

Background: CRP exerts antipneumococcal function in mice if administered during the early stages of pneumococcal infection.
Results: CRP loses such antipneumococcal function when its phosphocholine-binding pocket is blocked.
Conclusion: The phosphocholine-binding pocket on CRP participates in antipneumococcal function of CRP during the early stages of infection.
Significance: Remodeling of CRP may be required for successful treatment of mice during the late stages of infection.
Keywords: Host Defense, Inflammation, Innate Immunity, Pattern Recognition Receptor, Pentraxins, C-reactive Protein
Abstract
Human C-reactive protein (CRP) protects mice from lethal Streptococcus pneumoniae infection when injected into mice within the range of 6 h before to 2 h after the administration of pneumococci. Because CRP binds to phosphocholine-containing substances and subsequently activates the complement system, it has been proposed that the antipneumococcal function of CRP requires the binding of CRP to phosphocholine moieties present in pneumococcal cell wall C-polysaccharide. To test this proposal experimentally, in this study, we utilized a new CRP mutant incapable of binding to phosphocholine. Based on the structure of CRP-phosphocholine complexes, which showed that Phe66, Thr76, and Glu81 formed the phosphocholine-binding pocket, we constructed a CRP mutant F66A/T76Y/E81A in which the pocket was blocked by substituting Tyr for Thr76. When compared with wild-type CRP, mutant CRP bound more avidly to phosphoethanolamine and could be purified by affinity chromatography using phosphoethanolamine-conjugated Sepharose. Mutant CRP did not bind to phosphocholine, C-polysaccharide, or pneumococci. Mutant CRP was free in the mouse serum, and its rate of clearance in vivo was not faster than that of wild-type CRP. When either 25 μg or 150 μg of CRP was administered into mice, unlike wild-type CRP, mutant CRP did not protect mice from lethal pneumococcal infection. Mice injected with mutant CRP had higher mortality rates than mice that received wild-type CRP. Decreased survival was due to the increased bacteremia in mice treated with mutant CRP. We conclude that the phosphocholine-binding pocket on CRP is necessary for CRP-mediated initial protection of mice against lethal pneumococcal infection.
Introduction
C-reactive protein (CRP),3 a member of the pentraxin family of proteins, reacts with cell wall C-polysaccharide (PnC) of Streptococcus pneumoniae in a Ca2+-dependent manner (1–3). The binding specificity of CRP is for the phosphocholine (PCh) moieties present in PnC (4). CRP also binds to whole pneumococci in human and mouse sera and in Ca2+-containing buffers (5–7). Another member of the pentraxin family, serum amyloid P (SAP), which is structurally similar to CRP, displays Ca2+-dependent binding specificity for phosphoethanolamine (PEt) (8–10). CRP also binds to PEt but not as avidly as it does to PCh (4, 8–13).
CRP is composed of five identical noncovalently attached subunits. Each subunit has 206 amino acids, and the molecular mass of each subunit is ∼23 kDa (14). All five subunits have the same orientation in the pentamer, with a PCh-binding site located on the same face of each subunit (14, 15). The PCh-binding site consists of a hydrophobic pocket formed by several amino acids, including Phe66, Thr76, and Glu81, and two Ca2+ ions, which are bound to CRP by interactions with amino acids from other parts of the protein (Fig. 1). The phosphate group of PCh directly coordinates with the two Ca2+ ions. The choline group of PCh lies within the hydrophobic pocket. Phe66 provides hydrophobic interactions with the three methyl groups of choline. Thr76 is critical for creating the appropriately sized pocket to accommodate PCh. Glu81 interacts with the positively charged nitrogen atom of choline (14, 16). Previous mutational analysis of Thr76 in CRP has confirmed the significance of the hydrophobic pocket for PCh binding (17). In SAP, at the position corresponding to Thr76 in CRP, it is a Tyr (Tyr74) (18, 19).
FIGURE 1.

One subunit of CRP. A, structure of one of the five subunits of WT CRP bound to PCh (Protein Data Bank (PDB) ID 1B09) is shown. The side chains of Phe66, Thr76 and Glu81 involved in the formation of the PCh-binding pocket are highlighted. Calcium ions (Ca) are shown as cyan balls. B, molecular modeling of F66A/T76Y/E81A mutant CRP. The PDB coordinates of mutant CRP were generated from the PDB file 1B09 using SYBYL (Tripos, Inc.). The side chains of Phe66, Thr76 and Glu81 are substituted with Ala, Tyr, and Ala, respectively. One of the five subunits is shown. The figures were rendered using PyMOL (66).
Pneumococci remain the most common cause of community-acquired pneumonia world-wide (20–22). In humans, CRP is an acute phase protein; that is, its serum concentration is increased several hundredfold in response to pneumococcal infection (2). However, the functions of CRP in pneumococcal infection are not known (3). In mice, CRP is only a trace serum component and is not an acute phase protein (23). Therefore mice are used to explore the in vivo functions of human CRP. In mouse models of infection, passively administered human CRP has been shown to be protective against lethal pneumococcal infection, as determined by increased survival of and decreased bacteremia in the infected mice (24, 25). Interestingly, CRP was most effective in protecting mice from infection only when injected within the range of 6 h before to 2 h after administering pneumococci into mice (26). The protective function of CRP was not observed when mice received CRP 24 or 36 h after infection (7, 26). Thus, the CRP-mediated protection of mice requires the presence of CRP in the early stages of infection. Mice transgenic for human CRP were also protected from lethal pneumococcal infection and showed both decreased bacteremia and increased survival (27).
Because PnC-complexed CRP activates the complement system in both human and mouse sera (3, 28), it has been proposed that CRP is protective through a pathway in which CRP binds to pneumococci through PCh groups present on their surfaces, the pathogen-bound CRP activates the complement system, and bacteremia is then reduced through complement-dependent opsonophagocytosis (29–31). The aim of this study was to determine whether the binding of CRP to PCh on pneumococci was required for the protection of mice against pneumococcal infection. Employing site-directed mutagenesis, we generated a novel triple mutant of CRP, F66A/T76Y/E81A, incapable of binding to PCh, and used mutant CRP in mouse protection experiments. We hypothesized that if the binding of CRP to PCh was required for the protection of mice against pneumococcal infection, then mutant CRP should not be protective. In a similar previously published study, another CRP mutant, F66A/E81A, incapable of binding to PCh was used (7).
EXPERIMENTAL PROCEDURES
Construction and Expression of the CRP Triple Mutant F66A/T76Y/E81A
The construction of the double mutant F66A/E81A CRP cDNA has been described earlier (13). F66A/E81A CRP cDNA was used as the template for construction of the triple mutant F66A/T76Y/E81A cDNA (substitution of Phe66 and Glu81 with Ala and of Thr76 with Tyr). Mutagenic oligonucleotides, 5′-GGATACAGTTTTTACGTGGGTGGGTCTG-3′ and 5′-CAGACCCACCCACGTAAAAACTGTATCC-3′, which substitute Tyr for Thr76 (codons shown in bold and italicized letters), were designed according to the sequence of F66A/E81A cDNA template and obtained from Integrated DNA Technologies. Mutagenesis was conducted using the QuikChange site-directed mutagenesis kit (Stratagene). Mutations were verified by nucleotide sequencing, utilizing the services of the Molecular Biology Core Facility of our university. Stable transfection of F66A/T76Y/E81A CRP cDNA was carried out in CHO cells as described previously (7). A CHO cell line expressing the F66A/T76Y/E81A mutant CRP was isolated by a series of subcloning steps.
