

Background: Gp96 is the cellular receptor for Vip, a Listeria monocytogenes surface protein. The molecular details of Gp96-Vip interaction are unknown.
Results: Cell surface expression of Gp96 increases upon Listeria infection. Gp96 N terminus is required for Vip interaction.
Conclusion: Gp96 N-terminal domain is exposed to the extracellular milieu and is required for optimal Listeria entry.
Significance: Our study provides topological insights into Gp96 plasma membrane association.
Keywords: Bacterial Adhesion, Bacterial Pathogenesis, Cell Surface Receptor, Microbiology, Molecular Chaperone, Bacterial Invasion
Abstract
Listeria monocytogenes is an intracellular food-borne pathogen causing listeriosis in humans. This bacterium deploys an arsenal of virulence factors that act in concert to promote cellular infection. Bacterial surface proteins are of primary importance in the process of host cell invasion. They interact with host cellular receptors, inducing/modulating specific cellular responses. We previously identified Vip, a Listeria surface protein covalently attached to the bacterial cell wall acting as a key virulence factor. We have shown that Vip interacts with Gp96 localized at the surface of host cells during invasion and that this interaction is critical for a successful infection in vivo. To better understand the importance of Vip-Gp96 interaction during infection, we aimed to characterize this interaction at the molecular level. Here we demonstrate that, during infection, L. monocytogenes triggers the cellular redistribution of Gp96, inducing its exposure at the cell surface. Upon infection, Gp96 N-terminal domain is exposed to the extracellular milieu in L2071 fibroblasts and interacts with Vip expressed by Listeria. We identified Gp96 (Asp1–Leu170) as sufficient to interact with Vip; however, we also showed that the region Tyr179–Leu390 of Gp96 is important for the interaction. Our findings unravel the Listeria-induced surface expression of Gp96 and the topology of its insertion on the plasma membrane and improve our knowledge on the Vip-Gp96 interaction during Listeria infection.
Introduction
Listeria monocytogenes is a facultative intracellular human pathogen that causes listeriosis in immunocompromised individuals (1). L. monocytogenes enters the host via the consumption of contaminated food; it invades the small intestine, colonizes the liver and the spleen, and reaches the brain, and the fetus in pregnant women (2). To enter, survive, and multiply inside phagocytic and non-phagocytic cells, L. monocytogenes deploys an arsenal of virulence factors that act together to hijack cellular functions, promoting infection (3). Bacterial surface proteins play critical roles in the interaction with host cells and invasion. Importantly, the L. monocytogenes genome encodes a large repertoire of surface proteins that promote adhesion and/or invasion by binding and activating host membrane receptors (4, 5). We identified and characterized Vip as an L. monocytogenes surface protein covalently linked to the bacterial peptidoglycan via its C-terminal LPXTG motif (6). We have shown that Vip is required for infection in vivo and mediates invasion of specific cultured cell lines. In addition, we identified the host protein Gp96 as the cellular receptor for Vip (6).
Gp96 is a 96-kDa chaperone belonging to the Hsp90 family. This glycoprotein is constitutively and ubiquitously expressed. It localizes mainly within the lumen of the endoplasmic reticulum (ER)5 (7) and shares ≈50% homology at the amino acid level with human cytosolic Hsp90, the major differences being the N- and C-terminal extensions present in Gp96 but absent in Hsp90 (8). In its C terminus, Gp96 contains a KDEL sequence that is involved in retrograde transport from the Golgi apparatus to the ER and actively retains Gp96 within the ER (9). Through its N terminus, Gp96 binds/hydrolyzes ATP (8, 10) and chaperones multiple protein substrates. Consistent with this function, Gp96 expression is increased under stress conditions and accumulation of misfolded proteins (9). In addition to its central role as a chaperone in protein quality control, Gp96 has been implicated in innate and adaptive immunity (7, 11). Indeed, it can chaperone antigenic peptides, promoting their delivery to antigen-presenting cells; it activates and/or induces the maturation of dendritic cells (12, 13); and it has been shown to be a master chaperone for Toll-like receptors (TLRs) (11, 14, 15). Importantly, gp96-deficient macrophages failed to respond to ligands of both intracellular and cell surface-associated TLRs (11).
Although Gp96 is mainly localized at the ER, it may be exposed at the surface of certain cell types in particular conditions such as cell activation, infection, or necrotic cell death (9, 16). Gp96 was detected at the surface of human cells derived from tissues constituting tight human barriers that Listeria is able to cross during infection; thus, such cells (Caco-2 and human brain microvascular endothelial cells) should be preferentially used to address the role of Gp96 in infection. Besides its role as an L. monocytogenes receptor and because of its ability to bind a variety of bacterial pathogens or their products, Gp96 emerged recently as a key mediator in the establishment of various human infections. The Neisseria gonorrhoeae surface protein PorBIA interacts with Gp96, promoting bacterial adherence. Additionally, Gp96 sequestration through the binding of PorBIA leads to an impairment of the immune response and favors infection (17). Gp96 also serves as the cellular receptor for enterotoxin A from Clostridium difficile (18), OmpA expressed at the surface of Escherichia coli K1 (19–21), and Als3, a major invasin of Candida albicans (22). Interestingly, Gp96 is critical in C. albicans and E. coli K1 brain infections (22, 23). Very recently, Gp96 was also shown to interact directly with Bap, a Staphylococcus aureus protein involved in biofilm formation. Bap-Gp96 interaction provokes a significant reduction in the capacity of S. aureus to invade epithelial cells by interfering with the fibronectin-binding protein invasion pathway (24). Orientia tsutsugamushi and rotavirus directly modulate the expression of Gp96, disturbing innate and adaptive immune responses and thus providing the appropriate environment for pathogen survival and proliferation (25, 26).
Despite the massive progress in understanding the roles of Gp96 during pathogenesis, much remains to be learned. Although Gp96 is often hijacked as a membrane protein that serves as a receptor for bacterial virulence factors, the molecular mechanisms underlying its cellular membrane association are still unknown. This study aimed to characterize the interaction between Vip and the surface-associated Gp96 and identify the domains that are driving this interaction required for Listeria uptake into host cells.
Here we provide evidences showing that during infection L. monocytogenes triggers the Gp96 cell surface expression in a Vip-independent manner. We showed that the N-terminal domain of Gp96 is the domain exposed at the cell surface. In addition, we found that the first 170 amino acid residues of Gp96 are sufficient to interact with Vip expressed at the Listeria surface. Moreover, we demonstrated that Gp96 (Tyr179–Leu390) is also exposed to the extracellular milieu and is important for the interaction with Vip.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Cell Lines, and Culture Media
L. monocytogenes EGDe strain was grown in brain-heart infusion broth (Difco) at 37 °C with agitation. E. coli BL21(DE3) and E. coli BL21-CodonPlus(DE3) were grown in Luria-Bertani medium (Difco) supplemented when required with 100 μg/ml ampicillin (Calbiochem). J774 and L2071 cell lines were grown in DMEM with 4.5 g/liter glucose and l-glutamine supplemented with 10% fetal bovine serum (FBS) at 37 °C in a 7% CO2 humidified atmosphere. Caco-2 cells were grown in Eagle's minimal essential medium supplemented with sodium pyruvate, non-essential amino acids, and 20% FBS. Cell culture media and supplements were purchased from Lonza.
RNA Techniques
Total RNA was extracted from cell lines with RNAsolv reagent (Omega Bio-Tek) as recommended by the manufacturer. RNAs were treated with the TURBO DNA-free kit (Ambion). RNA quality was monitored on an Experion Automated Electrophoresis Station (Bio-Rad).
