). As seen with overexpressed BRAP in Figs. 2 and 3, endogenous BRAP presents as a ladder of higher molecular weight ubiquitylated forms that can be most easily detected in BRAP immunoprecipitates (Fig. 4C). Depletion of USP15 with two independent siRNA oligos does not interfere with this basic ubiquitylation ladder but promotes the appearance of an additional higher molecular weight ubiquitin smear above the 116-kDa marker that is indicative of the accumulation of distinct polyubiquitylated species of BRAP in the absence of USP15. This led us to test the idea that this hyperubiquitylated BRAP may be turned over by the proteasome. Application of the proteasome inhibitor epoxomicin is able to rescue BRAP protein levels (Fig. 4D), and the turnover of BRAP levels, observed upon inhibition of translation with cycloheximide, is significantly accelerated in USP15 depleted cells within the first two h of chase time (Fig. 4E). This restriction of the effect on turnover to the earliest time points can be explained by two models. (i) There are multiple pools of BRAP with differing stability, and only one pool is sensitive to USP15 depletion, which would be consistent with incomplete loss of BRAP observed in both Fig. 4, A and D. (ii) BRAP turnover requires the activity of a further factor that is itself turned over on a faster time scale and becomes depleted during the cycloheximide incubation period.
Importantly, we were able to rescue BRAP expression levels by co-expressing wild-type but not catalytically inactive siRNA resistant GFP-USP15 in USP15-depleted cells (Fig. 4F). Taken together, these data strongly suggest that USP15 directly deubiquitylates BRAP and thereby promotes its stability in mammalian cells.
USP15 Is a Positive Regulator of MAPK Signaling
After acute stimulation with EGF, HeLa cells show an increased level of phospho-MEK and phospho-ERK, canonical components of the RAS-activated MAPK pathway. Using pools of four oligos specific to an individual target, we have examined the requirement of BRAP, KSR-1, USP4, and USP15 for pMEK and pERK activity in response to EGF stimulation. First of all we titrated the EGF concentration to determine the dose necessary to elicit a degree of MEK phosphorylation within the dynamic component of the dose response curve. This range of EGF-doses (1–2 ng/ml), which we then used in all experiments, is lower than that used in many published experiments, which could render them insensitive to either inhibitory and stimulatory effects.
The specific activity of pMEK was not affected by BRAP or USP4 knockdown (Fig. 5, A and B). However, we observed a clear reduction after USP15 knockdown, which is mirrored by reduced pERK levels, upon which other oligos had no effect (Fig. 5A). To confirm this effect of USP15 depletion on the MAPK signaling cascade, we carried out further experiments with three individual oligos directed against USP15 that show a significant reduction in pMEK and pERK (Fig. 5, B and C).
The small effects of BRAP depletion on pMEK and pERK we observed upon stimulating HeLa cells with a 30-min pulse of EGF contrasted with the clear stimulatory effect reported by others, albeit at earlier time points (6). We, therefore, first assessed the impact of BRAP and USP15 depletion on MEK and ERK activation induced by a shorter stimulus (5 min EGF) (Fig. 6A). This experimental setup equally failed to reveal a significant effect of BRAP depletion while reconfirming our initial observation that USP15 knockdown inhibits MEK phosphorylation. Finally, we conducted a parallel set of experiments in cells that had not been serum-starved, conditions that are more comparable to those previously used by others (28). Under these conditions, BRAP depletion stimulates MEK phosphorylation significantly, whereas USP15 depletion has a reduced inhibitory effect on pMEK (Fig. 6B). In summary, despite markedly reducing BRAP protein levels, USP15 depletion does not promote but rather inhibits MEK activation in response to EGF.
FIGURE 6.