Preparation of PEt-conjugated Sepharose
PEt-conjugated Sepharose was prepared as described previously (32). In brief, 25 ml of packed ECH-Sepharose 4B beads (GE Healthcare) was first washed with water (pH 4.5) and subsequently with 0.5 m NaCl. Then, 180 mg of PEt (catalog number P0503, Sigma-Aldrich) was dissolved in 25 ml water (pH 4.5) and added to the washed beads. 500 mg of N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (catalog number E6383, Sigma-Aldrich) was then added, and the mixture was stirred for 1 h at room temperature. After monitoring the pH for 1 h to ensure that the pH stayed at 4.5, the mixture was left overnight at 4 °C with slow stirring. The beads were then washed with 0.1 m acetate buffer, pH 4.0, followed by washing with 100 mm Tris/HCl, pH 8.0, containing 0.5 m NaCl. Washing was repeated three times alternating between the acetate and Tris buffers. Finally, the beads were washed with water and then with TBS containing 2 mm CaCl2.
Purification of Native WT CRP
WT CRP was purified from discarded human pleural fluid by affinity chromatography on a PCh-Sepharose column (Pierce) followed by ion-exchange chromatography on a MonoQ column (GE Healthcare) and gel filtration chromatography on a Superose 12 column (GE Healthcare), as described previously (33), and stored frozen. On the day of the experiments, CRP was repurified by gel filtration chromatography on a Superose 12 column to remove any form of modified CRP that might have been generated due to storage of CRP. Repurified CRP was stored in TBS containing 2 mm CaCl2 at 4 °C and was used within a week. The purity of CRP was confirmed by using denaturing SDS-PAGE.
Purification of Mutant CRP
Purification of mutant CRP from the cell culture supernatants involved two steps: Ca2+-dependent affinity chromatography on a PEt-conjugated Sepharose column followed by gel filtration chromatography on a Superose 12 column. For affinity chromatography, the culture medium containing CRP was diluted (1:1) in 0.1 m borate buffered saline, pH 8.3, containing 3 mm CaCl2 and passed through the PEt-conjugated Sepharose column. After collecting the flow-through and washing the column with the same buffer, bound CRP was eluted with 0.1 m borate-buffered saline, pH 8.3, containing 5 mm EDTA. Eluted CRP was concentrated and further purified by gel filtration chromatography on a Superose 12 column. Gel filtration chromatography was carried out as described for WT CRP, except that the column was equilibrated and eluted with TBS containing 5 mm EDTA. It was necessary that the gel filtration chromatography of the mutant CRP be performed in the presence of EDTA because in the presence of Ca2+, the mutant CRP bound to Superose beads (data not shown). Eluted CRP was immediately dialyzed against TBS containing 2 mm CaCl2, stored at 4 °C, and used within a week. The purity of CRP was confirmed by using denaturing SDS-PAGE.
The concentration of purified WT and mutant CRP was determined by measuring absorbance at 280 nm. The extinction coefficient value of 19.5 at 280 nm for 1% CRP solution was used for calculations. For mouse protection experiments, both purified WT and mutant CRP were treated with the Detoxi-Gel endotoxin removing gel according to the manufacturer's instructions (Thermo). The concentration of endotoxin in CRP was determined by using the QCL-1000 limulus amebocyte lysate kit according to the manufacturer's instructions (Lonza).
PCh Binding Assay
Binding activity of CRP for PCh was evaluated by using PCh-conjugated BSA and PnC (Statens Serum Institut) as the ligands, as described previously (7, 33). Microtiter wells (96-well plates) were coated with 10 μg/ml PCh-BSA or PnC in TBS, overnight at 4 °C. The unreacted sites in the wells were blocked with TBS containing 0.5% gelatin for 45 min at room temperature. CRP diluted in TBS containing 5 mm CaCl2, 0.1% gelatin, and 0.02% Tween 20 (TBS-Ca) was added in duplicate wells. After incubating the plates for 2 h at 37 °C, the wells were washed with TBS-Ca. The assays were performed in duplicate plates. In one plate, anti-CRP monoclonal antibody (mAb) HD2.4, diluted in TBS-Ca, was used (1 h at 37 °C) to detect bound CRP. In the other plate, rabbit polyclonal anti-CRP antibody (Sigma-Aldrich), diluted in TBS-Ca, was used (1 h at 37 °C) to detect bound CRP. HRP-conjugated goat anti-mouse IgG and HRP-conjugated donkey anti-rabbit IgG (Thermo), diluted in TBS-Ca, were used (1 h at 37 °C) as secondary antibodies. Color was developed, and the A405 was read in a microtiter plate reader (Molecular Devices).
Anti-CRP mAb Binding Assay
The anti-CRP mAb HD2.4 and EA4.1 binding assays were performed as described previously (13). Microtiter wells (96-well plates) were coated with 10 μg/ml anti-CRP mAb HD2.4 (34, 35) or EA4.1 (34) in TBS, overnight at 4 °C. The unreacted sites in the wells were blocked with TBS containing 0.5% gelatin for 45 min at room temperature. CRP diluted in TBS-Ca was added in duplicate wells. After incubating the plates for 2 h at 37 °C, the wells were washed with TBS-Ca. Rabbit polyclonal anti-CRP antibody (Sigma-Aldrich), diluted in TBS-Ca, was used (1 h at 37 °C) to detect bound CRP. HRP-conjugated donkey anti-rabbit IgG (Thermo), diluted in TBS-Ca, was used (1 h at 37 °C) as secondary antibody. Color was developed, and the A405 was read in a microtiter plate reader.
Pneumococci
S. pneumoniae type 3, strain WU2, were made virulent by sequential intravenous passages in mice and were stored in aliquots at −80 °C in Todd-Hewitt broth containing 0.5% yeast extract and 10% glycerol, as described previously (7, 28). For each experiment, a separate aliquot of frozen pneumococci was thawed. Pneumococci were then grown in Todd-Hewitt broth containing 0.5% yeast extract and collected from mid-log phase cultures. Pneumococci were washed and resuspended in normal saline (A600 = 0.35 = 2.5 × 108 cfu/ml). The concentration, purity, and viability of pneumococci were confirmed by plating on sheep blood agar plates.
Pneumococci Binding Assay
Microtiter wells (96-well plates) were coated with pneumococci in TBS (107 cfu/100 μl/well) overnight at 4 °C. The unreacted sites in the wells were blocked with TBS containing 0.5% gelatin for 45 min at room temperature. CRP diluted in TBS-Ca was added in duplicate wells. After incubating the plates for 2 h at 37 °C, the wells were washed with TBS-Ca. The assays were performed in duplicate plates. The plates were then processed exactly as described for the PCh binding assay.
The binding of CRP to pneumococci in the fluid phase was investigated as follows. Pneumococci (2 × 107 cfu; final concentration 108 cfu/ml) were incubated with CRP (10 μg; final concentration 50 μg/ml) in TBS containing 2 mm CaCl2 and 0.02% Tween 20 at 37 °C in a shaking water bath. After 30 min, pneumococci were pelleted, washed three times with the same buffer, resuspended in TBS, and subjected to denaturing SDS-PAGE.
PEt Binding Assay
Binding activity of CRP for PEt was evaluated by using 1-oleoyl-2(1,2-biotinyl(amidodecanoyl)-sn-glycero-3-phosphoethanolamine (biotinylated PEt) (catalog number 860562C, Avanti Polar Lipids) as the ligand. Stock biotinylated PEt (1 mg/ml in chloroform) was nitrogen-bubbled for 5 min to evaporate chloroform and then air-dried for 1 h at room temperature to evaporate residual chloroform. Biotinylated PEt was resuspended in 1 ml of ethanol, aliquoted, and stored at −20 °C.