Quantitative Real Time RT-PCR Analysis
Real time RT-PCR was performed as described previously (27). Up to 1 μg of total RNA was reverse transcribed using the iScript cDNA Synthesis kit (Bio-Rad). Primers were designed to produce an amplicon length of 70–200 bp (Table 1). A standard curve was generated for each primer pair using four 10-fold dilutions of cDNA. Quantitative PCR was performed for 45 cycles with 2 μl of cDNA, 10 μl of SYBR® Green Supermix (Bio-Rad), and 0.25 pm (each) forward and reverse primers in a final volume of 20 μl. For each primer pair, a negative control (water) was included during cDNA quantification. After PCR amplification, a melting curve was generated for every PCR product to check the specificity of the PCR. Data were analyzed by the comparative Ct method, which provides the target gene expression value as unitless-fold change in the unknown sample compared with a calibrator sample (28). Both unknown and calibrator sample target gene expression data were normalized by the relative expression of a reference gene (GAPDH-encoding gene).
TABLE 1.
Sequences of the oligonucleotides used for quantitative RT-PCR
| Name | Primer sequences (5′ to 3′) |
|---|---|
| hGAPDHR | ctccgaccttcaccttcc |
| hGAPDHF | acagtcagcgcatcttc |
| Gp96-CL-fwd | ggaattctcaaaacacaacaacatacc |
| Gp96-CL- rev | ccctcgagttacaattcatctttttcagc |
| Gp96-N-fwd | ggaattcgacgatgaagttgatgtgg |
| Gp96-N-rev | ccctcgagtcaaatgtcagttgg |
| Gp96-C-fwd | ggaattcactagcctagacca |
| Gp96-C- rev | ccctcgagttacaattcatctttttcagc |
Biotinylation, Immunoprecipitations, and Pulldown Assays
Cell surface proteins were biotinylated using the EZ-Link Sulfo-NHS-Biotinylation kit (Pierce) following the manufacturer's protocol. Briefly, infected and non-infected live cells were washed in PBS (pH 8) and incubated with 2 mm Sulfo-NHS-biotin for 2 h at 4 °C. Sulfo-NHS-biotin was quenched with 100 mm glycine in PBS (pH 7.2) at 4 °C. Cells were lysed in radioimmune precipitation assay buffer, and Gp96 was immunoprecipitated from 600 μg of total protein extracts using 3 μg of anti-Gp96 (SPA-850, Stressgen). E-cadherin was immunoprecipitated from 500 μg of total protein extracts using 3 μg of anti-E-cadherin (H-108, sc-7870, Santa Cruz Biotechnology). Immunocomplexes were captured with 60 μl of Protein G-Sepharose 4 Fast Flow (GE Healthcare). Proteins were eluted and boiled in Laemmli buffer. Pulldown assays to capture biotinylated proteins were performed overnight at 4 °C with end-over-end rotation in a mixture of 200 μl of 50% slurry neutravidin-agarose resin (Thermo Scientific) and 500 μg of total protein extracts. Resin was washed three times with PBS at 4 °C, and captured proteins were eluted in Laemmli buffer. Samples were analyzed by SDS-PAGE followed by immunoblotting. Nitrocellulose membranes were blocked with 5% skim milk in buffer A (150 mm NaCl, 20 mm Tris-HCl (pH 7.4), and 0.1% Triton X-100) for 1 h at room temperature. Primary and secondary antibodies were diluted in 2.5% skim milk in buffer A as follows: anti-Gp96, 1:2000; anti-E-cadherin, 1:500; anti-β-catenin (H-102, sc-7199, Santa Cruz Biotechnology), 1:500; anti-actin (AC-15, Sigma-Aldrich), 1:5000; HRP-conjugated secondary antibodies (PARIS Anticorps), 1:2000; and HRP-streptavidin (Sigma-Aldrich), 1:2000.
Cloning, Expression, and Purification of Gp96 Variants
DNA encoding Gp96 variants (N terminus (N), C terminus (C), N terminus short (NS), and C terminus long (CL)) was amplified by PCR using the full-length Gp96-encoding sequence and primers listed in Table 2. PCR products were digested by EcoRI and XhoI and cloned into pGEX4T-1 (GE Healthcare). The insert was confirmed by PCR and sequencing. Plasmids were introduced into E. coli BL21(DE3) or E. coli BL21-CodonPlus(DE3) for protein expression. Bacteria were grown overnight in Luria-Bertani broth supplemented with ampicillin at 37 °C. The following day, a 1:40 subculture was incubated at 37 °C until it reached an absorbance at 600 nm (A600) of 0.3. Cultures were then transferred to 30 °C, and Gp96 expression was induced by addition of 0.3 mm isopropyl 1-thio-β-d-galactopyranoside (A600 = 0.5). Cultures were centrifuged at 4700 rpm for 15 min at 4 °C, and pellets were stored overnight at −20 °C. Pellets were resuspended in lysis buffer (PBS, 1 mm 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (Calbiochem), Complete protease inhibitor mixture (Roche Applied Science), and 1 mg/ml lysozyme (Calbiochem)) and centrifuged at 13,200 rpm for 15 min at 4 °C. Gp96 variants were captured from the supernatants with a glutathione-Sepharose 4B 50% slurry (GE Healthcare) overnight at 4 °C with end-over-end rotation. Glutathione-Sepharose beads were washed four times with PBS at 4 °C for 20 min. Three subsequent elutions were performed with 250 μl of elution buffer (50 mm Tris-HCl and 10 mm reduced glutathione). Eluted proteins were concentrated using Vivaspin 10-kDa-cutoff spin concentrators (Sartorius). Protein purity was assessed by SDS-PAGE and Western blot using anti-GST (sc-459, Santa Cruz Biotechnology; 1:1000), anti-Gp96 (H-212, sc-11402, Santa Cruz Biotechnology; 1:1000), and anti-Gp96 (Stressgen; 1:2000) antibodies.
TABLE 2.
Sequences of the oligonucleotides used to obtain the Gp96 variants
| Name | Primer sequences (5′ to 3′) |
|---|---|
| Gp96-NS-fwd | ggaattcgacgatgaagttgatgtgg |
| Gp96-NS- rev | ccctgcagtcacaattcagaagttgactgg |
| Gp96-CL-fwd | ggaattctcaaaacacaacaacatacc |
| Gp96-CL-rev | ccctcgagttacaattcatctttttcagc |
| Gp96-N-fwd | ggaattcgacgatgaagttgatgtgg |
| Gp96-N-rev | ccctcgagtcaaatgtcagttgg |
| Gp96-C-fwd | ggaattcactagcctagacca |
| Gp96-C-rev | ccctcgagttacaattcatctttttcagc |
Ligand Overlay Assay
Bacterial lysates from E. coli harboring a Vip expression vector (pET22b-Vip) cultured in non-induced and induced conditions were separated by SDS-PAGE and transferred onto a nitrocellulose membrane. The membrane was incubated for 48 h in TEN solution (10 mm Tris-HCl, 1 mm EDTA, and 50 mm NaCl) at 4 °C, blocked with 5% skim milk in buffer A, and then incubated with 25 μg/ml purified full-length Gp96 (Gp96-FL), Gp96-N, or Gp96-C. Bound Gp96 proteins were detected with anti-GST antibody (1:1000). The membrane was washed and incubated with HRP-conjugated anti-rabbit antibody. The signal was revealed using an ECL chemiluminescence detection system (Pierce).
In Vitro Invasion Assays
Invasion assays were performed in L2071 cells cultured on 24-well plates following the gentamicin survival assay as described previously (29). Bacterial suspensions were treated 1 h prior to infection at room temperature with purified Gp96-FL, Gp96-N, Gp96-C, Gp96-NS, and Gp96-CL (25 and 100 μg/ml in DMEM). Bacteria used in control infections were treated with elution buffer used to elute Gp96 variants. Bacteria were added to L2071 cells at a multiplicity of infection of 50 and incubated for 1 h at 37 °C and 7% CO2. After three washes with DMEM, cells were incubated for 90 min at 37 °C and 7% CO2 with DMEM containing 20 μg/ml gentamicin (Lonza). Cells were lysed with 0.2% Triton X-100, and serial dilutions of the lysates were plated in brain-heart infusion agar to assess the number of viable intracellular bacteria. When indicated, L2071 cells were treated (1 h prior to infection at 37 °C and 7% CO2) with 25 μg/ml rabbit polyclonal anti-Gp96 (H-212) or 25 μg/ml normal rabbit IgG (sc-2027, Santa Cruz Biotechnology).