siRNA depletion of BRAP, but not USP15, stimulates MEK phosphorylation in non-starved cells stimulated with a short pulse of EGF. A, HeLa cells were treated for 72 h with siRNA targeting KSR1, BRAP, USP4, or USP15 (oligo #17) and serum-starved for the last 16 h before stimulation with EGF (1 ng/ml) for 5 min. Knockdown efficiency as well as MEK and ERK phosphorylation were assessed by Western blotting (IB). B, cells were treated as in A but were not serum-starved. Graphs show results from four biological replicates (error bars, S.E., two-tailed paired t test compared with control; BRAP, p < 0.05; USP15: p < 0.01).
We wondered whether this positive regulatory role of USP15 was hard-wired into the canonical RAS-MAPK pathway and independent of the growth factor used to activate the cascade. We turned to assess USP15 depletion in the osteosarcoma U2OS cell line, which responds to PDGF. We found that USP15 depletion again significantly dampens PDGF-induced MEK phosphorylation while only marginally affecting BRAP levels in these cells (Fig. 7).
FIGURE 7.
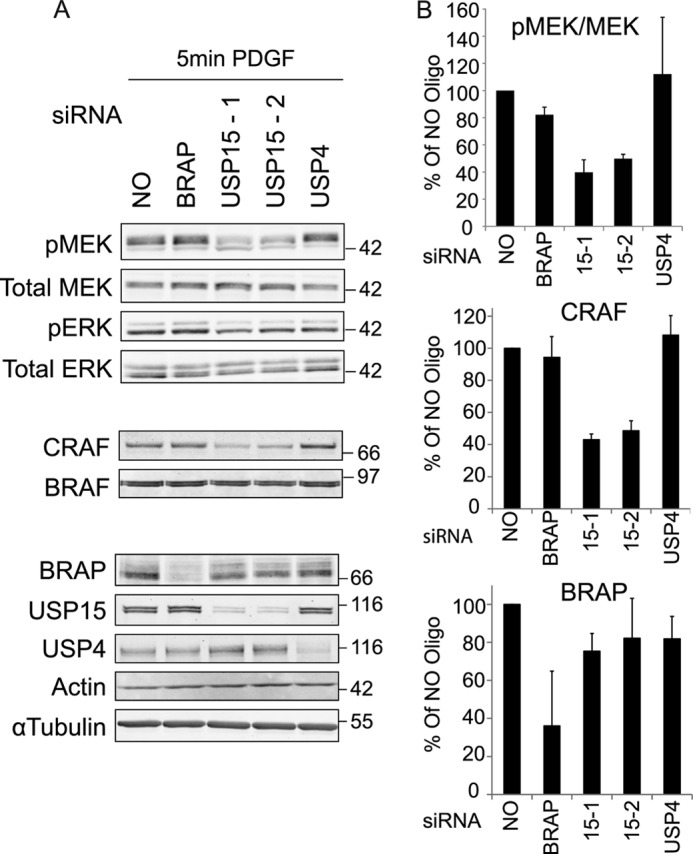
USP15 depletion in U2OS cells decreases CRAF expression levels and inhibits PDGF dependent pMEK activation. A, U2OS cells were treated >72 h with 20 nm of siRNA oligos targeting BRAP, USP4, and USP15 (two individual oligos). Cells were serum-starved for 16 h and then stimulated with 10 ng/ml PDGF for 5 min. RIPA lysates were probed with the indicated antibodies to assess knockdown efficiencies and expression levels or activation status of the key kinases of the MAPK cascade (CRAF, BRAF, MEK, and ERK). B, the ratio of pMEK/MEK, CRAF, and BRAP levels at 5 min post stimulation with PDGF, averaged from three independent experiments, are shown relative to control samples (error bars, S.D.; two-tailed paired t test compared with control; pMEK-USP15-1, p < 0.05; USP15-2, p < 0.05; CRAF-USP15-1, p < 0.01; USP15-2: p < 0.05).