Microtiter wells (96-well plates) were coated with 10 μg/ml avidin (catalog number A9275, Sigma-Aldrich) in TBS for 2 h at 37 °C. The unreacted sites in the wells were blocked with TBS containing 0.5% gelatin for 45 min at room temperature. After washing the wells with TBS, biotinylated PEt diluted in TBS (10 μg/ml) was added to the wells for 2 h at 37 °C. After washing the wells with TBS-Ca, CRP diluted in TBS-Ca was added in duplicate wells. After incubating the plates overnight at 4 °C, the wells were washed with TBS-Ca. The assays were performed in duplicate plates. The plates were then processed exactly as described for the PCh binding assay.
Streptavidin could not be used to capture biotinylated PEt on the wells because in preliminary experiments, CRP was found to bind to streptavidin (data not shown). This was not the case with avidin coating (data not shown).
Mice
Male C57BL/6J mice (Jackson ImmunoResearch Laboratories) were brought up and maintained according to protocols approved by the University Committee on Animal Care. Mice were 8–10 weeks old when used in experiments.
Mouse Protection Experiments
Two separate mouse protection experiments were performed using two batches of purified WT and mutant CRP. Mice were first injected intravenously with either 25 μg or 150 μg of WT or mutant CRP in 150 μl of TBS containing 2 mm CaCl2. The endotoxin content in 25 and 150 μg of WT CRP was 0.18 ± 0.09 and 1.08 ± 0.52 endotoxin units, respectively. The endotoxin content in 25 and 150 μg of mutant CRP was 0.16 ± 0.09 and 0.93 ± 0.54 endotoxin units, respectively. After 30 min, mice were injected intravenously with 5 × 107 cfu (based on A600) of pneumococci in 100 μl of saline. The actual number of pneumococci injected, based on the plating results obtained on the next day, was 5.15 ± 0.13 × 107 cfu. Survival of mice was recorded three times per day for 7 days. Survival curves were generated using the GraphPad Prism 4 software. To determine p values for the differences in the survival curves among various groups, the survival curves were compared using the software's log rank test. To determine bacteremia (cfu/ml) in the surviving mice, blood was collected daily for 5 days from the tip of the tail vein, diluted in normal saline, and plated on blood agar plates for colony counting. Bacteremia values for dead mice were taken as >108 cfu/ml because mice died when the bacteremia exceeded 108 cfu/ml. The plotting and statistical analyses of the bacteremia data were done using the GraphPad Prism 4 software and Mann-Whitney two-tailed t test.
Clearance of CRP from Mouse Circulation
Mice were injected intravenously with 100 μg of CRP in TBS containing 2 mm CaCl2 through the tail. Blood was collected from the tip of the tail vein at various times up to 30 h. The concentration of CRP in the serum was measured by ELISA (13). The statistical analyses of the data were performed using the Mann-Whitney two-tailed t test.
Repurification of Mutant CRP from Purified Mutant CRP-spiked Mouse Serum
Purified F66A/T76Y/E81A mutant CRP (400 μg) was added to 2 ml of C57BL/6 mouse serum (Innovative Research), and the final volume was made up to 10 ml by adding 0.1 m borate-buffered saline, pH 8.3, containing 3 mm CaCl2. The mixture was incubated for 30 min at 37 °C. Mutant CRP was repurified by Ca2+-dependent affinity chromatography using PEt-conjugated Sepharose beads whose capacity to bind mutant CRP was >400 μg, as described above. After collecting the flow-through and washing the column with the same buffer, bound CRP was eluted with 0.1 m borate-buffered saline, pH 8.3, containing 5 mm EDTA. To control the experiment, mouse serum alone (2 ml), without spiking with purified mutant CRP, was used. The EDTA eluates were subjected to denaturing SDS-PAGE. The concentration of CRP in the EDTA eluates was measured by ELISA to calculate the percentage of recovery.
RESULTS
Characterization of F66A/T76Y/E81A Mutant CRP
Mutant CRP cDNA was successfully expressed in CHO cells and could be purified by PEt affinity chromatography followed by gel filtration chromatography. The elution profiles of WT CRP and mutant CRP from the gel filtration column were almost overlapping; the elution volume of the mutant CRP was only 250 μl less than that of WT CRP (Fig. 2A). Denaturing SDS-PAGE analysis of purified WT CRP and mutant CRP showed single bands, and the molecular mass of the subunits of mutant CRP was the same as that of WT CRP (Fig. 2B). We also evaluated the epitope for anti-CRP mAb HD2.4, which is a pentameric CRP-specific antibody and whose epitope is located on the face opposite to the PCh-binding face of the CRP pentamer (34, 36). As shown in Fig. 2C, both WT and mutant CRP recognized mAb HD2.4 equally well. These data demonstrated that the substitution of Phe66, Thr76, and Glu81 with Ala, Tyr, and Ala, respectively, did not affect the overall structure of CRP and that mutant CRP was pentameric.
FIGURE 2.

Overall pentameric structure of CRP. A, elution profiles of WT and mutant CRP from the Superose 12 gel filtration chromatography column are shown. WT CRP (1.0 mg) in TBS containing 2 mm CaCl2 was applied to the equilibrated column and eluted with the same buffer. Mutant CRP (0.95 mg) in TBS containing 5 mm EDTA (see “Experimental Procedures”) was applied to the equilibrated column and eluted with the same buffer. Sixty fractions (0.25 ml) were collected, and protein was measured (A280) to determine the elution volume of CRP from the column. A representative of three experiments is shown. B, denaturing SDS-PAGE (4–20% gel) of CRP (5 μg). A representative gel, stained with Coomassie Brilliant Blue, is shown. C, microtiter wells were coated with anti-CRP mAb HD2.4. The unreacted sites in the wells were blocked with gelatin. Purified CRP diluted in TBS-Ca was then added to the wells. Bound CRP was detected by using rabbit polyclonal anti-CRP antibody and HRP-conjugated donkey anti-rabbit IgG. Color was developed, and the absorbance was read at 405 nm. A representative of three experiments is shown.
The PCh binding activity of mutant CRP was assessed by using two different PCh-containing ligands, PCh-BSA and PnC. WT CRP bound to both ligands in a CRP concentration-dependent manner, but mutant CRP bound neither to PCh-BSA (Fig. 3A) nor to PnC (Fig. 3B). We also characterized the PCh-binding site using anti-CRP mAb EA4.1. The binding of EA4.1 mAb to CRP is Ca2+-dependent and can be inhibited by PCh, indicating that EA4.1 binds at or near the PCh-binding site (34). As shown in Fig. 3C, the mutations decreased the binding of CRP to EA4.1 by ∼99%; for equivalent binding to EA4.1, 100 ng/ml mutant CRP was required when compared with 1 ng/ml WT CRP, indicating that the EA4.1-binding epitope, and hence the PCh-binding site, on mutant CRP was lost. We also determined the binding of mutant CRP to whole pneumococci that were used in the mouse protection experiments. In the solid phase pneumococci binding assay, mutant CRP did not bind to whole pneumococci (Fig. 4A). Mutant CRP also did not bind to pneumococci in the fluid phase binding assay (Fig. 4B, compare lanes 4 and 5). Thus, F66A/T76Y/E81A mutant CRP binds neither PCh nor pneumococci.
FIGURE 3.