RESULTS
Total Levels of Gp96 Are Not Affected by L. monocytogenes Cellular Infection
We have previously shown that Gp96 serves as receptor for Vip, an L. monocytogenes surface protein. In addition, our data showed that Gp96-Vip interaction has a key role in Listeria infection in vivo and in vitro (6). Gp96 plays an important role in pathogenesis, and its expression is directly modulated by different pathogens such as O. tsutsugamushi (25) and rotavirus (30). Thus, we investigated whether Listeria interferes with Gp96 expression during infection. We used three different cell lines that are relevant in the context of Listeria infection: human epithelial colorectal adenocarcinoma cells (Caco-2) are commonly used to mimic the intestinal barrier, L2071 cells are mouse fibroblasts used as an in vitro model for Listeria invasion, and J774 cells are murine macrophages frequently used for Listeria intracellular multiplication assays. Listeria invasion of Caco-2 and L2071 was shown to be dependent on Vip-Gp96 interaction (6). We first quantified gp96 transcripts in infected cells as compared with non-infected controls (Fig. 1A). Caco-2 cells and J774 macrophages were infected with L. monocytogenes wild type (WT) or the deletion mutant Δvip. At different times post-infection, total RNAs were extracted, and gp96 expression, relative to the expression of gapdh, was evaluated by quantitative RT-PCR. Our results showed that in Caco-2 cells the levels of gp96 mRNA do not change in response to infection. In J774, the levels of gp96 mRNA appeared slightly increased in infected cells as compared with non-infected cells (Fig. 1A). This increase was independent from Vip expression. To verify these results, we assessed by Western blot the expression of Gp96 in total protein extracts from Caco-2, L2071, and J774 infected by L. monocytogenes WT or Δvip for various time points (Fig. 1, B and C). We observed that Listeria infection does not interfere with the total levels of Gp96 (quantifications are provided in Fig. 1C). These results indicate that the minor increase observed in gp96 mRNA levels in Listeria-infected J774 did not result in an increase of the total Gp96 protein levels. Altogether these data show that L. monocytogenes does not adjust Gp96 expression.
FIGURE 1.

Expression levels of Gp96 are not affected by L. monocytogenes cellular infection. A, levels of gp96 expression were assessed by real time RT-PCR in Caco-2 and J774 non-infected (NI) or infected with L. monocytogenes WT or Δvip for different periods of time. Represented values are relative to the expression of gapdh in the same conditions. Error bars represent S.D. B, Caco-2, L2071, and J774 cells were left non-infected or were infected with L. monocytogenes WT or Δvip for 1, 2, and 4 h. Cells were lysed at different time points, and levels of Gp96 were assessed by Western blot. Actin was used as loading control. C, for each condition, expression levels of Gp96 relative to the expression of actin were quantified. p.i., post-infection.
L. monocytogenes Cellular Infection Induces the Surface Expression of Gp96
Taking into account our findings showing that Gp96 acts as a receptor for Listeria at the surface of host cells (6) and considering that under stress conditions Gp96 localizes at the cell surface, we hypothesized that Listeria could induce Gp96 cellular redistribution, promoting its exposure to the extracellular milieu. To evaluate whether Gp96 becomes surface-exposed upon infection, L2071 and Caco-2 cells were left uninfected or infected with L. monocytogenes for 1 h after which cell surface proteins were biotinylated. Cells were harvested and lysed, and the total Gp96 was immunoprecipitated from whole-cell extracts containing biotinylated surface proteins. Immunoprecipitates were revealed for Gp96 and biotin by Western blot (Fig. 2A). Streptavidin revealed a protein at the molecular weight of Gp96 only in infected cells (Fig. 2A, upper panel). We also observed that Gp96 was immunoprecipitated to a similar extent from lysates of infected and non-infected cells (Fig. 2A, middle panel). In accordance, the levels of Gp96 in total cell lysates were similar in infected and non-infected cells (Fig. 2A, lower panel). Together these results suggest that Gp96 becomes localized at the surface of the host cell in response to Listeria infection. To further confirm these findings, we performed pulldown assays using neutravidin-coupled beads to recover only biotinylated proteins from infected and non-infected Caco-2 whole-cell extracts. Proteins bound to neutravidin beads were analyzed by Western blot. The anti-Gp96 antibody revealed a band in Listeria-infected Caco-2 cells, thus confirming that Gp96 is exposed to the extracellular milieu upon infection (Fig. 2B, upper panel). As a control, we used an anti-E-cadherin antibody and showed that levels of biotinylation of this host transmembrane protein are identical in infected and non-infected cells (Fig. 2B, lower panel). To show that the biotin labeling was restricted to the surface proteins and confirm that cytoplasmic proteins were not biotin-labeled in our experiments, we performed control experiments using whole-cell lysates containing biotinylated surface proteins from Caco-2 in infected and non-infected conditions. E-cadherin was immunoprecipitated, and immunoprecipitates were revealed using specific antibodies for E-cadherin (transmembrane protein), its ligand β-catenin (an intracellular protein), and streptavidin (Fig. 2C). We observed that E-cadherin and β-catenin were similarly expressed in infected and non-infected Caco-2 cells. In addition, we confirmed that our immunoprecipitates contained both E-cadherin and β-catenin. However, only E-cadherin was detected by streptavidin in immunoprecipitates from infected and non-infected cell lysates (Fig. 2C). These results indicate that in our experimental conditions biotin labeling is restricted to the cell surface proteins, thus reinforcing our findings and definitively indicating that Gp96 is addressed to the host cell surface in response to Listeria infection.
FIGURE 2.

L. monocytogenes infection induces the cellular redistribution of Gp96 and its exposure to the extracellular milieu. A, L2071 and Caco-2 cells were infected (I) with L. monocytogenes WT or left uninfected (NI). Surface-exposed proteins of infected and non-infected cells were biotinylated, and cells were lysed. Gp96 was immunoprecipitated (IP:Gp96) from total cell lysates, and immunoprecipitates were revealed for Gp96 and streptavidin. As a control, Gp96 was revealed in whole-cell extracts (WCE). B, Caco-2 cells were infected (I) with L. monocytogenes WT or left uninfected (NI). Surface-exposed proteins of infected and non-infected cells were biotinylated. Total cell extracts were subjected to a pulldown assay using neutravidin beads. Biotinylated proteins recovered by the neutravidin beads were revealed for Gp96 and as a control for E-cadherin (E-cad), a 120-kDa transmembrane protein. C, total extracts from surface biotin-labeled infected (I) or non-infected (NI) Caco-2 cells were immunoprecipitated with an anti-E-cadherin antibody. Immunoprecipitates were revealed for E-cadherin, β-catenin (β-cat; a 92-kDa intracellular protein), and streptavidin (Strep). D, surface localization of Gp96 induced by Listeria infection is Vip-independent. Cells were infected with L. monocytogenes WT or Δvip for 1 or 2 h. Surface proteins were labeled with biotin, and cells were lysed. Immunoprecipitates obtained with an anti-Gp96 antibody were revealed for Gp96 and streptavidin. The asterisk (*) indicates the band corresponding to Gp96. p.i., post-infection.
Because Gp96 plays a key role as a Listeria cellular receptor in Vip-mediated bacterial entry (6), we investigated whether Gp96 surface expression was driven by the bacterial Vip protein. To address this question, Caco-2 cells were infected for 1 or 2 h with L. monocytogenes WT or Δvip. After infection, host cell surface proteins were biotinylated (as above). Gp96 immunoprecipitates were analyzed and revealed with both an anti-Gp96 antibody and streptavidin. A band revealed by streptavidin and corresponding to Gp96 appeared in immunoprecipitates from cells infected by L. monocytogenes WT or Δvip (Fig. 2D). These results indicate that the surface localization of Gp96 induced by Listeria infection is independent of Vip.
