

Abstract
Telomere shortening is associated with cellular senescence and aging. Dyskeratosis congenita (DC) is a premature aging syndrome caused by mutations in genes for telomerase components or telomere proteins. DC patients have very short telomeres and exhibit aging-associated pathologies including epidermal abnormalities and bone marrow failure. Here, we show that DC skin fibroblasts are defective in their ability to support the clonogenic growth of epidermal keratinocytes. Conditioned media transfer experiments demonstrated that this defect was largely due to lack of a factor or factors secreted from the DC fibroblasts. Compared to early passage normal fibroblasts, DC fibroblasts express significantly lower transcript levels of several genes that code for secreted proteins, including Insulin-like Growth Factor 1 (IGF1) and Hepatocyte Growth Factor (HGF). Aged normal fibroblasts with short telomeres also had reduced levels of IGF1 and HGF, similar to early passage DC fibroblasts. Knockdown of IGF1 or HGF in normal fibroblasts caused a reduction in the capacity of conditioned media from these fibroblasts to support keratinocyte clonogenic growth. Surprisingly, reconstitution of telomerase in DC fibroblasts did not significantly increase transcript levels of IGF1 or HGF or substantially increase the ability of the fibroblasts to support keratinocyte growth, indicating that the gene expression defect is not readily reversible. Our results suggest that telomere shortening in dermal fibroblasts leads to reduction in expression of genes such as IGF1 and HGF and that this may cause a defect in supporting normal epidermal proliferation.
Keywords: Telomerase, telomeres, IGF1, HGF, dyskeratosis, skin
The aging of skin is a complex process involving interplay between different cell types that comprise the dermis and epidermis that is further regulated by hormonal factors (1). The shortening of telomeres has been demonstrated to play an important role in aging, skin maintenance, and other phenotypes associated with aging (2). In vertebrates, telomeres consist of thousands of copies of the TTAGGG repeat plus associated proteins in what is referred to as the shelterin complex (3). The shelterin complex protects the telomere end and prevents the cellular machinery from recognizing the telomere as a double-strand break (4). Telomeres shorten during cell proliferation due to the end-replication problem, which occurs because DNA polymerase cannot completely replicate new 5′ ends (5). Accelerated telomere shortening can also occur through DNA damage including that caused by reactive oxygen species (ROS) (6). When telomeres shorten, the shelterin complex is disrupted and the telomeres become dysfunctional, leading to cellular senescence or genetic instability (4). Telomeres can be maintained by the enzyme telomerase which adds telomere repeats back to the chromosome end (7, 8). Telomerase is active in germline cells and in greater than 90% of cancers (9, 10). It is also active, but at lower levels, in some somatic cells such as lymphocytes and the basal cells of the epidermis (11–14). Telomerase activity might provide some measure of telomere maintenance in these normal cells, though telomere shortening is still a natural consequence of the aging process (13, 15, 16).
Studies in mice have provided insight into how telomere shortening is involved in aging. Most inbred strains of mice have extremely long telomeres. Knockout of telomerase genes in these mice leads to telomere shortening and, after 3 to 5 generations, defects in highly proliferative tissues such as bone marrow and skin (e.g. early hair loss, alopecia, defects in wound healing) (17, 18). Mice that have been engineered to overexpress TERT (the reverse transcriptase component of telomerase) in their skin have improved wound healing and a thickened epidermis (19, 20). However, they are more susceptible to skin cancer (19). Transgenic expression of TERT also causes increased epidermal stem cell mobilization and hair follicle stimulation (21, 22). Knockout of proteins that are found in the telomere shelterin complex also leads to epidermal abnormalities, including skin degeneration and impaired hair follicle morphogenesis (23–25). Thus, telomerase and telomeres appear to be important for maintenance of skin function in mice.