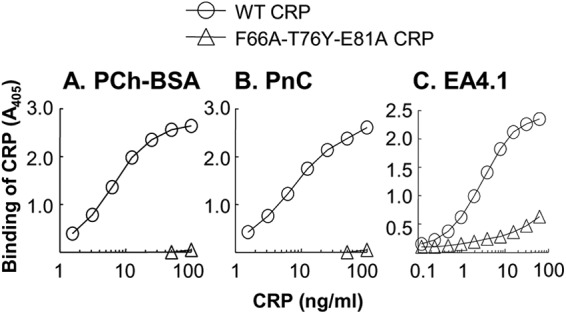
PCh-binding site of CRP. A and B, microtiter wells were coated with PCh-BSA (A) and PnC (B). The unreacted sites in the wells were blocked with gelatin. Purified CRP diluted in TBS-Ca was then added to the wells. Bound CRP was detected by using anti-CRP mAb HD2.4 and HRP-conjugated goat anti-mouse IgG. Color was developed, and the absorbance was read at 405 nm. A representative of four experiments is shown. Similar results were obtained when polyclonal anti-CRP antibody was used to detect ligand-bound CRP (data not shown). C, microtiter wells were coated with anti-CRP mAb EA4.1. The unreacted sites in the wells were blocked with gelatin. Purified CRP diluted in TBS-Ca was then added to the wells. Bound CRP was detected by using rabbit polyclonal anti-CRP antibody and HRP-conjugated donkey anti-rabbit IgG. Color was developed, and the absorbance was read at 405 nm. A representative of three experiments is shown.
FIGURE 4.

Binding of CRP to pneumococci. A, microtiter wells were coated with pneumococci. The unreacted sites in the wells were blocked with gelatin. Purified CRP diluted in TBS-Ca was then added to the wells. Bound CRP was detected by using anti-CRP mAb HD2.4 and HRP-conjugated goat anti-mouse IgG. Color was developed, and the absorbance was read at 405 nm. A representative of four experiments is shown. Similar results were obtained when polyclonal anti-CRP antibody was used to detect ligand-bound CRP (data not shown). B, a representative denaturing SDS-PAGE gel (4–20%) stained with Coomassie Brilliant Blue is shown. Lane 1, purified WT CRP (5 μg); lane 2, purified mutant CRP (5 μg); lane 3, pneumococci alone; lane 4, pneumococci after mixing with WT CRP; lane 5, pneumococci after mixing with mutant CRP.
Because the substitution of Tyr for Thr76 was based on the structure of SAP, and SAP binds to PEt (8–10), we next evaluated the effects of the mutations on the binding of CRP to PEt. Both WT CRP and mutant CRP bound to PEt in a CRP concentration-dependent manner (Fig. 5). However, mutant CRP was much more efficient than WT CRP in binding to PEt. For equivalent binding to PEt, 10 μg/ml WT CRP was required when compared with 170 ng/ml mutant CRP. By repeating the PEt binding assays four times, we found that ∼98% less mutant CRP was required when compared with WT CRP for equivalent binding to PEt. Thus, F66A/T76Y/E81A mutant CRP binds to PEt more avidly than WT CRP does.
FIGURE 5.

Binding of CRP to PEt. Microtiter wells were coated with avidin in TBS. The unreacted sites in the wells were blocked with gelatin. Biotinylated PEt diluted in TBS was then added to the wells. After washing the wells with TBS, purified CRP diluted in TBS-Ca was added to the wells. Bound CRP was detected by using anti-CRP mAb HD2.4 and HRP-conjugated goat anti-mouse IgG. Color was developed, and the absorbance was read at 405 nm. A representative of four experiments is shown. Similar results were obtained when polyclonal anti-CRP antibody was used to detect PEt-bound CRP (data not shown).
In the ligand binding assays (Figs. 3, A and B, 4A, and 5), similar results were obtained irrespective of which anti-CRP antibody, the anti-CRP mAb HD2.4 (data shown) or polyclonal anti-CRP antibody (data not shown), was used to detect ligand-bound CRP. Because mAb HD2.4 is a pentameric CRP-specific antibody (34, 36), the ligand binding data further suggested that the overall structure of mutant CRP was not different from that of WT CRP.
F66A/T76Y/E81A Mutant CRP Does Not Protect Mice from Pneumococcal Infection
Fig. 6A shows the combined results of two separate protection experiments using 25 μg of CRP and six mice in each group in each experiment. The median survival time (the time taken for the death of 50% of mice) for mice injected with bacteria alone (control group A) was 40 h. The median survival time for mice injected with bacteria and WT CRP (group B) was 88 h. The median survival time for mice injected with bacteria and mutant CRP (group C) was 44 h. In the WT CRP-treated group, no deaths occurred in 44 h, and 25% of mice survived up to 7 days. By the end of the 3rd day, all mice had died in the mutant CRP-treated group, whereas the survival was 58% in the WT CRP-treated group. Fig. 6B shows the combined results of two separate protection experiments using 150 μg of CRP and six mice in each group in each experiment. The median survival time for mice injected with bacteria and WT CRP (group D) was 92 h. The median survival time for mice injected with bacteria and mutant CRP (group E) was 46 h. In the WT CRP-treated group, no deaths occurred in 46 h, and 17% of mice survived up to 7 days. By the end of the 3rd day, 75% of mice survived in the WT CRP-treated group when compared with only 25% in the mutant CRP-treated group. By the end of the 4th day, all mice died in the mutant CRP-treated group, whereas the survival was 25% in the WT CRP-treated group. Thus, similar results were obtained when mice were given either 25 μg of CRP or 150 μg of CRP; in contrast to WT CRP, mutant CRP did not decrease mortality and did not prolong survival of infected mice. Because 25 μg of WT CRP was protective and mutant CRP was not protective even when used at 150 μg, that is, at six times greater than the protective dose, we conclude that CRP-mediated protection of mice from infection is dependent upon the PCh binding activity of CRP.
FIGURE 6.

Survival curves of mice infected with S. pneumoniae with or without 25 and 150 μg of either WT or mutant CRP. CRP was injected first; bacteria (5 × 107 cfu) were injected 30 min later. Deaths were recorded three times a day for 7 days. The data are combined from two separate experiments with six mice in each group in both experiments. The p values for the difference in the survival curves between groups A and B, A and C, B and C, A and D, A and E, and D and E are <0.005, 0.79, <0.005, <0.005, 0.31, and <0.005, respectively.
Fig. 7 shows the bacteremia values in mice from the protection experiment shown in Fig. 6. Based on the median bacteremia values, in mice injected with bacteria alone (group A), 1 day after infection, bacteremia was ∼5.8 × 104 cfu/ml blood. In mice injected with bacteria and 25 μg of WT CRP (group B), bacteremia was 8.4 × 102 cfu/ml 1 day after infection. In mice injected with bacteria and 150 μg of WT CRP (group D), bacteremia was 2.5 × 103 cfu/ml 1 day after infection. However, in mice injected with bacteria and 25 μg of mutant CRP (group C), 1 day after infection, bacteremia was 5.9 × 105 cfu/ml. In mice injected with bacteria and 150 μg of mutant CRP (group E), 1 day after infection, bacteremia was 2.3 × 105 cfu/ml. In groups A, C, and E, bacteremia increased dramatically after day 1, and mice died once bacteremia exceeded 108 cfu/ml. In mice administered with WT CRP, there was an increase in bacteremia past day 1, but it took another 2 days to exceed 108 cfu/ml when those mice died, when compared with <1 day for the WT CRP-treated group. Statistically significant differences in bacteremia were observed between the control (group A) and the WT CRP-treated groups (groups B and D) and between the WT CRP-treated and the mutant CRP-treated groups (groups C and E), until day 2. These results indicated that the increased resistance to infection in WT CRP-treated mice was associated with a slower increase in bacteremia and that the PCh binding activity of CRP was critical in this process.
FIGURE 7.