Listeria Invasion Is Impaired in Cells Treated with Anti-Gp96 Antibody
Although we showed here that Listeria infection induces membrane relocalization of Gp96 and other studies report the presence of Gp96 at the cellular membrane, the molecular mechanisms governing the Gp96 membrane localization remain largely unknown. We used L. monocytogenes as a tool to identify the domain of Gp96 exposed at the cell surface. We assumed that the Gp96 domain engaged in the interaction with Vip, which allows bacterial internalization, might be exposed at the cell surface. Regarding this, we hypothesized that an antibody recognizing such domain should compete with Vip and impair Listeria invasion. In accordance with this assumption, we have previously shown that Listeria invasion is impaired in cells treated with an anti-Gp96 antibody raised against the entire protein (6). Here we evaluated the capacity of the anti-Gp96 antibody (H-212), raised against amino acid residues 179–390 of human Gp96, to interfere with Vip-Gp96 interaction and block bacterial internalization. L2071 cells were incubated with the anti-Gp96 H-212 antibody described above and then infected with L. monocytogenes WT or Δvip. Levels of bacterial internalization were assessed by gentamicin survival assays. We observed that the entry level of Listeria WT was reduced to ≈50% in anti-Gp96 H-212-treated cells as compared with the control non-treated (NT) cells. Incubation of cells with control rabbit IgG had no effect on the number of intracellular bacteria (Fig. 3). In addition, in NT cells, L. monocytogenes Δvip showed a ≈40% invasion defect as compared with the WT strain, thus confirming the involvement of Vip in L2071 invasion. Importantly, we noticed that anti-Gp96 antibody or control rabbit IgG cellular pre-treatments do not significantly affect the remaining invasion capacity of Listeria Δvip in L2071 cells (Fig. 3). Together these data show that L. monocytogenes requires available Gp96 at the surface of host cells to be fully invasive and that the exploitation of Gp96 as a receptor is entirely dependent on the expression of bacterial Vip. In addition, these results suggest that Gp96 (Tyr179–Leu390) is exposed outside of the cell and appears to be important in the interaction with Vip, thus participating in the Vip-dependent entry process.
FIGURE 3.

Saturation of membrane-exposed Gp96 reduces Listeria invasion in a Vip-dependent manner. NT L2071 cells or those pre-treated with a 25 μg/ml concentration of either anti-Gp96 (H-212) or normal rabbit IgG were infected with WT or Δvip L. monocytogenes. Bacterial entry levels in NT, rabbit IgG control-, and anti-Gp96-treated cells were assessed by gentamicin survival assays. Values are given relative to the invasion of the WT strain into L2071 NT cells arbitrarily fixed to 100. Experiments were repeated twice in triplicate for each condition. Error bars represent S.D. Statistically significant differences are indicated: **, p < 0.01; ***, p < 0.001.
Identification of a Putative Transmembrane Domain in Gp96
Mature Gp96 encompasses several conserved domains: the N terminus contains the nucleotide-binding site followed by the acidic domain, whereas the peptide-binding domain and the dimerization domain are located at the C terminus (Fig. 4A). Considering the results described above showing that a Gp96 fragment encompassing amino acid residues 179–390 is, at least partially, exposed at the cell surface and is important for the interaction with Vip, we searched for a Gp96 transmembrane domain. We performed an in silico analysis in an attempt to identify a hydrophobic sequence that could suggest a putative transmembrane domain. This would give us an insight into the Gp96 domain exposed at the cell surface and consequently available to interact with Vip. The amino acid sequence of Gp96 was analyzed in two different transmembrane domain prediction software programs (TMpred and Mobyle TopPred). Both hydrophobicity profiles obtained revealed a hydrophobic (putative transmembrane) domain in the region between amino acids 166 and 190. In addition, both analyses suggested the same transmembrane topology in which the Gp96 N terminus (amino acids 1–166) would be exposed to the extracellular medium and the C terminus (amino acids 190–781) would be cytoplasmic.
FIGURE 4.
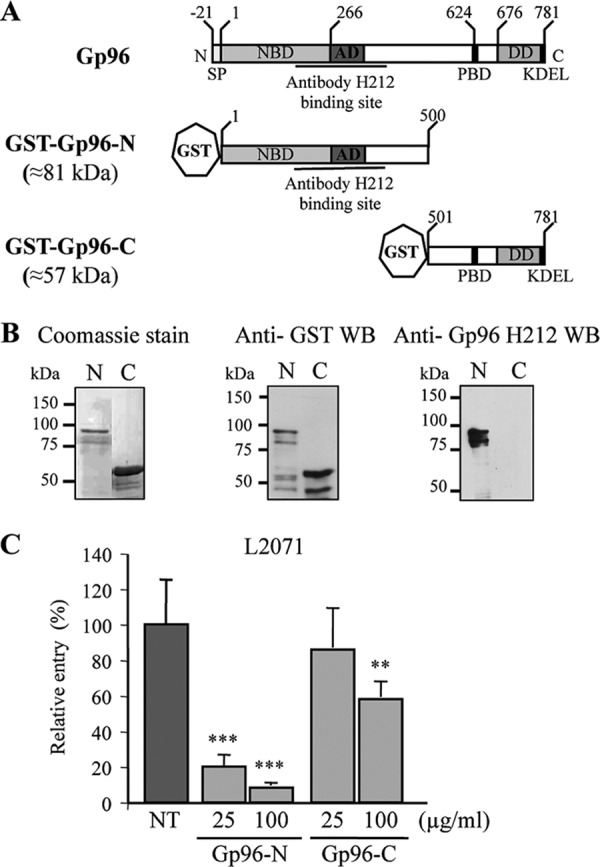
The N-terminal domain of Gp96 blocks Listeria invasion of L2071 cells. A, schematic representation of full-length and truncations of Gp96. Domains and amino acid residue positions are indicated. Gp96 possesses an N-terminal signal peptide and a C-terminal KDEL motif responsible for ER retention. Gp96 contains several conserved domains: a nucleotide-binding site (NBD), an acidic domain (AD), a peptide-binding domain (PBD), and a region crucial for dimerization (DD). The recognition site of H-212 antibody is indicated. B, the N and C termini of Gp96 were expressed in E. coli as GST fusion proteins (GST-Gp96-N and GST-Gp96-C) and purified using a glutathione affinity chromatography approach. Purified truncated proteins (N and C) were analyzed by SDS-PAGE and Coomassie Blue staining. Western blots (WB) using anti-GST and anti-Gp96 antibodies were also performed. Molecular masses are indicated. C, WT L. monocytogenes was incubated with either 25 or 100 μg/ml purified Gp96-N or Gp96-C and used to infect L2071 cells. Entry levels of treated and control NT bacteria were assessed by gentamicin survival assays. Values are given relative to the invasion of the NT bacteria into L2071 cells arbitrarily fixed to 100. Experiments were repeated twice in triplicate for each condition. Error bars represent S.D. Statistically significant differences are indicated: **, p < 0.01; ***, p < 0.001.
These predictions appeared to be in disagreement with our results. To further characterize the extracellular region of Gp96 and its domain interacting with Vip, we considered both the in silico predictions and the experimental data showed in Fig. 3. We designed various Gp96 truncations, purified the corresponding proteins as GST fusions, and evaluated their capacity to bind Vip and block infection of L2071 cells.