The contribution of the dermal component of skin to the function and proliferation of epidermal cells is now being more fully appreciated. Dermal fibroblasts secrete a wide variety of factors that can influence the growth, inflammatory, and wound healing response of keratinocytes (26). In addition, the dermal extracellular matrix (ECM) is essential for skin structure, function, and homeostasis. The aging of fibroblasts significantly affects their phenotype, mostly resulting from global changes in gene expression (27). These changes affect normal epidermal cell proliferation. In addition to effects on normal physiology, numerous studies have indicated that aged fibroblasts secrete factors that promote the growth of transformed epithelial cells and that this may lead to the development of cancer (28–31).
The premature aging syndrome dyskeratosis congenita (DC) is caused by mutations in genes that code for telomerase components or telomere binding proteins (32). Along with a high risk for bone marrow failure and cancer, DC patients also suffer from numerous epidermal abnormalities including dyskeratotic nails, early hair graying and loss, dyspigmented and reticulated skin, poor wound healing, and skin atrophy. We have shown previously that DC T lymphocytes, hematopoietic stem cells, skin fibroblasts, and skin keratinocytes are defective in proliferation (33–37). DC cells provide a relevant human model for studying how telomere shortening affects the function of different tissues. In the current study, we wanted to determine how telomere shortening in dermal fibroblasts affected the growth of epidermal keratinocytes. We found that DC fibroblasts are deficient in a secreted factor or factors that support the clonogenic growth of keratinocytes. Both DC fibroblasts and aged normal fibroblasts exhibited reduction in expression of the genes that code for IGF1 and HGF. Knockdown of these genes in normal fibroblasts also resulted in a reduced ability of the cells to support clonogenic growth of keratinocytes. We propose a link between telomere shortening and defective expression of secreted factors that contribute to epidermal growth and function.
MATERIALS AND METHODS
Cell culture
Human skin keratinocytes (HSK) and fibroblasts (HSF) were isolated as previously described from skin punch biopsies (36–38). DC-HSF-1 (DC-1) and DC-HSF-4 (DC-4) were isolated from third and second generation adults, respectively, both with an autosomal dominant form of DC caused by a large 3′-deletion of one copy of TERC (36, 37). Normal skin fibroblasts (N-1 and N-2) were isolated from normal individuals. The studies were approved by the University of Iowa Institutional Review Board for human studies and conformed to protocols outlined by the Declaration of Helsinki. Donors gave their written, informed consent for collection of tissue. Fibroblasts were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco by Life Technologies, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin. Fibroblasts were split 1:8 when less than 90% confluent. Early passage was considered to between passages 3 and 7, whereas late passage was considered to be greater than 20. Keratinocytes were cultured in Keratinocyte serum-free media (KSFM; Gibco) supplemented with bovine pituitary extract (BPE), epidermal growth factor (EGF), and 1% penicillin and streptomycin. Normal HSK were considered early passage between 2 and 4. HaCaT cells were kindly provided by Jackie Bickenbach who obtained them as early passage cultures from Petra Boukamp (39). Later passage transformed HaCaT cells were obtained from Sheila Stewart (40). TERT and TERT/TR transduced DC and normal fibroblasts have been described previously (36).