Bacteremia in mice treated with or without 25 and 150 μg of either WT or mutant CRP. Blood samples were collected from each surviving mouse shown in Fig. 6 for the first 5 days after infection. Bacteremia was determined by plating. Each dot represents one mouse. The horizontal line in each group of mice represents the median value of bacteremia in that group. A bacteremia value of >108 indicates a dead mouse. The p values for the difference between groups A and B, A and C, B and C, A and D, A and E, and D and E, on day 1, are 0.011, 0.74, 0.025, <0.005, 0.09, and 0.17, respectively. The p values for the difference between groups A and B, A and C, B and C, A and D, A and E, and D and E, on day 2, are <0.005, 0.72, <0.005, <0.005, 0.39, and 0.02, respectively.
F66A/T76Y/E81A Mutant CRP Stays Free in the Mouse Serum, and Its Clearance Rate in Vivo Is Not Faster than That of WT CRP
Although mutant CRP did not protect mice from infection even when used at an amount that was six times higher than the protective dose for WT CRP, we determined the rate of clearance of mutant CRP from mouse circulation and compared it with that of WT CRP to confirm that mutant CRP was not protective because of its inability to bind pneumococci and not because of its faster clearance in vivo. The average slope of the four clearance curves for WT CRP was −0.67 ± 0.02 (Fig. 8A), and the average slope of the four clearance curves for mutant CRP was −0.33 ± 0.03 (Fig. 8B). Although the rate of clearance of mutant CRP (0.33 μg/ml/h) was significantly different (p = 0.03) from that of WT CRP (0.67 μg/ml/h), the clearance of mutant CRP was not faster than that of WT CRP. These data suggested that the mutations did not confer instability to mutant CRP in vivo.
FIGURE 8.

Clearance of CRP from mouse circulation. Mice were injected with 100 μg of CRP in TBS containing 2 mm CaCl2. Blood was collected at various time points, sera were separated, and the concentration of CRP was measured.
Although the clearance data also suggested that mutant CRP was free in the circulation and available for functions because it reacted with anti-CRP mAb HD2.4, we used another approach to confirm that mutant CRP was free in the mouse serum and was not sequestered by any other serum protein. As shown in Fig. 9, mutant CRP present in the mouse serum bound to PEt in a Ca2+-dependent manner and could be eluted with EDTA (lane 3). The recovery of mutant CRP was 96%. Besides CRP, no additional protein bands were found when compared with the nonspecific bands seen with the serum alone control (compare lanes 3 and 4). Successful repurification of mutant CRP from mutant CRP-spiked mouse serum further suggested that mutant CRP was free in the mouse serum and was not sequestered by any other serum protein.
FIGURE 9.

Repurification of mutant CRP from purified mutant CRP-spiked mouse serum. A representative denaturing SDS-PAGE gel (4–20%) stained with Coomassie Brilliant Blue is shown. Lane 1, molecular mass markers; lane 2, purified mutant CRP (5 μg); lane 3, EDTA eluate (25 μl, A280 1.13) from the PEt affinity chromatography column through which mouse serum containing mutant CRP was passed in the presence of CaCl2; lane 4, EDTA eluate (25 μl, A280 0.29) from the PEt affinity chromatography column through which mouse serum alone was passed in the presence of CaCl2.
DISCUSSION
In this study, we tested the hypothesis that the CRP-mediated protection of mice from pneumococcal infection is dependent upon the PCh binding activity of CRP. We mutated three amino acids in the PCh-binding pocket of CRP to generate a CRP mutant incapable of binding to PCh. We then compared the protective ability of WT CRP with that of mutant CRP. Our major findings were as follows. 1) Substitution of Phe66, Thr76, and Glu81 with Ala, Tyr, and Ala, respectively, abolished the PCh binding, PnC binding, and pneumococcus binding activity of CRP. 2) F66A/T76Y/E81A mutant CRP was more efficient than WT CRP in binding to PEt. 3) Mutations in the PCh-binding pocket of CRP did not affect the overall pentameric structure of CRP. 4) F66A/T76Y/E81A mutant CRP stayed free in the mouse serum, and its clearance rate in vivo was not faster than that of WT CRP. 5) At both 25 μg and 150 μg, F66A/T76Y/E81A mutant CRP did not protect mice from pneumococcal infection.
Previously, we tested the same hypothesis that the CRP-mediated protection of mice from pneumococcal infection is dependent upon the PCh binding activity of CRP (7). The CRP mutant used in the earlier investigation was a CRP double mutant, F66A/E81A, which also does not bind to PCh, PnC, or pneumococci (7, 13). We reported that F66A/E81A mutant CRP was as capable as WT CRP in protecting mice from pneumococcal infection, which was a surprising finding (7). This and the following three points prompted us to make another CRP mutant incapable of binding to PCh and repeat the mouse protection experiments. First, we wanted to use a mutant CRP, incapable of binding to PCh, with more drastic changes in the PCh-binding pocket. We wanted to block the pocket. In the earlier F66A/E81A double mutant, although two residues were mutated, the pocket was not blocked. Second, we wanted to use freshly purified CRP in all experiments. Third, because we were now successful in establishing a mouse model of pneumococcal infection in which 25 μg of CRP was protective, as has been shown by others (25, 26), we wanted to use two different doses of CRP (25 and 150 μg) in protection experiments. Previously (7), we only used one dose of CRP, 150 μg.
As shown in Fig. 1A, Phe66, Thr76, and Glu81 participate in the CRP-PCh interaction (16). To generate a CRP mutant incapable of binding to PCh, our mutagenesis plan included mutating all three amino acids. Based on the structure of SAP (18), we mutated Thr to Tyr and added the T76Y mutation to our previously published CRP mutant, F66A/E81A. Molecular modeling of F66A/T76Y/E81A CRP triple mutant showed that the large side chain of Tyr could partially block the PCh-binding pocket (Fig. 1B). Loss of binding of mutant CRP to PCh, but dramatically enhanced binding to PEt, further indicated that the PCh-binding pocket was blocked in mutant CRP. In WT CRP, the amino group of PEt makes an electrostatic interaction with Glu81 (10). In SAP, the amino group of PEt is situated within H-bonding distance of the Tyr74 hydroxyl group; such H-bonding is not possible for WT CRP as the corresponding residue is Thr (10). It has also been shown that SAP binds to immobilized PEt much more avidly than CRP does, although the Kd values of CRP and SAP for PEt are similar (10). In mutant CRP, the presence of Tyr at position 76 might have facilitated H-bonding between the amino group of PEt and hydroxyl group of Tyr. Mutation of Glu81 might have further facilitated this interaction and increased the avidity of the binding of mutant CRP to PEt. Also, due to stronger avidity of mutant CRP for PEt, it was easier to purify mutant CRP by PEt affinity chromatography. Interestingly, like SAP (9, 10, 37, 38), mutant CRP exhibited carbohydrate binding property; in the presence of Ca2+, mutant CRP bound to agarose beads used for gel filtration chromatography (data not shown).
CRP binds to FcγR (39–41), and FcγR have been implicated in protection from pneumococcal infection in mice (42). CRP has been shown to enhance uptake and presentation of pneumococcal antigens through FcγR on dendritic cells in vitro (43). However, we did not characterize mutant CRP for binding to FcγR because FcγR do not participate in CRP-mediated protection of mice against pneumococcal infection (44). CRP was found to be equally protective in WT and FcγR knock-out mice (44). We did not characterize mutant CRP for activating the complement system because CRP activates complement only when ligand-complexed (30), but mutant CRP was unable to complex with PCh-BSA, PnC, or pneumococci.