USP15 Controls CRAF Levels
What then is the relevant BRAP-independent target of USP15? Analyzing the key upstream kinases of the cascade, we found that CRAF, but not BRAF expression levels, are strongly reduced in USP15-depleted U2OS as well as in the HeLa cells (Figs. 7 and 8A). In contrast to our observations on BRAP, CRAF levels cannot be rescued by the addition of proteasome inhibitors. Likewise, USP15 depletion does not alter the ubiquitylation pattern associated with CRAF immunoprecipitates or enhance the turnover of CRAF as assessed by cycloheximide chase experiments (Fig. 8, B–D). Myc-CRAF, heterologously expressed under the control of a CMV promoter, is not affected by USP15 knockdown, further arguing against a post-translational regulatory role of USP15 on CRAF levels (Fig. 8E).
FIGURE 8.

USP15 regulates CRAF expression levels. A, samples shown in Fig. 4A were probed with the indicated antibodies. Quantitation shows the mean of three biological replicates (error bars, S.E.; two-tailed paired t test compared with control; USP15-1, p < 0.05; USP15-17, p < 0.025). IB, immunoblot. B, CRAF was immunoprecipitated (IP) from lysates shown in Fig. 4B and probed with anti-ubiquitin antibodies (Ab). C, samples from epomoxicin (Epo) and vehicle-treated cells (Fig. 4C) were probed with anti-CRAF antibodies. D, HeLa cells were treated with control reagents or USP15 siRNA (Oligo #17) for 66 h before cycloheximide (CHX; 100 μg/ml) treatment. Cells were lysed at indicated time points in Nonidet P-40 lysis buffer, and lysates were probed with anti-CRAF antibody. E, HEK293T cells were treated with control reagents or USP15 siRNA (Oligo#1) for 24 h before transfection with myc-CRAF or an empty vector for another 48 h. Cell lysates were probed with anti-USP15 and anti-CRAF antibodies. Endogenous CRAF levels are only visible in the darker exposure.
RT-PCR analysis of mRNA levels showed that USP15 depletion with two independent oligos causes a reduction of CRAF transcript levels (Fig. 9A). We wondered whether this may be caused by a decrease in transcriptional activity at the CRAF promoter. We used a previously described CRAF promoter-driven luciferase reporter construct (25) to address this question. Our results argue against this interpretation; we do not see a decrease in CRAF promoter-driven transcription (Fig. 9B). USP15 shares with USP4 several binding partners linked to mRNA-processing and USP4 has previously been shown to regulate components of the U4/U6-spliceosome (9, 29). We, therefore, considered altered splicing efficiency as one potential mechanism that might yield reduced transcript levels in the absence of a reduction in transcriptional activity. We first verified our quantitative real-time RT-PCR results with a second CRAF primer set designed to anneal to exon boundaries that are predicted to be included in all four major annotated isoforms (CRAFe/e, Fig. 9, A and C). Secondly, we also designed primer sets to exon-intron boundaries aimed at amplifying unspliced pre-mRNA (CRAFe/i). Although we see some trend toward increased pre-mRNA levels in USP15-depleted cells, these results are not statistically significant (Fig. 9C). Finally, we asked the question of whether USP15 depletion may alter CRAF mRNA stability via its 3′-UTR. To this end we cloned the CRAF 3′-UTR downstream of the luciferase coding sequence in pGL3-control to assess whether this generates a USP15-sensitive reporter construct. The CRAF 3′-UTR had no impact on luciferase reporter activity in control cells; however, in cells depleted of USP15, with two independent oligos, we observed a significant, reproducible decrease in luciferase activity (Fig. 9D). These data suggest that the CRAF 3′-UTR renders mRNA stability or mRNA competence sensitive to USP15 depletion.