Co-culture of human skin keratinocytes with epidermal fibroblasts to assess colony forming efficiency (CFE)
Clonogenic assays, also referred to as colony forming efficiency (CFE) assays, were performed using co-cultures of keratinocytes with fibroblasts. Fibroblasts and keratinocytes were trypsinized using 0.05% Trypsin/EDTA. After the cells were rounded and detached, the trypsin was inhibited with 2% FBS in PBS. The cells were counted using a hemocytometer and resuspended in KSFM at appropriate concentrations (see below). The desired fibroblast-keratinocyte combinations were mixed in a conical tube and plated in 60 mm dishes (in at least triplicate, per condition) in 4 mls KSFM with supplements and penicillin/streptomycin using the following cell numbers per dish: primary fibroblasts (N or DC), 2×105; primary keratinocytes, 1×104; HaCaT, 500 cells per dish. Lower numbers of HaCaT cells were used because they had much higher CFE as compared to normal primary keratinocytes. For the experiment to test the effects for more DC fibroblasts on keratinocyte growth (shown in figure 1C), the same number of HaCaT cells were used (i.e. 500) but with increasing numbers (2 × 105, 5 × 105, and 1 × 106) of DC fibroblasts. Co-cultures of fibroblasts and keratinocytes were maintained at 37°C, 5% CO2 for 7–9 days, with fresh KSFM media (with EGF, BPE, and penicillin/streptomycin) changes every other day. After 7–9 days of co-culture, dishes were washed with 1X PBS, and then fixed in 4% Paraformaldehyde for 10 minutes at room temperature. To differentiate between fibroblasts and keratinocyte colonies, the cells were then stained with 1% Rhodanile blue (in water) (Sigma-Aldrich, St. Louis, MO, USA) for 25 minutes at room temperature as described (41). Dishes were de-stained briefly in a basin of water, until colonies of keratinocytes were bright pink or purple and surrounding fibroblasts were gray. Dishes were dried overnight at room temperature, and colonies of approximately 25 or more keratinocytes were counted visually.
Figure 1.
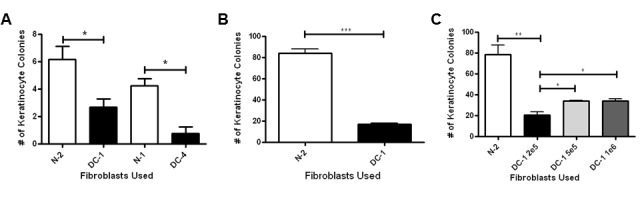
Fibroblasts from dyskeratosis congenita (DC) skin are defective in supporting colony growth of epidermal keratinocytes. (A) Age- and sex-matched primary skin fibroblasts isolated from normal donors (N-2 vs. DC-1 and N-1 vs. DC-4) were grown in co-culture with normal skin keratinocytes, as described in the Materials and Methods. After 8 days in culture, cells were stained with Rhodanile blue to differentially stain keratinocyte colonies. Colonies were counted and averaged. (B) HaCaT cells were grown in co-culture with N-2 or DC-1 fibroblasts as described in the Materials and Methods and cultured and counted as above. (C) Increasing numbers of DC-1 (2e5=2 × 105, 5e5=5 × 105, or 1e6=1 × 106) cells were plated in co-culture with HaCaT cells and compared to separate cultures of N-2 (2e5=2 × 105) cells plated with HaCaT cells. For all experiments, error bars shown represent the SEM from at least 3 replicates per condition. Asterisks represent statistical significance (1, p<0.05; 2, p<0.01; 3, p<0.005) as calculated by Student’s t-test.
Conditioned media transfer in keratinocyte cultures
For the CFE experiments to assess the effects of conditioned media on keratinocyte growth, co-cultures of normal or DC fibroblasts with 500 HaCaT cells were plated as described above. Additional plates of DC and normal fibroblasts were plated at the same time in KSFM with supplements (to maintain similar culture conditions). Daily transfers of conditioned media from the DC or normal fibroblast only plates to the co-culture plates were performed. Co-culture plates that did not get conditioned media received fresh media daily. In the CFE experiments where there was no co-culture, conditioned media alone (i.e. with no cells) was transferred daily from DC or normal fibroblast cultures to dishes that had been seeded with 500 HaCaT cells. After 10 days, the HaCaT dishes were stained with Methylene Blue to identify colonies of keratinocytes.
siRNA transfection of cells
Normal early passage fibroblasts were transfected with siRNA to knockdown expression of factors found to be important to their support of keratinocyte colony growth. 100 nM or 200 nM concentrations of the following siRNAs were transfected using Lipofectamine 2000 (Invitrogen) for 72 or 96 hours. 100 nM concentrations of negative control (scrambled) siRNAs from Qiagen and Invitrogen were also used. HGF siRNA#1: AAGCCUUGCAAGUGAAUGGAAGUCC, siRNA#2: CCGCUGGGAGUACUGUGCAAUUAAA (Invitrogen Stealth™ siRNA).