We used a high dose, 5 × 107 cfu, of pneumococci in mouse protection experiments. This dose of bacteria was necessary because endogenous mouse CRP might have been protective against lower doses of bacteria. It has been reported that endogenous mouse CRP is also protective against pneumococcal infection in mice; the LD50 of pneumococci was drastically reduced in CRP KO mice (45). We used 25 and 150 μg of CRP in mouse protection experiments; however, we also found that even 10 μg of WT CRP was protective (data not shown), as has been reported earlier (24–26). Our finding that WT CRP was protective, whereas mutant CRP was not protective even at a 150-μg dose, clearly indicated that the PCh-binding pocket in CRP was critical for the initial protection of mice from pneumococcal infection. Our findings are consistent with other studies that showed that the PCh on pneumococci mediated the function of CRP to block the attachment of pneumococci to platelet-activating factor receptors on human pharyngeal epithelial cells and that the PCh binding activity of CRP was required for the protection of mice from challenge with platelet-activating factor (46, 47).
Because CRP protects mice from infection only when injected within the range of 6 h before to 2 h after administering pneumococci, and not when injected 24 or 36 h after infection (7, 26), our data suggest that CRP is able to use the PCh binding-based mechanism for the protection of mice only during the early stages of infection. It has been shown that WT CRP undergoes structural transformations under several different experimental conditions (3, 33, 48–53). It has also been shown that structurally altered, or slightly to completely denatured, CRP is capable of binding to immobilized factor H (3, 33, 51–56). Factor H is a regulator of complement activation and has been implicated in the resistance of pneumococci to complement attack (3, 57–59). It remains to be investigated whether such CRP-factor H interaction plays any role in CRP-mediated protection of mice from pneumococcal infection. However, the combined data indicate that the CRP-mediated protection of mice requires the presence of CRP during the early stages of infection.
The outcome of the in vivo experiments obtained with the use of CRP triple mutant in this investigation was different from the results obtained with the use of CRP double mutant reported earlier (7). Both mutants fail to bind to PCh, PnC, or pneumococci. The only difference in the ligand binding properties of the two mutants was their binding avidity for PEt; the binding of CRP double mutant to PEt was not different from that of WT CRP (data not shown). However, there were two major differences in the methods used in the two in vivo experiments. The first difference was in the purity of CRP. Unlike CRP double mutant, CRP triple mutant was freshly purified and never stored frozen. Whether the difference in the results of the two in vivo experiments was due to the difference in the structures of the PCh-binding pocket of the two CRP mutants, or due to the difference in the ages of purified CRP, remains to be explored. The second difference was in the infection models, regarding the protective ability of WT CRP. In the infection model used earlier, <100 μg of WT CRP was not protective and, therefore, we had to use 150 μg of both WT and double mutant CRP (7). In the infection model used in the current study, even <25 μg of WT CRP was protective, and thus, we could use two doses, 25 and 150 μg, of WT and triple mutant CRP. The data shown in Fig. 6A with the use of 25 μg of CRP clearly indicated that the PCh-binding pocket was necessary and required for protection of mice during the initial stage of infection. Statistical analysis of the data shown in Fig. 6B with the use of 150 μg of CRP showed that even 150 μg of CRP triple mutant was not protective. However, although statistically not significant, there was a slight protective effect of CRP triple mutant when used at 150 μg, raising the possibility that >150 μg of CRP triple mutant might have produced results similar to those obtained with 150 μg of CRP double mutant reported earlier (7). Alternatively, 25 μg of CRP double mutant might not have been protective as opposed to 150 μg. Because administering a higher dose of CRP allows CRP to stay longer in the host, we hypothesize that CRP mutants, when given at the beginning of the experiment, undergo remodeling to gain other ligand binding properties, as yet undefined, during their stay in the host, and that may be responsible for the protection of mice from infection independent of the PCh binding activity of CRP. This hypothesis also provides insight into why CRP does not protect mice from infection if administered later and how CRP can be engineered so that it can protect mice from infection even if administered during the late stages of infection (33, 51). However, this is just a hypothesis because we have no data.
We conclude that the PCh-binding pocket on CRP is necessary for CRP-mediated initial protection of mice against lethal pneumococcal infection. Development of infection models involving passively administered human WT and mutant CRP in CRP KO mice (45, 60–62) and SAP KO mice (63–65), and also the development of mice transgenic for CRP mutants, may provide more information on the mechanisms of antipneumococcal function of CRP in protecting mice against early and late stages of infection.
Acknowledgments
We thank Dr. David E. Briles (University of Alabama at Birmingham, Birmingham, AL) for the gift of S. pneumoniae, Dr. John E. Volanakis (University of Alabama at Birmingham) for the gift of mAb EA4.1, Dr. Darrel Pilling (Texas A&M University, Houston, TX) for the protocol for preparing PEt-Sepharose, Dr. Scott M. Reed (University of Colorado, Denver, CO) for suggestions regarding the use of biotinylated PEt, Dr. Gregory A. Hanley of our Division of Laboratory and Animal Resources for help with the mouse experiments, and Avinash Thirumalai from our laboratory for help with the statistical analysis of some data.
This work was supported, in whole or in part, by National Institutes of Health Grant R01HL071233 (to A. A.).
- CRP
- C-reactive protein
- PnC
- pneumococcal C-polysaccharide
- PCh
- phosphocholine
- PEt
- phosphoethanolamine
- SAP
- serum amyloid P
- TBS-Ca
- TBS, pH 7.2, containing 5 mm CaCl2, 0.1% gelatin, and 0.02% Tween 20
- FcγR
- Fcγ receptor.
REFERENCES
- 1. Kushner I., Samols D. (2011) Oswald Avery and the pneumococcus. Pharos Alpha Omega Alpha Honor Med. Soc. 74, 14–18 [PubMed] [Google Scholar]
- 2. Abernethy T. J., Avery O. T. (1941) The occurrence during acute infections of a protein not normally present in the blood. I. Distribution of the reactive protein in patients' sera and the effect of calcium on the flocculation reaction with C polysaccharide of pneumococcus. J. Exp. Med. 73, 173–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Agrawal A., Suresh M. V., Singh S. K., Ferguson D. A., Jr. (2008) The protective function of human C-reactive protein in mouse models of Streptococcus pneumoniae infection. Endocr. Metab. Immune Disord. Drug Targets 8, 231–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Volanakis J. E., Kaplan M. H. (1971) Specificity of C-reactive protein for choline phosphate residues of pneumococcal C-polysaccharide. Proc. Soc. Exp. Biol. Med. 136, 612–614 [DOI] [PubMed] [Google Scholar]