Purified Gp96-N (Asp1–Ile500) Impairs Listeria Cellular Invasion
In accordance with the results obtained in cells treated with the anti-Gp96 antibody and our published data showing that purified Gp96 blocks Listeria invasion (6), two expression vectors harboring truncated variants of Gp96 fused to a GST tag were constructed (Fig. 4A): Gp96-N (Asp1–Ile500) and Gp96-C (Thr501–Leu781). Note that the Gp96-N construct encompasses the extracellular domain predicted by in silico analysis and the region that is recognized by the blocking antibody (H-212). The GST fusion proteins (Gp96-N and Gp96-C) were expressed in E. coli and purified by reduced glutathione affinity chromatography. Purified proteins were analyzed by Coomassie staining and Western blot (Fig. 4B). In Coomassie-stained polyacrylamide gels, we observed that the purified proteins displayed the expected molecular weight, and no major contamination was detected (Fig. 4B, left panels). The Western blot analysis using an anti-GST antibody revealed a major band corresponding to the GST-Gp96 variants, indicating as expected that the anti-GST antibody recognized the recombinant proteins. Some weaker bands were visible below the band corresponding to the full-length recombinant variants, suggesting protein degradation or the existence of incomplete translation products with an intact GST tag (Fig. 4B, middle panels). Recombinant proteins were also analyzed by Western blot using the anti-Gp96 antibody (H-212). As expected, this antibody only recognized the N-terminal variant (Fig. 4B, right panels).
The purified Gp96 variants described above were used to determine the region of Gp96 engaged in Vip-Gp96 interaction during Listeria cellular infection. We hypothesized that the incubation of Listeria with Gp96 purified variants able to bind Vip before infection would saturate Vip at the bacterial surface, thus reducing Listeria invasion capacity. We performed in vitro gentamicin survival assays using L2071 cells that we infected with L. monocytogenes WT preincubated with 25 and 100 μg/ml purified Gp96 variants (Gp96-N or Gp96-C). As shown in Fig. 4C, bacteria treated with 25 μg/ml Gp96-N were ∼5-fold less invasive as compared with the control. This effect was even greater when Listeria were incubated with 100 μg/ml protein, revealing a dose-dependent effect. In contrast, the incubation of bacteria with 25 μg/ml purified Gp96-C had no inhibitory effect on the invasion of L2071 cells, and the 100 μg/ml treatment caused a 1.5-fold decrease in bacterial invasion. This defect appears to be minor as compared with the 10-fold reduction in the uptake of bacteria treated with 100 μg/ml purified Gp96-N. Together these results suggest that Gp96-N interacts with Vip expressed at the surface of Listeria, preventing its interaction with Gp96 at the cell membrane and thus impairing bacterial invasion.
Gp96 N-terminal (Asp1–Ile500) Domain Directly Interacts with Vip
To further demonstrate that Gp96-N directly interacts with Vip, we performed a ligand overlay assay using purified Gp96 variants. Total extracts of E. coli expressing or not expressing Vip fused to a His tag (Vip-His) were membrane-immobilized and incubated with 25 μg/ml purified GST-Gp96-FL, GST-Gp96-N, or GST-Gp96-C. The bound GST-Gp96 variants were detected with an anti-GST antibody. In membranes incubated with GST-Gp96-FL or GST-Gp96-N, the anti-GST revealed a protein band with the molecular weight of Vip-His only in Vip-His-containing extracts (Fig. 5A, upper panel). No band was detected in membranes incubated with Gp96-C. Therefore, these data indicate that Gp96-FL and Gp96-N, but not Gp96-C, directly interact with Vip-His in vitro. To confirm this observation, membranes were stripped and immunoblotted with anti-His antibody. The same band corresponding to the molecular weight of Vip-His was detected in all the lanes containing extracts of E. coli expressing Vip-His (Fig. 5A, middle panel), thus confirming that only Gp96-FL and Gp96-N directly bind Vip. To ensure that the total amount of E. coli extracts was the same in the different lanes, the samples were adjusted according to the A600 of the bacterial cultures. We used as a loading control a nonspecific band revealed by the anti-His antibody in all E. coli total protein extracts (Fig. 5A, lower panel).
FIGURE 5.

The N-terminal part of Gp96 directly interacts with Vip in vitro. A, E. coli BL21(DE3) harboring a Vip-His tag expression vector were grown in the absence (−) or presence (+) of 0.1 mm isopropyl 1-thio-β-d-galactopyranoside to induce the expression of Vip-His. Total extracts of E. coli were separated on a polyacrylamide gel, transferred onto a membrane, and probed successively with 25 μg/ml purified GST-Gp96 variants (Gp96-FL, Gp96-N, or Gp96-C) and anti-GST (Overlay Anti-GST). As control, the same whole protein extracts were revealed with an anti-His antibody (WB Anti-His), showing the presence of Vip-His only in isopropyl 1-thio-β-d-galactopyranoside-induced (+) E. coli total extracts. Loading controls are shown for each condition. B, purified GST-Gp96-N and -C were separated on a polyacrylamide gel, transferred to a membrane, and probed successively with 25 μg/ml purified Vip-His and anti-His antibody (Overlay Vip-His). For the Gp96-N variant, 3 and 7 μg of purified protein were used. Higher amounts (5 and 12 μg) of Gp96-C were used. Western blot using anti-GST antibody was performed to control for the presence of GST-Gp96 proteins in the membrane (WB Anti-GST).
We performed the reverse experiment to assess the capacity of purified Vip to bind membrane-immobilized purified Gp96 variants (Fig. 5B). Different quantities of purified GST-Gp96-N (3 and 7 μg) and GST-Gp96-C (5 and 12 μg) were transferred to a membrane, and the membrane was incubated with 25 μg/ml purified Vip-His. To detect membrane-bound Vip, we used an anti-His antibody. Bands at the molecular mass of Gp96-N (81 kDa) were detected in Gp96-N-containing lanes (Fig. 5B, upper left panel), confirming the direct interaction between Vip and Gp96-N. No bands were detected in Gp96-C-containing lanes (Fig. 5B, upper right panel), confirming that Vip does not interact with Gp96-C. As a control, the membrane was incubated with anti-GST antibody, which revealed both Gp96-N and Gp96-C at their expected molecular masses (81 and 57 kDa, respectively) (Fig. 5B, lower panel).
These data confirm the direct interaction between the Listeria surface protein Vip and the host protein Gp96. In addition, our results show that the domain of Gp96 that interacts with Vip comprises the Gp96 N-terminal region (Asp1–Ile500).
Purified Gp96 (Asp1–Leu170) Is Sufficient to Impair Listeria Invasion of L2071 Cells
In accordance with the in silico predictions, two other truncated variants of Gp96 fused to a GST tag were constructed (Fig. 6A): Gp96-NS (Asp1–Leu170) and Gp96-CL (Val190–Leu781). GST fusion proteins were expressed in E. coli and purified by reduced glutathione affinity chromatography (GST-Gp96-FL, GST-Gp96-NS, and GST-Gp96-CL). Purified proteins were analyzed by Coomassie staining and Western blot (Fig. 6B). In Coomassie-stained polyacrylamide gels, we observed that purified proteins displayed the expected molecular weight, and no major contamination was detected (Fig. 6B, left panels). Western blot analysis using an anti-GST specific antibody revealed a major band corresponding to the three GST variants showing as expected that recombinant proteins were recognized by the anti-GST antibody (Fig. 6B, middle panels).
FIGURE 6.