IGF-1 siRNA: Qiagen #sI02664060 (Hs_IGF1_5 FlexiTube siRNA) (Qiagen)
Conditioned media from transfected fibroblasts was collected at 72, 96, and 120 hours and frozen at −20°C. CFE assays using conditioned media were performed as described above with new conditioned media added daily.
Quantitative PCR
q-RT-PCR:
Collection of cells for RNA was performed when the cells were 80–90% confluent. Collection of RNA from siRNA transfection experiments was performed at 72 and 96 hours post transfection. Briefly, total RNA was extracted from cells using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions and purified with RNeasy Mini Columns (Qiagen). RNA was reverse transcribed using a Retroscript kit (Ambion, Austin, TX, USA) with random decamers as recommended by the manufacturer. Quantitative PCR was performed in triplicate using the ABI PRISM 7900 Sequence Detection System with standard cycling in SYBR-Green PCR Master Mix (Applied Biosystems) with the following primers at 50 μM each: Hepatocyte growth factor (HGF), Forward: 5′-CAAATGTCAGCCCTGGAGTT-3′, Reverse: 5′-TCGATAACTCTCCCCATTGC-3′ (42); Insulin-like growth factor-1 (IGF-1), Forward: 5′-GCCAAGTCAGCTCGCTCTGT-3′, Reverse: 5′-TTTCCTTCTCTGAGACTTCGTGTTC-3′ (43); keratinocyte growth factor (KGF), Forward: 5′-AGTTGGAATTGTGGCAATCA-3′, Reverse: 5′-CCGTTGTGTGTCCATTTAGC-3′ (44); interleukin-6 (IL-6), Forward: 5′-ATGAACTCCT TCTCCACAAGC-3′, Reverse: 5′-CTACATTTGCCGAAGAGCCC-3′ (44); stem cell factor (SCF), Forward: 5′-CCAAAAGACTACATGATAACCCTCAA-3′, Reverse: 5′-CATCTCGCTTATCCAACAATGACT-3′ (45); osteopontin (OPN), Forward: 5′-CCTGATGC TACAGACGAGGAC-3′, Reverse: 5′-CTGACTATCAATCACATCGGAAT-3′ (46); glyceraldehyde 3-phosphate dehydrogenase (GAPDH), Forward: 5′-AAGGTCATCCATGACAACTTTG-3′, Reverse: 5′-GTAGAGGCAGGGATGATGTTCT-3′ (37). Expression levels were normalized to GAPDH.
Statistical Analysis
For growth studies and q-RT-PCR analyses, Student t-test was performed on 3 replicates.
RESULTS
Dyskeratosis congenita fibroblasts are deficient in supporting clonogenic growth of keratinocytes in co-culture
Previous studies demonstrated that DC fibroblasts have extremely short telomeres and that they only proliferate for approximately half the lifespan of normal fibroblasts (36). To begin to address whether the DC fibroblasts are functionally defective, we developed an assay to determine how co-culture with fibroblasts affects the clonogenic growth, also referred to as colony forming efficiency (CFE), of keratinocytes. In this assay, a large number of fibroblasts (2 × 105 in a 60 mm dish) are plated with a low number of keratinocytes, and keratinocyte colonies are counted after 8–10 days. The growth of the fibroblasts is kept in “check” in our assay by using keratinocyte serum free media (KSFM), which allows growth of keratinocyte colonies but only minimal growth of fibroblasts. Rhodanile-Blue was used to differentially stain keratinocytes as described (41). We first performed co-cultures using normal primary human skin keratinocytes (N-HSK-1) at early passage. These primary keratinocytes had low CFE, in general. However, CFE was significantly lower when the N-HSK-1 cells were grown with DC fibroblasts as compared to growth with normal fibroblasts (age- and sex-matched pairs, N-2 vs. DC-1 and N-1 vs. DC-4) (Figure 1A). Because the primary keratinocytes had such low CFE, we decided to perform further assays using the spontaneously immortalized keratinocyte cell line HaCaT, which are known to have higher CFE (39). We also focused our studies on DC-1 and N-2 fibroblasts (both from 21-year old females). HaCaT cells had much higher CFE compared to primary keratinocytes when grown with normal (N-2) fibroblasts but, as with primary keratinocytes, colony numbers were greatly reduced when co-cultured with DC-1 fibroblasts (Figure 1B). To address whether the lower number of colonies in co-culture with DC fibroblasts simply had to do with cell density, we performed an experiment with increasing numbers of DC fibroblasts (Figures 1C). Increasing the number of DC fibroblasts slightly increased keratinocyte colony number but it was still much less than that seen in co-cultures with normal fibroblasts. Overall, our results indicate that the DC fibroblasts are defective in their ability to support the growth of keratinocytes.