- 5. Mold C., Rodgers C. P., Kaplan R. L., Gewurz H. (1982) Binding of human C-reactive protein to bacteria. Infect. Immun. 38, 392–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mold C., Edwards K. M., Gewurz H. (1982) Effect of C-reactive protein on the complement-mediated stimulation of human neutrophils by Streptococcus pneumoniae serotypes 3 and 6. Infect. Immun. 37, 987–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Suresh M. V., Singh S. K., Ferguson D. A., Jr., Agrawal A. (2007) Human C-reactive protein protects mice from Streptococcus pneumoniae infection without binding to pneumococcal C-polysaccharide. J. Immunol. 178, 1158–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schwalbe R. A., Dahlbäck B., Coe J. E., Nelsestuen G. L. (1992) Pentraxin family of proteins interacts specifically with phosphorylcholine and/or phosphorylethanolamine. Biochemistry 31, 4907–4915 [DOI] [PubMed] [Google Scholar]
- 9. Christner R. B., Mortensen R. F. (1994) Binding of human serum amyloid P-component to phosphocholine. Arch. Biochem. Biophys. 314, 337–343 [DOI] [PubMed] [Google Scholar]
- 10. Mikolajek H., Kolstoe S. E., Pye V. E., Mangione P., Pepys M. B., Wood S. P. (2011) Structural basis of ligand specificity in the human pentraxins, C-reactive protein, and serum amyloid P component. J. Mol. Recognit. 24, 371–377 [DOI] [PubMed] [Google Scholar]
- 11. Anderson J. K., Stroud R. M., Volanakis J. E. (1978) Studies on the binding specificity of human C-reactive protein for phosphorylcholine. Fed. Proc. 37, 1495 [Google Scholar]
- 12. Oliveira E. B., Gotschlich E. C., Liu T.-Y. (1980) Comparative studies on the binding properties of human and rabbit C-reactive proteins. J. Immunol. 124, 1396–1402 [PubMed] [Google Scholar]
- 13. Agrawal A., Simpson M. J., Black S., Carey M.-P., Samols D. (2002) A C-reactive protein mutant that does not bind to phosphocholine and pneumococcal C-polysaccharide. J. Immunol. 169, 3217–3222 [DOI] [PubMed] [Google Scholar]
- 14. Shrive A. K., Cheetham G. M., Holden D., Myles D. A., Turnell W. G., Volanakis J. E., Pepys M. B., Bloomer A. C., Greenhough T. J. (1996) Three-dimensional structure of human C-reactive protein. Nat. Struct. Biol. 3, 346–354 [DOI] [PubMed] [Google Scholar]
- 15. Roux K. H., Kilpatrick J. M., Volanakis J. E., Kearney J. F. (1983) Localization of the phosphocholine-binding sites on C-reactive protein by immunoelectron microscopy. J. Immunol. 131, 2411–2415 [PubMed] [Google Scholar]
- 16. Thompson D., Pepys M. B., Wood S. P. (1999) The physiological structure of human C-reactive protein and its complex with phosphocholine. Structure 7, 169–177 [DOI] [PubMed] [Google Scholar]
- 17. Agrawal A., Lee S., Carson M., Narayana S. V., Greenhough T. J., Volanakis J. E. (1997) Site-directed mutagenesis of the phosphocholine-binding site of human C-reactive protein: Role of Thr76 and Trp67. J. Immunol. 158, 345–350 [PubMed] [Google Scholar]
- 18. Emsley J., White H. E., O'Hara B. P., Oliva G., Srinivasan N., Tickle I. J., Blundell T. L., Pepys M. B., Wood S. P. (1994) Structure of pentameric human serum amyloid P component. Nature 367, 338–345 [DOI] [PubMed] [Google Scholar]
- 19. Srinivasan N., White H. E., Emsley J., Wood S. P., Pepys M. B., Blundell T. L. (1994) Comparative analyses of pentraxins: Implications for protomer assembly and ligand binding. Structure 2, 1017–1027 [DOI] [PubMed] [Google Scholar]
- 20. Tuomanen E. I., Austrian R., Masure H. R. (1995) Pathogenesis of pneumococcal infection. N. Engl. J. Med. 332, 1280–1284 [DOI] [PubMed] [Google Scholar]
- 21. van der Poll T., Opal S. M. (2009) Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet 374, 1543–1556 [DOI] [PubMed] [Google Scholar]
- 22. Malley R., Anderson P. W. (2012) Serotype-independent pneumococcal experimental vaccines that induce cellular as well as humoral immunity. Proc. Natl. Acad. Sci. U.S.A. 109, 3623–3627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Whitehead A. S., Zahedi K., Rits M., Mortensen R. F., Lelias J. M. (1990) Mouse C-reactive protein: Generation of cDNA clones, structural analysis, and induction of mRNA during inflammation. Biochem. J. 266, 283–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mold C., Nakayama S., Holzer T. J., Gewurz H., Du Clos T. W. (1981) C-reactive protein is protective against Streptococcus pneumoniae infection in mice. J. Exp. Med. 154, 1703–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yother J., Volanakis J. E., Briles D. E. (1982) Human C-reactive protein is protective against fatal Streptococcus pneumoniae infection in mice. J. Immunol. 128, 2374–2376 [PubMed] [Google Scholar]
- 26. Nakayama S., Gewurz H., Holzer T., Du Clos T. W., Mold C. (1983) The role of the spleen in the protective effect of C-reactive protein in Streptococcus pneumoniae infection. Clin. Exp. Immunol. 54, 319–326 [PMC free article] [PubMed] [Google Scholar]
- 27. Szalai A. J., Briles D. E., Volanakis J. E. (1995) Human C-reactive protein is protective against fatal Streptococcus pneumoniae infection in transgenic mice. J. Immunol. 155, 2557–2563 [PubMed] [Google Scholar]
- 28. Suresh M. V., Singh S. K., Ferguson D. A., Jr., Agrawal A. (2006) Role of the property of C-reactive protein to activate the classical pathway of complement in protecting mice from Streptococcus pneumoniae infection. J. Immunol. 176, 4369–4374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Horowitz J., Volanakis J. E., Briles D. E. (1987) Blood clearance of Streptococcus pneumoniae by C-reactive protein. J. Immunol. 138, 2598–2603 [PubMed] [Google Scholar]
- 30. Volanakis J. E. (1982) Complement activation by C-reactive protein complexes. Ann. N.Y. Acad. Sci. 389, 235–250 [DOI] [PubMed] [Google Scholar]
- 31. Volanakis J. E. (2001) Human C-reactive protein: Expression, structure, and function. Mol. Immunol. 38, 189–197 [DOI] [PubMed] [Google Scholar]
- 32. Pilling D., Roife D., Wang M., Ronkainen S. D., Crawford J. R., Travis E. L., Gomer R. H. (2007) Reduction of bleomycin-induced pulmonary fibrosis by serum amyloid P. J. Immunol. 179, 4035–4044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hammond D. J., Jr., Singh S. K., Thompson J. A., Beeler B. W., Rusiñol A. E., Pangburn M. K., Potempa L. A., Agrawal A. (2010) Identification of acidic pH-dependent ligands of pentameric C-reactive protein. J. Biol. Chem. 285, 36235–36244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kilpatrick J. M., Kearney J. F., Volanakis J. E. (1982) Demonstration of calcium-induced conformational change(s) in C-reactive protein by using monoclonal antibodies. Mol. Immunol. 19, 1159–1165 [DOI] [PubMed] [Google Scholar]
- 35. Suresh M. V., Singh S. K., Agrawal A. (2004) Interaction of calcium-bound C-reactive protein with fibronectin is controlled by pH: In vivo implications. J. Biol. Chem. 279, 52552–52557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ying S.-C., Gewurz H., Kinoshita C. M., Potempa L. A., Siegel J. N. (1989) Identification and partial characterization of multiple native and neoantigenic epitopes of human C-reactive protein by using monoclonal antibodies. J. Immunol. 143, 221–228 [PubMed] [Google Scholar]