If a reduction in CRAF levels is the cause of the decrease in MEK phosphorylation observed in USP15-depleted cells, then we should be able to recapitulate this effect by depleting CRAF directly using siRNA. CRAF depletion mimics the effect of USP15 depletion and results in a marked decrease of pMEK levels in EGF-stimulated HeLa cells comparable to that obtained with USP15 knockdown (Fig. 10, A and B). To further corroborate the identification of CRAF as a key target for USP15 function, we made use of a melanoma cell line expressing constitutively active BRAF V600D, which renders the MAPK cascade in these cells independent of CRAF (30). Concordantly, depletion of USP15 in these cells caused a reduction of CRAF expression levels but did not affect the MEK or ERK phosphorylation status (Fig. 10C).
FIGURE 10.

CRAF is the key target for USP15 regulation of ERK signaling. A, HeLa cells were treated with control reagents or CRAF siRNA for 60 h before starvation for 12 h. EGF (1 ng/ml) was added for various times before lysis. IB, immunoblot. B, direct comparison of the effect of USP15 and CRAF depletion in HeLa cells stimulated for 5 min with EGF (n = 3, error bars; S.E., paired two-tailed t test compared with 5 min; control, CRAF (5 min), p < 0.01; USP15 (5 min), p < 0.025; CRAF (10 min), p < 0.005; USP15 (10 min), p < 0.001). C, WM266-4 BRAF (V600D) expressing melanoma cells were treated with control reagents or target specific siRNA, and Nonidet P-40 cell lysates were probed with antibodies indicated. Depletion of USP15 by two individual oligos (#1 and #17) leads to reduced CRAF levels without affecting pMEK and pERK levels. * indicates a nonspecific band detected by the CRAF antibody. D, a working model is shown. i, USP15 positively controls signal output by regulating CRAF levels. ii, USP15 may also negatively regulate MAPK signaling by directly stabilizing BRAP.
DISCUSSION
The interplay between the ubiquitin system and classical signal transduction cascades involving phosphorylation is an emerging area of study (31). This is best exemplified by the NFκB pathway, which uses ubiquitin both as a signaling entity and a degradation signal (1, 32). Using a comprehensive yeast two hybrid matrix screen of 55 DUBs against a representative panel of 133 RING E3 ligases, we identified 159 interactions between 47 RING E3 ligases and 27 DUBs. Of these, the interaction between BRAP and two related USPs, USP4 and USP15, was of compelling physiological interest given the established role of BRAP/IMP in the regulation of the MAP kinase signaling cascade (8). We confirmed this interaction by immunoprecipitation and mapped its determinants to an N-terminal region encompassing the DUSP and a UBL domain of USP15 (annotated in PFAM as DUF1055) with the C-terminal, extended coiled-coil region of BRAP. The DUSP domain is a 3-fold α-helical bundle supporting a triple-stranded anti-parallel β-sheet (α/β tripod) found in seven DUB family members (16, 33). Neither the DUSP nor the UBL was capable of sustaining the interaction independently, suggesting that they may combine to produce a single interaction surface. The crystal structure of the USP15 DUSP-UBL double domain was recently solved and shows that this double domain forms a single, rigid unit in support of this hypothesis (34). A similarly unified DUSP-UBL interaction surface was also suggested for the USP4 interaction with the U4/U6 recycling protein SART3 (29).
Our data indicate that BRAP can undergo autoubiquitylation, consistent with previous findings, which have led to the suggestion that this modification may be permissive for productive RAF-MEK assembly (8). We have been able to show that USP4 and USP15 can oppose this BRAP ubiquitylation and that BRAP can promote ubiquitylation of catalytically inactive USP15. This fits with the notion that one major facet of E3-DUB interactions is their reciprocal regulation of each other (11, 16), perhaps best exemplified by the interplay between HAUSP and MDM2, which jointly influences p53 ubiquitylation status (13). In contrast to the promiscuous behavior of BRAP with respect to USP4 and USP15 in a heterologous overexpression system, endogenous BRAP protein levels are exclusively sensitive to USP15 depletion, revealing the physiologically relevant interaction partner as USP15. Importantly, the observed decrease in BRAP-levels can be rescued by catalytically competent USP15. Endogenous BRAP appears to be constitutively poly- or multiply monoubiquitylated in our system; however, depletion of USP15 promotes the appearance of additional high molecular weight ubiquitylated species of BRAP. The most parsimonious interpretation is that USP15 plays a direct role in regulating the basal levels of BRAP by preventing its targeting to the proteasome for degradation, although indirect mechanisms cannot be fully excluded. Interestingly, BRAP falls within the top 20 genes whose expression profile most closely correlates with that of USP15 across a large panel of cancer cell lines (35).