DC Fibroblasts are Deficient in Secretion of a Factor or Factors that Supports Keratinocyte Growth
The defect in the ability of the DC cells to support keratinocyte growth could be due to several possibilities including 1) deficiency in secretion of a factor or factors important for keratinocyte proliferation, 2) secretion of a factor or factors by DC cells that inhibits keratinocyte proliferation, or 3) defective cell-cell interactions. To address these possibilities, we performed conditioned media transfer experiments. First, keratinocytes were plated in co-cultures with DC or normal fibroblasts as above. The media was changed on a daily basis with conditioned media from DC or normal fibroblast cultures. The results clearly demonstrated that the DC fibroblasts do not secrete an inhibitor of keratinocyte proliferation since DC fibroblast media did not inhibit keratinocyte growth (Figure 2A). Normal fibroblast conditioned media, however, significantly increased keratinocyte growth when in co-culture with DC fibroblasts. In order to assess the contribution of cell-cell contact in our assay, we grew HaCaT cells without fibroblasts but with and without fibroblast conditioned media. Our results indicated that conditioned media from DC fibroblasts was defective in supporting keratinocyte clonogenic growth (Figure 2B and 2C). This difference was especially apparent in number of cells per colony (Figure 2C). These findings indicate that DC fibroblasts are defective in secreting a factor (or factors) important for keratinocyte clonogenic growth.
Figure 2.

Conditioned media from normal fibroblasts rescues the ability of DC fibroblasts to support epidermal clonogenic growth. Error bars represent the SEM of the average of at least 3 dishes of cells per condition. Asterisks represent statistical significance (1, p<0.05; 2, p<0.01; 3, p<0.005) as calculated by Student t-test. (A) N-2 or DC-1 cells were co-cultured with HaCaT cells. To some of these plates, media was also transferred daily from either N-2 or DC-1 cultures alone, creating co-cultures of N-2 cells and HaCaT cells with DC-1 conditioned media, or DC-1 cells and HaCaT cells with N-2 conditioned media. (B) Conditioned media alone from cultures of N-2 or DC-1 cells was added daily to cultures of HaCaT cells and colonies were counted. (C) Cells per colony in the conditioned media experiments were quantified. Asterisks represent statistical significance as described in Figure 1.
DC Fibroblasts Exhibit Aberrant Expression of Genes that Code for Secreted Cytokines
Various studies have demonstrated that aged fibroblasts either downregulate or upregulate genes that code for secreted proteins. We therefore examined the transcript levels of a panel of these genes in DC and normal fibroblasts. We found that DC fibroblasts express similar amounts of the housekeeping gene GAPDH but significantly lower amounts of HGF and IGF-1, significantly higher amounts of osteopontin, slightly lower amounts of KGF (Keratinocyte Growth Factor), and slightly higher amounts of IL-6 (Interleukin 6) and SCF (Stem Cell Factor) (Figure 3A). Differences were noted in expression of a subset of these genes as compared to late passage, near senescent normal fibroblasts (Figure 3B). For example, both DC and late passage normal fibroblasts had much lower expression of IGF1 and HGF and higher expression of osteopontin. However, in contrast to DC fibroblasts, late passage normal fibroblasts had much lower levels of IL-6. In support of a role for fibroblast-expressed IGF1 and HGF in maintaining keratinocyte growth, we found that the late passage fibroblasts were also unable to support epidermal clonogenic growth (Figure 3C).