- 37. Hind C. R., Collins P. M., Renn D., Cook R. B., Caspi D., Baltz M. L., Pepys M. B. (1984) Binding specificity of serum amyloid P component for the pyruvate acetal of galactose. J. Exp. Med. 159, 1058–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Loveless R. W., Floyd-O'Sullivan G., Raynes J. G., Yuen C.-T., Feizi T. (1992) Human serum amyloid P is a multispecific adhesive protein whose ligands include 6-phosphorylated mannose and the 3-sulphated saccharides galactose, N-acetylgalactosamine and glucuronic acid. EMBO J. 11, 813–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bharadwaj D., Stein M.-P., Volzer M., Mold C., Du Clos T. W. (1999) The major receptor for C-reactive protein on leukocytes is Fcγ receptor II. J. Exp. Med. 190, 585–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chi M., Tridandapani S., Zhong W., Coggeshall K. M., Mortensen R. F. (2002) C-reactive protein induces signaling through FcγRIIa on HL-60 granulocytes. J. Immunol. 168, 1413–1418 [DOI] [PubMed] [Google Scholar]
- 41. Bang R., Marnell L., Mold C., Stein M.-P., Du Clos K. T., Chivington-Buck C., Du Clos T. W. (2005) Analysis of binding sites in human C-reactive protein for FcγRI, FcγRIIA, and C1q by site-directed mutagenesis. J. Biol. Chem. 280, 25095–25102 [DOI] [PubMed] [Google Scholar]
- 42. Bitsaktsis C., Iglesias B. V., Li Y., Colino J., Snapper C. M., Hollingshead S. K., Pham G., Gosselin D. R., Gosselin E. J. (2012) Mucosal immunization with an unadjuvanted vaccine that targets Streptococcus pneumoniae PspA to human Fcγ receptor type I protects against pneumococcal infection through complement- and lactoferrin-mediated bactericidal activity. Infect. Immun. 80, 1166–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thomas-Rudolph D., Du Clos T. W., Snapper C. M., Mold C. (2007) C-reactive protein enhances immunity to Streptococcus pneumoniae by targeting uptake to FcγR on dendritic cells. J. Immunol. 178, 7283–7291 [DOI] [PubMed] [Google Scholar]
- 44. Mold C., Rodic-Polic B., Du Clos T. W. (2002) Protection from Streptococcus pneumoniae infection by C-reactive protein and natural antibody requires complement but not Fcγ receptors. J. Immunol. 168, 6375–6381 [DOI] [PubMed] [Google Scholar]
- 45. Loeffler J. M., Simons J. P., Al-Shawi R. A., Hutchinson W. L., Mangione P., Pepys M. B. (2009) C-reactive protein is essential for host resistance to pneumococci. Q. J. Med. 102, 644 [Google Scholar]
- 46. Gould J. M., Weiser J. N. (2002) The inhibitory effect of C-reactive protein on bacterial phosphorylcholine platelet-activating factor receptor-mediated adherence is blocked by surfactant. J. Infect. Dis. 186, 361–371 [DOI] [PubMed] [Google Scholar]
- 47. Black S., Wilson A., Samols D. (2005) An intact phosphocholine binding site is necessary for transgenic rabbit C-reactive protein to protect mice against challenge with platelet-activating factor. J. Immunol. 175, 1192–1196 [DOI] [PubMed] [Google Scholar]
- 48. Chudwin D. S., Artrip S. G., Korenblit A., Schiffman G., Rao S. (1985) Correlation of serum opsonins with in vitro phagocytosis of Streptococcus pneumoniae. Infect. Immun. 50, 213–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang M.-Y., Ji S.-R., Bai C.-J., El Kebir D., Li H.-Y., Shi J.-M., Zhu W., Costantino S., Zhou H.-H., Potempa L. A., Zhao J., Filep J. G., Wu Y. (2011) A redox switch in C-reactive protein modulates activation of endothelial cells. FASEB J. 25, 3186–3196 [DOI] [PubMed] [Google Scholar]
- 50. Zhang J., Yang L., Anand G. S., Ho B., Ding J. L. (2011) Pathophysiological condition changes the conformation of a flexible FBG-related protein, switching it from pathogen-recognition to host-interaction. Biochimie 93, 1710–1719 [DOI] [PubMed] [Google Scholar]
- 51. Singh S. K., Thirumalai A., Hammond D. J., Jr., Pangburn M. K., Mishra V. K., Johnson D. A., Rusiñol A. E., Agrawal A. (2012) Exposing a hidden functional site of C-reactive protein by site-directed mutagenesis. J. Biol. Chem. 287, 3550–3558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Okemefuna A. I., Nan R., Miller A., Gor J., Perkins S. J. (2010) Complement factor H binds at two independent sites to C-reactive protein in acute phase concentrations. J. Biol. Chem. 285, 1053–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hakobyan S., Harris C. L., van den Berg C. W., Fernandez-Alonso M. C., de Jorge E. G., de Cordoba S. R., Rivas G., Mangione P., Pepys M. B., Morgan B. P. (2008) Complement factor H binds to denatured rather than to native pentameric C-reactive protein. J. Biol. Chem. 283, 30451–30460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mold C., Kingzette M., Gewurz H. (1984) C-reactive protein inhibits pneumococcal activation of the alternative pathway by increasing the interaction between factor H and C3b. J. Immunol. 133, 882–885 [PubMed] [Google Scholar]
- 55. Sjöberg A. P., Trouw L. A., Clark S. J., Sjölander J., Heinegård D., Sim R. B., Day A. J., Blom A. M. (2007) The factor H variant associated with age-related macular degeneration (His-384) and the non-disease-associated form bind differentially to C-reactive protein, fibromodulin, DNA, and necrotic cells. J. Biol. Chem. 282, 10894–10900 [DOI] [PubMed] [Google Scholar]
- 56. Deban L., Jarva H., Lehtinen M. J., Bottazzi B., Bastone A., Doni A., Jokiranta T. S., Mantovani A., Meri S. (2008) Binding of the long pentraxin PTX3 to factor H: Interacting domains and function in the regulation of complement activation. J. Immunol. 181, 8433–8440 [DOI] [PubMed] [Google Scholar]
- 57. Zipfel P. F., Hallström T., Hammerschmidt S., Skerka C. (2008) The complement fitness factor H: Role in human diseases and for immune escape of pathogens, like pneumococci. Vaccine 26, I67-I74 [DOI] [PubMed] [Google Scholar]
- 58. Ferreira V. P., Pangburn M. K., Cortés C. (2010) Complement control protein factor H: The good, the bad, and the inadequate. Mol. Immunol. 47, 2187–2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kopp A., Hebecker M., Svobodová E., Józsi M. (2012) Factor H: A complement regulator in health and disease, and a mediator of cellular interactions. Biomolecules 2, 46–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jones N. R., Pegues M. A., McCrory M. A., Kerr S. W., Jiang H., Sellati R., Berger V., Villalona J., Parikh R., McFarland M., Pantages L., Madwed J. B., Szalai A. J. (2011) Collagen-induced arthritis is exacerbated in C-reactive protein-deficient mice. Arthritis Rheum. 63, 2641–2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Teupser D., Weber O., Rao T. N., Sass K., Thiery J., Fehling H. J. (2011) No reduction of atherosclerosis in C-reactive protein (CRP)-deficient mice. J. Biol. Chem. 286, 6272–6279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rutemark C., Alicot E., Bergman A., Ma M., Getahun A., Ellmerich S., Carroll M. C., Heyman B. (2011) Requirement for complement in antibody responses is not explained by the classic pathway activator IgM. Proc. Natl. Acad. Sci. U.S.A. 108, E934-E942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Togashi S., Lim S.-K., Kawano H., Ito S., Ishihara T., Okada Y., Nakano S., Kinoshita T., Horie K., Episkopou V., Gottesman M. E., Costantini F., Shimada K., Maeda S. (1997) Serum amyloid P component enhances induction of murine amyloidosis. Lab. Invest. 77, 525–531 [PubMed] [Google Scholar]
- 64. Noursadeghi M., Bickerstaff M. C., Gallimore J. R., Herbert J., Cohen J., Pepys M. B. (2000) Role of serum amyloid P component in bacterial infection: Protection of the host or protection of the pathogen. Proc. Natl. Acad. Sci. U.S.A. 97, 14584–14589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yuste J., Botto M., Bottoms S. E., Brown J. S. (2007) Serum amyloid P aids complement-mediated immunity to Streptococcus pneumoniae. PLoS Pathog. 3, 1208–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.3r1, Schrödinger, LLC, New York [Google Scholar]