Our results lead to the prediction that USP15 knockdown may recapitulate the effect of BRAP knockdown by destabilizing and thereby reducing the pool of BRAP that is able to sequester key components of the MAPK module, such as KSR1 (8). We set out to investigate the contributions of these various components to the amplitude of MAPK activation, which is known to influence cellular responses to growth factors. Using our standard experimental conditions, we were unable to show a significant impact of BRAP depletion upon EGF-induced MEK phosphorylation in serum-starved cells despite excellent knockdown efficiencies. Further investigation led us to reassess the effect of BRAP depletion without cell starvation, conditions that were used in previous studies by others. We then detected a significant and reproducible stimulation of MEK phosphorylation in line with prior literature (6, 36). Unexpectedly, we observed a highly significant reduction in both MEK and ERK phosphorylation after knockdown of USP15, but not USP4, which was most obvious in serum-starved cells. This inhibition could not be explained by the observed loss of BRAP, which is proposed to be a negative regulator. Furthermore, similar effects were also observed in U2OS cells for which BRAP levels were largely insensitive to USP15 depletion.
Having demonstrated an inhibitory effect of USP15 depletion in two cell lines and in response to two growth factors, we examined the remaining two core components involved in MEK activation that lie downstream of BRAP, BRAF, and CRAF. Remarkably, CRAF levels are reduced by ∼50% after USP15 knockdown, whereas BRAF levels are unaffected.
We were unable to obtain any evidence for a role of USP15 in the stabilization of CRAF protein. Instead, our data show a marked decrease of CRAF mRNA levels in USP15-depleted cells, suggesting that USP15 may regulate CRAF transcription. USP15 has recently been shown to deubiquitylate and thereby activate R-SMAD (Sma and Mad Related Family) transcription factors (37). Although SMADs have not been implicated in CRAF expression, it is conceivable that USP15 may regulate another transcriptional activator. However, we were unable to observe an effect of USP15 depletion on luciferase transcription driven from a previously described CRAF-promoter (25, 38), although we cannot fully exclude that USP15 may regulate CRAF transcription via an upstream or downstream element not contained in this 1.2-kb promoter construct. Alternatively, USP15 may regulate either processing or turnover of CRAF mRNA. Our results did not reveal a significant effect on mRNA processing. Rather we found a specific and significant impact of USP15 depletion on a luciferase reporter cloned upstream of the CRAF 3′-UTR, suggesting that USP15 may modulate CRAF protein levels via an as yet identified mechanism involving its 3′-UTR.
Previous studies have shown that MEK activation is highly sensitive to CRAF depletion and indicated that the observed effects of BRAP on MEK activation are critically dependent on the presence of CRAF (7). In this study we show that CRAF expression levels are controlled by the BRAP binding partner USP15, which therefore acts as a positive regulator of MAPK signaling. Using mutant BRAF-expressing melanoma cells, we confirmed that USP15 depletion only affects MEK phosphorylation in the context of a CRAF-dependent system. Our data suggest that the most consequential means by which USP15 depletion affects MAPK signaling is through control of CRAF mRNA via its 3′-UTR.