Figure 3.

DC and late passage fibroblasts exhibit aberrant expression of genes that code for secreted cytokines. Quantitative RT-PCR was done on samples from normal and DC fibroblasts to examine the transcript levels of candidate genes as described in Materials and Methods. Expression levels were normalized to GAPDH. Error bars represent the SEM of the average of triplicate wells. (A) Transcript levels of selected genes in early passage normal and DC fibroblasts. (B) Transcript levels in late passage (LP) normal fibroblasts compared to early passage (EP) normal fibroblasts and early passage DC fibroblasts. Asterisks represent statistical significance as described in Figure 1. (C) Comparison of colony forming efficiency of keratinocytes grown with EP normal and DC fibroblasts and LP normal fibroblasts.
Knockdown of IGF1 or HGF in normal fibroblasts leads to a defect in their ability to support keratinocyte growth
To further assess the role of fibroblast-derived IGF1 and HGF in keratinocyte growth, we knocked down these genes in normal fibroblasts by siRNA transfection strategies. For both genes, we were able to achieve approximately 75% downregulation using gene specific siRNAs (Figures 4A and 4B). Conditioned media from these fibroblasts were collected starting at days 3 to 5 and used in growth experiments with HaCaT cells. Knockdown of IGF1 or HGF in fibroblasts led to significantly less keratinocyte clonogenic growth (Figure 4C and 4D), supporting the idea that less IGF1 and HGF in DC and aged fibroblasts may contribute to their ability to support keratinocyte growth.
Figure 4.

Downregulation of IGF1 and HGF in normal fibroblasts causes repression of keratinocyte clonogenic growth. Normal fibroblasts were transfected with siRNA against either (A) IGF1 or (B) HGF, and cells were then analyzed by qRT-PCR for IGF1 and HGF knockdown either 72 or 96 hours post-transfection. Values are relative to no siRNA treatment. Concentrations of siRNAs are shown on the X-axis of each graph. Conditioned media from IGF1 (C) or HGF (D) siRNA transfected normal fibroblasts was collected as described in the Materials and Methods and applied daily to HaCaT cells. Scrambled siRNA was included as a negative control for siRNA transfections. Colonies were counted after 8 days. Error bars represent the SEM of the average of at least 3 dishes of cells per condition. Asterisks represent statistical significance as in figure 1. (E) Colony forming efficiency of HaCaT cells co-cultured with DC fibroblasts that had been transduced with TERT or TERT/TR to activate telomerase. (F) Transcript levels of IGF1 and HGF in TERT or TERT/TR DC fibroblasts as compared to untransduced and normal fibroblasts.
Telomerase reconstitution in DC fibroblasts does not fully correct ability to support keratinocyte growth
Our previous studies demonstrated that transduction of TERT, either alone or with the RNA component TR, allowed maintenance or elongation of telomeres and rescued the ability of DC fibroblasts to proliferate (36). Unexpectedly, we did not observe a significant rescue of the ability of the telomerase reconstituted DC fibroblasts to support keratinocyte clonogenic growth (Figure 4E). Accordingly, we also did not see large increases in IGF1 or HGF in the telomerase reconstituted cells (Figure 4F). These results suggest that certain defects in DC fibroblasts are not readily reversed by telomerase reconstitution alone.