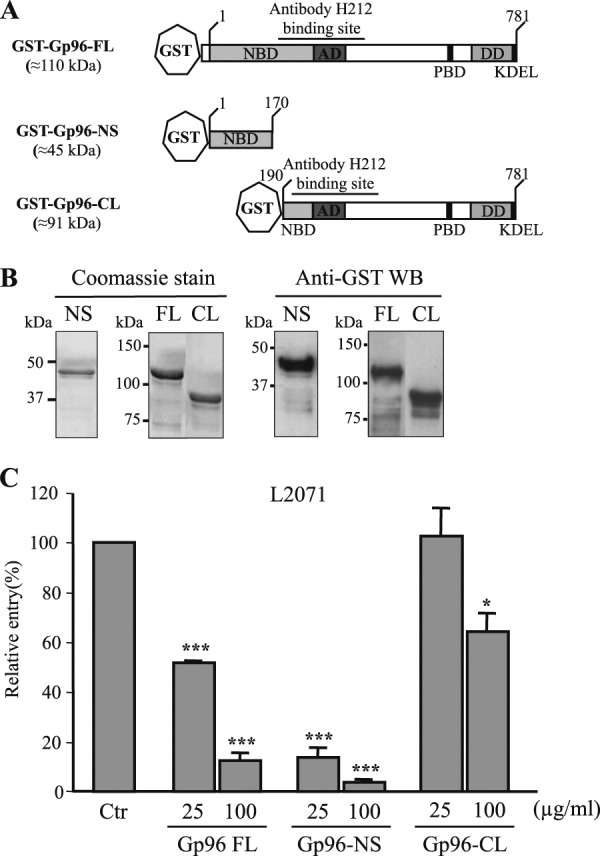
The N-terminal Gp96 (Asp1–Leu170) is sufficient to block Listeria invasion of L2071 cells. A, schematic representation of full-length and new truncated GST-Gp96 fusion proteins. Domains and amino acid residue positions are indicated. The N-terminal construct is shorter (GST-Gp96-NS). On the contrary, the C-terminal variant is longer (GST-Gp96-CL) and includes the H-212 antibody-binding site. B, Gp96-NS and -CL constructs were expressed in E. coli as GST fusion proteins and purified using a glutathione affinity chromatography approach. Purified truncated proteins (NS and CL) were analyzed by SDS-PAGE and Coomassie Blue staining. Western blots (WB) using anti-GST were also performed. Molecular masses are indicated. C, wild type L. monocytogenes were incubated with either 25 or 100 μg/ml purified GST-Gp96-FL, Gp96-NS, or Gp96-CL and used to infect L2071 cells. Entry levels of treated and NT bacteria were assessed by gentamicin survival assays. Values are given relative to the invasion of the non-treated bacteria into L2071 cells arbitrarily fixed to 100. Experiments were repeated three times in triplicate for each condition. Error bars represent S.D. Statistically significant differences are indicated: *, p < 0.05; ***, p < 0.001. NBD, nucleotide-binding site; AD, an acidic domain; PBD, peptide-binding domain; DD, region crucial for dimerization; Ctr, control.
The purified recombinant proteins described above were used to determine the minimal region of Gp96 engaged in the Vip-Gp96 interaction during Listeria cellular infection. We performed in vitro invasion assays using L2071 cells infected with L. monocytogenes preincubated with 25 and 100 μg/ml Gp96 recombinant proteins (Gp96-FL, Gp96-NS, and Gp96-CL). The capacity of Gp96-treated bacteria to enter the cells was assessed. As shown in Fig. 6C, bacteria treated with 25 μg/ml recombinant Gp96-NS were ∼7-fold less invasive as compared with the control. This effect was even greater (15-fold) when bacteria were incubated with 100 μg/ml protein, revealing a dose-dependent effect. As expected, a reduction of the invasive capacity was also observed for bacteria preincubated with Gp96-FL protein. In contrast, the incubation of bacteria with 25 μg/ml purified Gp96-CL had no effect on the invasion of L2071 cells. The 100 μg/ml treatment induced a 1.5-fold decrease in bacterial invasion that appears to be minor as compared with the effects observed for Gp96-FL- or Gp96-NS-treated bacteria. Together these results suggest that Gp96-NS (Asp1–Leu170) is sufficient to interact with Vip expressed at the Listeria surface, saturating the bacterial surface, preventing Vip interaction with Gp96 at the cell membrane, and impairing bacterial invasion.
DISCUSSION
We have previously shown that Vip, a surface protein of L. monocytogenes, interacts with Gp96 and uses it as a cellular receptor to invade cultured cell lines (6). In this study, we undertook the molecular characterization of the Vip-Gp96 interaction in an attempt to better understand how Gp96, thought to be essentially localized at the ER, could serve as a cellular receptor for a bacterial pathogen. We show here that Listeria infection triggers the cell surface localization of Gp96 in cultured epithelial cells without inducing an increase of the total expression level of Gp96.
Gp96 has been reported to be the membrane-associated eukaryotic ligand for various bacterial surface proteins, although the outcome of this interaction appear to be different depending on the bacterial partner (6, 17, 21, 24, 31–33). Commonly, pathogen ligands interact with surface-exposed Gp96, and presumably this interaction leads to the activation of host signaling cascades that facilitate bacterial invasion or adhesion. However, the mechanisms that govern the cell surface expression of Gp96 and overcome its ER retention remain largely unknown. Possibly, cellular infection triggers stress signals favoring Gp96-specific protein interactions that could mask the KDEL motif or induce its cleavage, thus circumventing Gp96 ER localization. We show here for the first time that Listeria infection promotes the cell surface expression of Gp96 in epithelial cells. This cell surface expression seems to be due to a relocalization of Gp96 and not to an increase of its total expression level. Another heat shock protein (Hsp60) was also shown to be expressed at a greater level at the surface of Caco-2 cells infected by Listeria. Hsp60 interacts with Listeria adhesion protein (LAP) (34), promoting transepithelial translocation (35). Recently, it was shown that E. coli K1 infection of neutrophils increases the cell surface expression of Gp96, which acts as a receptor for bacterial entry (23). In contrast with our results, in both cases, the cell surface expression of Hsp60 and Gp96 is the result of a general increased expression of these proteins in response to infection.
We found that at least part of the N-terminal domain (Asp1–Leu390) of Gp96 is exposed to the extracellular milieu, and we identified this domain as encompassing the minimal region involved in the direct interaction with Vip. Our experimental data indicate that Gp96 residues 1–170 (Asp1–Leu170) are sufficient to bind Vip, and thus, this region is likely to be exposed at the cell membrane. This assumption is in agreement with the in silico predictions that reveal a transmembrane domain between residues 166 and 190. Nevertheless our data also suggest that the amino acid 179–390 region (Tyr179–Leu390) is exposed to the extracellular milieu and may contribute to the Vip-Gp96 interaction. Indeed, bacteria treated with 100 μg/ml purified Gp96-CL showed a 30–40% decrease in invasion, suggesting that Gp96-CL interacts with Vip probably through the amino acid residue 190–390 region. However, the same results were observed for bacteria treated with 100 μg/ml Gp96-C, suggesting that the reduced invasion capacity observed in these conditions could be due to a nonspecific effect induced by high protein concentration. Taking into account the in silico predictions, the Gp96 region between amino acid residues 179 and 390 would be intracellular. Our results suggest the existence of other transmembrane domains that are not predictable.
The interaction between OmpA, an outer membrane virulence factor of E. coli K1, and Gp96 expressed in human brain microvascular endothelial cells is to date the best characterized (19, 21, 32, 36–40) and is usually used as a working model for the characterization of interactions involving Gp96 and pathogen products. OmpA interacts with the N terminus of Gp96 at the surface of human brain microvascular endothelial cells to promote infection (38). Here we show that despite the lack of homology shared between OmpA and Vip the listerial protein also interacts with the N terminus of Gp96. Interestingly, as compared with the Hsp90 sequence, the N-terminal extension of Gp96 is specific for Gp96 (8) and probably accounts for Gp96-specific functions such as its role in the infectious process of different human pathogens. Whether other pathogen proteins that bind Gp96 such as PorB from N. gonorrhoeae (17), Als3 from C. albicans (22), and Bap from S. aureus (24) also interact with the N terminus of Gp96 remains unknown.