The MAPK kinase pathway may use various scaffolding complexes depending upon physiological context (39). The KSR/BRAP axis may be more important in T cells where KSR has a profound effect upon MAPK activation (36, 40). We anticipate that in some instances USP15 may play a dual role, promoting signaling through CRAF expression and dampening signals through stabilization of the E3 ligase, BRAP. The ability to switch between these two activities would allow fine control of the system. Recent studies have shown the induction of signaling through CRAF activation after application of clinically relevant BRAF inhibitors; concomitant inhibition of USP15 may present an opportunity for ameliorating this effect (41, 42).
Supplementary Material
Acknowledgments
We thank Richard Marais, Rohan Baker, and Siobhan Corbett for providing reagents, Michael White for very helpful discussions, and Rebecca Eccles, Sara Cadeco, and Jennifer Martin for assistance with this project.
This work was supported by a Cancer Research United Kingdom Senior Fellowship.

This article contains supplemental Table 1.
- MAP
- mitogen-activated protein
- DUB
- deubiquitylating enzyme
- USP
- ubiquitin-specific protein
- DUSP
- domain in USP
- UBL
- ubiquitin-like domain.
REFERENCES
- 1. Chiu Y. H., Zhao M., Chen Z. J. (2009) Ubiquitin in NF-κB signaling. Chem. Rev. 109, 1549–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kolch W. (2005) Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 6, 827–837 [DOI] [PubMed] [Google Scholar]
- 3. Jura N., Scotto-Lavino E., Sobczyk A., Bar-Sagi D. (2006) Differential modification of Ras proteins by ubiquitination. Mol. Cell 21, 679–687 [DOI] [PubMed] [Google Scholar]
- 4. Xu L., Lubkov V., Taylor L. J., Bar-Sagi D. (2010) Feedback regulation of Ras signaling by Rabex-5-mediated ubiquitination. Curr. Biol. 20, 1372–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lu Z., Xu S., Joazeiro C., Cobb M. H., Hunter T. (2002) The PHD domain of MEKK1 acts as an E3 ubiquitin ligase and mediates ubiquitination and degradation of ERK1/2. Mol. Cell 9, 945–956 [DOI] [PubMed] [Google Scholar]
- 6. Matheny S. A., Chen C., Kortum R. L., Razidlo G. L., Lewis R. E., White M. A. (2004) Ras regulates assembly of mitogenic signalling complexes through the effector protein IMP. Nature 427, 256–260 [DOI] [PubMed] [Google Scholar]
- 7. Chen C., Lewis R. E., White M. A. (2008) IMP modulates KSR1-dependent multivalent complex formation to specify ERK1/2 pathway activation and response thresholds. J. Biol. Chem. 283, 12789–12796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Matheny S. A., White M. A. (2009) Signaling threshold regulation by the Ras effector IMP. J. Biol. Chem. 284, 11007–11011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sowa M. E., Bennett E. J., Gygi S. P., Harper J. W. (2009) Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ventii K. H., Wilkinson K. D. (2008) Protein partners of deubiquitinating enzymes. Biochem. J. 414, 161–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sacco J. J., Coulson J. M., Clague M. J., Urbé S. (2010) Emerging roles of deubiquitinases in cancer-associated pathways. IUBMB Life 62, 140–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smith F. D., Scott J. D. (2002) Signaling complexes. Junctions on the intracellular information super highway. Curr. Biol. 12, R32–R40 [DOI] [PubMed] [Google Scholar]
- 13. Brooks C. L., Li M., Hu M., Shi Y., Gu W. (2007) The p53-Mdm2-HAUSP complex is involved in p53 stabilization by HAUSP. Oncogene 26, 7262–7266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wertz I. E., O'Rourke K. M., Zhou H., Eby M., Aravind L., Seshagiri S., Wu P., Wiesmann C., Baker R., Boone D. L., Ma A., Koonin E. V., Dixit V. M. (2004) De-ubiquitination and ubiquitin ligase domains of A20 down-regulate NF-κB signalling. Nature 430, 694–699 [DOI] [PubMed] [Google Scholar]
- 15. Heyninck K., Beyaert R. (2005) A20 inhibits NF-κB activation by dual ubiquitin-editing functions. Trends Biochem. Sci. 30, 1–4 [DOI] [PubMed] [Google Scholar]
- 16. Komander D., Clague M. J., Urbé S. (2009) Breaking the chains. Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 10, 550–563 [DOI] [PubMed] [Google Scholar]
- 17. Nijman S. M., Luna-Vargas M. P., Velds A., Brummelkamp T. R., Dirac A. M., Sixma T. K., Bernards R. (2005) A genomic and functional inventory of deubiquitinating enzymes. Cell 123, 773–786 [DOI] [PubMed] [Google Scholar]
- 18. Deshaies R. J., Joazeiro C. A. (2009) RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434 [DOI] [PubMed] [Google Scholar]
- 19. Markson G., Kiel C., Hyde R., Brown S., Charalabous P., Bremm A., Semple J., Woodsmith J., Duley S., Salehi-Ashtiani K., Vidal M., Komander D., Serrano L., Lehner P., Sanderson C. M. (2009) Analysis of the human E2 ubiquitin conjugating enzyme protein interaction network. Genome Res. 19, 1905–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ye Y., Scheel H., Hofmann K., Komander D. (2009) Dissection of USP catalytic domains reveals five common insertion points. Mol. Biosyst. 5, 1797–1808 [DOI] [PubMed] [Google Scholar]
- 21. Faronato M., Urbé S., Coulson J. C. (2010) USP15 (ubiquitin-specific peptidase 15). Atlas Genet. Cytogenet. Oncol. Hematol. url: http://atlasgeneticsoncology.org/Genes/USP15ID44585ch12q14.html
- 22. Lehner B., Sanderson C. M. (2004) A protein interaction framework for human mRNA degradation. Genome Res. 14, 1315–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Asada M., Ohmi K., Delia D., Enosawa S., Suzuki S., Yuo A., Suzuki H., Mizutani S. (2004) Brap2 functions as a cytoplasmic retention protein for p21 during monocyte differentiation. Mol. Cell. Biol. 24, 8236–8243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abramoff M. D., Magalhaes P. J., Ram S. J. (2004) Image processing with ImageJ. Biophotonics Int. 11, 36–42 [Google Scholar]
- 25. Winters B. S., Raj B. K., Robinson E. E., Foty R. A., Corbett S. A. (2006) Three-dimensional culture regulates Raf-1 expression to modulate fibronectin matrix assembly. Mol. Biol. Cell 17, 3386–3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Joazeiro C. A., Wing S. S., Huang H., Leverson J. D., Hunter T., Liu Y. C. (1999) The tyrosine kinase negative regulator c-Cbl as a RING-type, E2-dependent ubiquitin-protein ligase. Science 286, 309–312 [DOI] [PubMed] [Google Scholar]
- 27. Zheng N., Wang P., Jeffrey P. D., Pavletich N. P. (2000) Structure of a c-Cbl-UbcH7 complex. RING domain function in ubiquitin-protein ligases. Cell 102, 533–539 [DOI] [PubMed] [Google Scholar]
- 28. Matheny S. A., White M. A. (2006) Ras-sensitive IMP modulation of the Raf/MEK/ERK cascade through KSR1. Methods Enzymol. 407, 237–247 [DOI] [PubMed] [Google Scholar]
- 29. Song E. J., Werner S. L., Neubauer J., Stegmeier F., Aspden J., Rio D., Harper J. W., Elledge S. J., Kirschner M. W., Rape M. (2010) The Prp19 complex and the Usp4Sart3 deubiquitinating enzyme control reversible ubiquitination at the spliceosome. Genes Dev. 24, 1434–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Karasarides M., Chiloeches A., Hayward R., Niculescu-Duvaz D., Scanlon I., Friedlos F., Ogilvie L., Hedley D., Martin J., Marshall C. J., Springer C. J., Marais R. (2004) B-RAF is a therapeutic target in melanoma. Oncogene 23, 6292–6298 [DOI] [PubMed] [Google Scholar]