In E. coli K1 infection, the interaction of OmpA with the N terminus of Gp96 triggers a series of host intracellular signaling events that stimulate actin polymerization at the bacterial entry site and assist internalization. OmpA-induced signaling is dependent on the C-terminal domain of Gp96 and involves focal adhesion kinase, Stat-3, PI3K, phospholipase C-γ, and PKC-α activation (36–40). During invasion, several L. monocytogenes surface proteins interact with and activate their mammalian receptors, inducing local membrane rearrangements and actin polymerization that result in the tight engulfment of the bacteria (41). Similarly to what was described for OmpA, we investigated the capacity of Vip to trigger host intracellular signaling events through its interaction with Gp96. In this context, the activation status of focal adhesion kinase and Stat-3 in response to L. monocytogenes infection was addressed. We did not detect modifications in the activation profile of focal adhesion kinase and Stat-3 in infected cells as compared with non-infected cells (Ref. 6 and data not shown). These data suggest that although OmpA and Vip bind to the same domain of Gp96 and both interactions favor bacterial invasion of host cells, the events triggered downstream are different, indicating that the receptor can be exploited in different ways by different pathogens. To date, the putative signaling events triggered by the Vip-Gp96 interaction remain to be elucidated, thus limiting our understanding of the Vip-dependent entry mechanism. In the absence of Vip-induced signaling, we consider two scenarios. 1) Vip-Gp96 interaction could cooperate with other ligand-receptor interactions, contributing to the stable bacterial adhesion to host cells and consequently favoring bacterial invasion, or 2) besides interacting with Vip, Gp96 chaperone activity could be critical in the surface localization of other host proteins involved in Listeria invasion. The identification of proteins associated to Gp96 at the cell surface in response to infection is critical to fully understand the role of Gp96 in the invasion process of Listeria.
In addition to its role as a cellular receptor for diverse pathogens, Gp96 can be exploited to modulate the host immune response developed to fight the infection. Indeed, Gp96 has been implicated in innate and adaptive immunity (7, 11). In particular, it is a master chaperone for the proper localization and function of TLRs (11, 14, 15) and specifically binds and activates neutrophils (42). During cellular infection, L. monocytogenes is exposed to plasma membrane and endosomal TLRs (43). The recognition of L. monocytogenes by TLR2 is mediated by listerial lipoproteins, and the lipidation of prelipoproteins in L. monocytogenes is required to promote NFκB activation via TLR2 (44). During Listeria infection, TLR2 was also shown to be an important factor promoting the production of interferon-β, which constitutes an innate response to cytoplasmic infection (45). Neutrophils are among the first inflammatory cells to migrate toward the site of inflammation and are critical for the early eradication of L. monocytogenes (43). Because Gp96 acts as a TLR2 chaperone (11) and binds neutrophils (42), we hypothesized that one of the functions of Vip-Gp96 interaction may lie in the sequestration of Gp96 by Listeria to impair an immune response dependent either on TLR2 activation/signaling or on neutrophil activation. We tested this hypothesis and confirmed the role of Vip for Listeria virulence (6), the crucial involvement of neutrophils in the immune response against Listeria, and the weak implication of TLR2 in the in vivo resistance to Listeria (data not shown). However, our results also indicate that the Vip-Gp96 interaction does not seem to play a critical role in neutrophil resistance and TLR2-dependent bacterial sensing (data not shown).
In conclusion, we have demonstrated that infection by L. monocytogenes triggers the cellular redistribution of Gp96, inducing its surface exposure in epithelial cells. The Gp96 N terminus is then exposed to the extracellular milieu and interacts with Vip expressed by L. monocytogenes via its first 170 amino acid residues, facilitating Vip-mediated invasion. However, the role of Gp96 in the broader context of Listeria infection is still unknown. Interestingly, a recent report shows that the TLR-MyD88 pathway was implicated in the increase of cell surface expression of Gp96, providing a new molecular mechanism underlying TLR-mediated Gp96 regulation (46). Upon infection, epithelial cells could thus via the TLR-MyD88 pathway induce the surface expression of Gp96 that would then be subverted by Listeria to promote its Vip-dependent internalization. In addition, as Gp96 is known to participate in the folding and assembly of many secretory and membrane proteins, we therefore wonder whether it participates in the cell membrane targeting of other proteins involved in Listeria infection.
Acknowledgments
We thank Filipe Carvalho and Francisco S. Mesquita for comments on the manuscript and members of the Cabanes laboratory for helpful discussions.
This work was supported in part by Fundo Europeu de Desenvolvimento Regional (FEDER)-Programa Operacional Factores de Competitividade (COMPETE) and Fundação para a Ciência e a Tecnologia (FCT) Grants PTDC/SAU-MII/65407/2006, FCOMP-01-0124-FEDER-007512, ERA-PTG/0003/2010, and PTDC/SAU-MIC/111581/2009FCOMP-01-0124-FEDER-015844.
- ER
- endoplasmic reticulum
- TLR
- Toll-like receptor
- NHS
- N-hydroxysuccinimide
- N
- N terminus
- C
- C terminus
- NS
- N terminus short
- CL
- C terminus long
- FL
- full length
- NT
- non-treated.
REFERENCES
- 1. Swaminathan B., Gerner-Smidt P. (2007) The epidemiology of human listeriosis. Microbes Infect. 9, 1236–1243 [DOI] [PubMed] [Google Scholar]
- 2. Cossart P. (2011) Illuminating the landscape of host-pathogen interactions with the bacterium Listeria monocytogenes. Proc. Natl. Acad. Sci. U.S.A. 108, 19484–19491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Camejo A., Carvalho F., Reis O., Leitão E., Sousa S., Cabanes D. (2011) The arsenal of virulence factors deployed by Listeria monocytogenes to promote its cell infection cycle. Virulence 2, 379–394 [DOI] [PubMed] [Google Scholar]
- 4. Bierne H., Cossart P. (2007) Listeria monocytogenes surface proteins: from genome predictions to function. Microbiol. Mol. Biol. Rev. 71, 377–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cabanes D., Dehoux P., Dussurget O., Frangeul L., Cossart P. (2002) Surface proteins and the pathogenic potential of Listeria monocytogenes. Trends Microbiol. 10, 238–245 [DOI] [PubMed] [Google Scholar]
- 6. Cabanes D., Sousa S., Cebriá A., Lecuit M., García-del Portillo F., Cossart P. (2005) Gp96 is a receptor for a novel Listeria monocytogenes virulence factor, Vip, a surface protein. EMBO J. 24, 2827–2838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang Y., Li Z. (2005) Roles of heat shock protein gp96 in the ER quality control: redundant or unique function? Mol. Cells 20, 173–182 [PubMed] [Google Scholar]
- 8. Argon Y., Simen B. B. (1999) GRP94, an ER chaperone with protein and peptide binding properties. Semin. Cell Dev. Biol. 10, 495–505 [DOI] [PubMed] [Google Scholar]
- 9. Marzec M., Eletto D., Argon Y. (2012) GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim. Biophys. Acta 1823, 774–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Soldano K. L., Jivan A., Nicchitta C. V., Gewirth D. T. (2003) Structure of the N-terminal domain of GRP94. Basis for ligand specificity and regulation. J. Biol. Chem. 278, 48330–48338 [DOI] [PubMed] [Google Scholar]
- 11. Yang Y., Liu B., Dai J., Srivastava P. K., Zammit D. J., Lefrançois L., Li Z. (2007) Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity 26, 215–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hilf N., Singh-Jasuja H., Schild H. (2002) The heat shock protein Gp96 links innate and specific immunity. Int. J. Hyperthermia 18, 521–533 [DOI] [PubMed] [Google Scholar]
- 13. Hilf N., Singh-Jasuja H., Schwarzmaier P., Gouttefangeas C., Rammensee H. G., Schild H. (2002) Human platelets express heat shock protein receptors and regulate dendritic cell maturation. Blood 99, 3676–3682 [DOI] [PubMed] [Google Scholar]