- 31. Hunter T. (2007) The age of crosstalk. Phosphorylation, ubiquitination, and beyond. Mol. Cell 28, 730–738 [DOI] [PubMed] [Google Scholar]
- 32. Liu H., Urbé S., Clague M. J. (2012) Selective protein degradation in cell signalling. Semin. Cell Dev. Biol. 23, 509–514 [DOI] [PubMed] [Google Scholar]
- 33. de Jong R. N., Ab E., Diercks T., Truffault V., Daniëls M., Kaptein R., Folkers G. E. (2006) Solution structure of the human ubiquitin-specific protease 15 DUSP domain. J. Biol. Chem. 281, 5026–5031 [DOI] [PubMed] [Google Scholar]
- 34. Elliott P. R., Liu H., Pastok M. W., Grossmann G. J., Rigden D. J., Clague M. J., Urbé S., Barsukov I. L. (2011) Structural variability of the ubiquitin specific protease DUSP-UBL double domains. FEBS Lett. 585, 3385–3390 [DOI] [PubMed] [Google Scholar]
- 35. Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A. A., Kim S., Wilson C. J., Lehár J., Kryukov G. V., Sonkin D., Reddy A., Liu M., Murray L., Berger M. F., Monahan J. E., Morais P., Meltzer J., Korejwa A., Jané-Valbuena J., Mapa F. A., Thibault J., Bric-Furlong E., Raman P., Shipway A., Engels I. H., Cheng J., Yu G. K., Yu J., Aspesi P., Jr., de Silva M., Jagtap K., Jones M. D., Wang L., Hatton C., Palescandolo E., Gupta S., Mahan S., Sougnez C., Onofrio R. C., Liefeld T., MacConaill L., Winckler W., Reich M., Li N., Mesirov J. P., Gabriel S. B., Getz G., Ardlie K., Chan V., Myer V. E., Weber B. L., Porter J., Warmuth M., Finan P., Harris J. L., Meyerson M., Golub T. R., Morrissey M. P., Sellers W. R., Schlegel R., Garraway L. A. (2012) The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Czyzyk J., Chen H. C., Bottomly K., Flavell R. A. (2008) p21 Ras/impedes mitogenic signal propagation regulates cytokine production and migration in CD4 T cells. J. Biol. Chem. 283, 23004–23015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Inui M., Manfrin A., Mamidi A., Martello G., Morsut L., Soligo S., Enzo E., Moro S., Polo S., Dupont S., Cordenonsi M., Piccolo S. (2011) USP15 is a deubiquitylating enzyme for receptor-activated SMADs. Nat. Cell Biol. 13, 1368–1375 [DOI] [PubMed] [Google Scholar]
- 38. Beck T. W., Brennscheidt U., Sithanandam G., Cleveland J., Rapp U. R. (1990) Molecular organization of the human Raf-1 promoter region. Mol. Cell. Biol. 10, 3325–3333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dhanasekaran D. N., Kashef K., Lee C. M., Xu H., Reddy E. P. (2007) Scaffold proteins of MAP-kinase modules. Oncogene 26, 3185–3202 [DOI] [PubMed] [Google Scholar]
- 40. Lin J., Harding A., Giurisato E., Shaw A. S. (2009) KSR1 modulates the sensitivity of mitogen-activated protein kinase pathway activation in T cells without altering fundamental system outputs. Mol. Cell. Biol. 29, 2082–2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Poulikakos P. I., Zhang C., Bollag G., Shokat K. M., Rosen N. (2010) RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464, 427–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Heidorn S. J., Milagre C., Whittaker S., Nourry A., Niculescu-Duvas I., Dhomen N., Hussain J., Reis-Filho J. S., Springer C. J., Pritchard C., Marais R. (2010) Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 140, 209–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.