- 14. Vabulas R. M., Braedel S., Hilf N., Singh-Jasuja H., Herter S., Ahmad-Nejad P., Kirschning C. J., Da Costa C., Rammensee H. G., Wagner H., Schild H. (2002) The endoplasmic reticulum-resident heat shock protein Gp96 activates dendritic cells via the Toll-like receptor 2/4 pathway. J. Biol. Chem. 277, 20847–20853 [DOI] [PubMed] [Google Scholar]
- 15. Liu B., Yang Y., Qiu Z., Staron M., Hong F., Li Y., Wu S., Li Y., Hao B., Bona R., Han D., Li Z. (2010) Folding of Toll-like receptors by the HSP90 paralogue gp96 requires a substrate-specific cochaperone. Nat. Commun. 1, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Altmeyer A., Maki R. G., Feldweg A. M., Heike M., Protopopov V. P., Masur S. K., Srivastava P. K. (1996) Tumor-specific cell surface expression of the-KDEL containing, endoplasmic reticular heat shock protein gp96. Int. J. Cancer 69, 340–349 [DOI] [PubMed] [Google Scholar]
- 17. Rechner C., Kühlewein C., Müller A., Schild H., Rudel T. (2007) Host glycoprotein Gp96 and scavenger receptor SREC interact with PorB of disseminating Neisseria gonorrhoeae in an epithelial invasion pathway. Cell Host Microbe 2, 393–403 [DOI] [PubMed] [Google Scholar]
- 18. Na X., Kim H., Moyer M. P., Pothoulakis C., LaMont J. T. (2008) gp96 is a human colonocyte plasma membrane binding protein for Clostridium difficile toxin A. Infect. Immun. 76, 2862–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maruvada R., Blom A. M., Prasadarao N. V. (2008) Effects of complement regulators bound to Escherichia coli K1 and Group B Streptococcus on the interaction with host cells. Immunology 124, 265–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Prasadarao N. V., Blom A. M., Villoutreix B. O., Linsangan L. C. (2002) A novel interaction of outer membrane protein A with C4b binding protein mediates serum resistance of Escherichia coli K1. J. Immunol. 169, 6352–6360 [DOI] [PubMed] [Google Scholar]
- 21. Prasadarao N. V., Srivastava P. K., Rudrabhatla R. S., Kim K. S., Huang S. H., Sukumaran S. K. (2003) Cloning and expression of the Escherichia coli K1 outer membrane protein A receptor, a gp96 homologue. Infect. Immun. 71, 1680–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu Y., Mittal R., Solis N. V., Prasadarao N. V., Filler S. G. (2011) Mechanisms of Candida albicans trafficking to the brain. PLoS Pathog. 7, e1002305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mittal R., Prasadarao N. V. (2011) gp96 expression in neutrophils is critical for the onset of Escherichia coli K1 (RS218) meningitis. Nat. Commun. 2, 552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Valle J., Latasa C., Gil C., Toledo-Arana A., Solano C., Penadés J. R., Lasa I. (2012) Bap, a biofilm matrix protein of Staphylococcus aureus prevents cellular internalization through binding to GP96 host receptor. PLoS Pathog. 8, e1002843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cho N. H., Choi C. Y., Seong S. Y. (2004) Down-regulation of gp96 by Orientia tsutsugamushi. Microbiol. Immunol. 48, 297–305 [DOI] [PubMed] [Google Scholar]
- 26. Greenberg H. B., Estes M. K. (2009) Rotaviruses: from pathogenesis to vaccination. Gastroenterology 136, 1939–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lebreton A., Lakisic G., Job V., Fritsch L., Tham T. N., Camejo A., Matteï P. J., Regnault B., Nahori M. A., Cabanes D., Gautreau A., Ait-Si-Ali S., Dessen A., Cossart P., Bierne H. (2011) A bacterial protein targets the BAHD1 chromatin complex to stimulate type III interferon response. Science 331, 1319–1321 [DOI] [PubMed] [Google Scholar]
- 28. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔCT) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 29. Reis O., Sousa S., Camejo A., Villiers V., Gouin E., Cossart P., Cabanes D. (2010) LapB, a novel Listeria monocytogenes LPXTG surface adhesin, required for entry into eukaryotic cells and virulence. J. Infect Dis. 202, 551–562 [DOI] [PubMed] [Google Scholar]
- 30. Xu A., Bellamy A. R., Taylor J. A. (1998) BiP (GRP78) and endoplasmin (GRP94) are induced following rotavirus infection and bind transiently to an endoplasmic reticulum-localized virion component. J. Virol. 72, 9865–9872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rolhion N., Barnich N., Bringer M. A., Glasser A. L., Ranc J., Hébuterne X., Hofman P., Darfeuille-Michaud A. (2010) Abnormally expressed ER stress response chaperone Gp96 in CD favours adherent-invasive Escherichia coli invasion. Gut 59, 1355–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Prasadarao N. V. (2002) Identification of Escherichia coli outer membrane protein A receptor on human brain microvascular endothelial cells. Infect. Immun. 70, 4556–4563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mittal R., Prasadarao N. V. (2010) Nitric oxide/cGMP signalling induces Escherichia coli K1 receptor expression and modulates the permeability in human brain endothelial cell monolayers during invasion. Cell. Microbiol. 12, 67–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wampler J. L., Kim K. P., Jaradat Z., Bhunia A. K. (2004) Heat shock protein 60 acts as a receptor for the Listeria adhesion protein in Caco-2 cells. Infect. Immun. 72, 931–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Burkholder K. M., Bhunia A. K. (2010) Listeria monocytogenes uses Listeria adhesion protein (LAP) to promote bacterial transepithelial translocation and induces expression of LAP receptor Hsp60. Infect. Immun. 78, 5062–5073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krishnan S., Chen S., Turcatel G., Arditi M., Prasadarao N. V. (2012) Regulation of Toll-like receptor 2 interaction with Ecgp96 controls Escherichia coli K1 invasion of brain endothelial cells. Cell. Microbiol., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Krishnan S., Fernandez G. E., Sacks D. B., Prasadarao N. V. (2012) IQGAP1 mediates the disruption of adherens junctions to promote Escherichia coli K1 invasion of brain endothelial cells. Cell. Microbiol. 14, 1415–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maruvada R., Argon Y., Prasadarao N. V. (2008) Escherichia coli interaction with human brain microvascular endothelial cells induces signal transducer and activator of transcription 3 association with the C-terminal domain of Ec-gp96, the outer membrane protein A receptor for invasion. Cell. Microbiol. 10, 2326–2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Reddy M. A., Prasadarao N. V., Wass C. A., Kim K. S. (2000) Phosphatidylinositol 3-kinase activation and interaction with focal adhesion kinase in Escherichia coli K1 invasion of human brain microvascular endothelial cells. J. Biol. Chem. 275, 36769–36774 [DOI] [PubMed] [Google Scholar]
- 40. Reddy M. A., Wass C. A., Kim K. S., Schlaepfer D. D., Prasadarao N. V. (2000) Involvement of focal adhesion kinase in Escherichia coli invasion of human brain microvascular endothelial cells. Infect. Immun. 68, 6423–6430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sousa S., Lecuit M., Cossart P. (2005) Microbial strategies to target, cross or disrupt epithelia. Curr. Opin. Cell Biol. 17, 489–498 [DOI] [PubMed] [Google Scholar]
- 42. Radsak M. P., Hilf N., Singh-Jasuja H., Braedel S., Brossart P., Rammensee H. G., Schild H. (2003) The heat shock protein Gp96 binds to human neutrophils and monocytes and stimulates effector functions. Blood 101, 2810–2815 [DOI] [PubMed] [Google Scholar]
- 43. Stavru F., Archambaud C., Cossart P. (2011) Cell biology and immunology of Listeria monocytogenes infections: novel insights. Immunol. Rev. 240, 160–184 [DOI] [PubMed] [Google Scholar]
- 44. Machata S., Tchatalbachev S., Mohamed W., Jänsch L., Hain T., Chakraborty T. (2008) Lipoproteins of Listeria monocytogenes are critical for virulence and TLR2-mediated immune activation. J. Immunol. 181, 2028–2035 [DOI] [PubMed] [Google Scholar]
- 45. Aubry C., Corr S. C., Wienerroither S., Goulard C., Jones R., Jamieson A. M., Decker T., O'Neill L. A., Dussurget O., Cossart P. (2012) Both TLR2 and TRIF contribute to interferon-beta production during Listeria infection. PLoS One 7, e33299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim G., Han J. M., Kim S. (2010) Toll-like receptor 4-mediated c-Jun N-terminal kinase activation induces gp96 cell surface expression via AIMP1 phosphorylation. Biochem. Biophys. Res. Commun. 397, 100–105 [DOI] [PubMed] [Google Scholar]