DISCUSSION
The aging of skin is a well-documented phenomenon that can lead to significant problems in the elderly. The mechanisms by which human skin ages are not completely clear but are likely due to a variety of factors. One factor that has been speculated to be involved in the aging of skin is telomere shortening (2, 47, 48). Mouse models of telomere dysfunction and the observation that DC patients suffer from epidermal abnormalities and prematurely aged skin support this hypothesis. Our previous studies indicated that telomere shortening in human keratinocytes leads to defective proliferation and a decreased ability to close in vitro scratch wounds (37). These results suggested that telomere shortening in keratinocytes could be involved in epidermal abnormalities associated with DC. Another possibility, and the one we have addressed here, is that telomere shortening and premature aging in dermal support cells leads to a defect in their ability to support normal epidermal proliferation and repair. Certainly, these two mechanisms are not mutually exclusive and both could be involved in causing the observed phenotypes.
Our results suggest that aged dermal fibroblasts have altered expression of genes that code for secreted proteins that are important for regulation of normal keratinocyte growth. Two genes that were found to be significantly downregulated in DC fibroblasts and in aged normal fibroblasts were IGF1 and HGF. Both of these genes have been implicated previously as being important for normal skin keratinocyte proliferation and growth. For example, IGF1 was found to be downregulated in senescent fibroblasts and this led to an inappropriate UVB response in keratinocytes(49). HGF is known to be involved in normal keratinocyte response to apoptotic agents and has been shown to stimulate keratinocyte migration and proliferation during wound healing (42, 50, 51). While our depletion experiment pointed to the possibility that reduction of each factor on its own has an effect on growth, we did not perform add-back experiments and have not ruled out the possibility that downregulation of other factors in DC fibroblasts may be important for the observed phenotype. One could speculate that telomere shortening and induction of a senescent phenotype leads to a global change in expression of a large number of genes. Other studies have indicated that the alterations in expression of genes during senescence are due to global epigenetic alterations that change the pattern of methylation and heterochromatin in cells (28). Interestingly, we were unable to reverse these effects by reconstitution of telomerase (and telomeres) alone in DC cells. This was a somewhat surprising finding given that our previous studies demonstrated that telomerase activation in DC cells leads to amelioration of their proliferation defect (36, 37) and a decrease in elevated reactive oxygen species levels (52). Our results indicate that some of the changes that occur during the aging of fibroblasts may not be telomere dependent and are not reversible by simply activating telomerase. Such findings have implications for proposed therapies for aging that rely on telomerase activation as a means to rejuvenate tissue, as telomerase activation alone may not be sufficient to reverse the aging phenotype.
Several studies have demonstrated that aged fibroblasts can increase expression of certain genes that code for secreted proteins such as osteopontin that stimulate proliferation of transformed epithelial cells (53). This phenomenon has been referred to as the “tumor microenvironment” and is believed to be important for carcinogenesis (29, 30, 40). One study demonstrated that senescent fibroblasts have higher expression of and secrete more osteopontin, and that this stimulates the growth of pre-malignant cells (40). It is interesting to note that we also found an increase in osteopontin expression in DC cells and aged normal fibroblasts, thus validating published findings. However, we did not observe improved clonogenic growth of normal or immortal keratinocytes, including HaCaT cells. A possible reason for this apparent discrepancy is that normal and transformed keratinocytes respond differently to factors secreted by fibroblasts, and that the HaCaT cells that we used in our assays had a more normal phenotype than those utilized by others. Indeed, our HaCaT cells were obtained from a relatively early passage stock (39) and HaCaT cells are well-known to evolve and become more highly transformed in culture (54). To address this possibility, similar co-culture experiments with DC or aged fibroblasts carried out with transformed HaCaT cells obtained from another source (40) demonstrated an increase in clonogenic growth of the HaCaT cells (Figure 5). Thus, it is possible that aging of fibroblasts may cause a decrease in their ability to support the growth of normal keratinocytes but may cause an increase in their ability to support more transformed, pre-malignant keratinocytes. This observation, if validated by further studies, would fit well with the hypothesis that aging is associated with a decrease in normal skin function and an increase in epidermal cancers.
Figure 5.
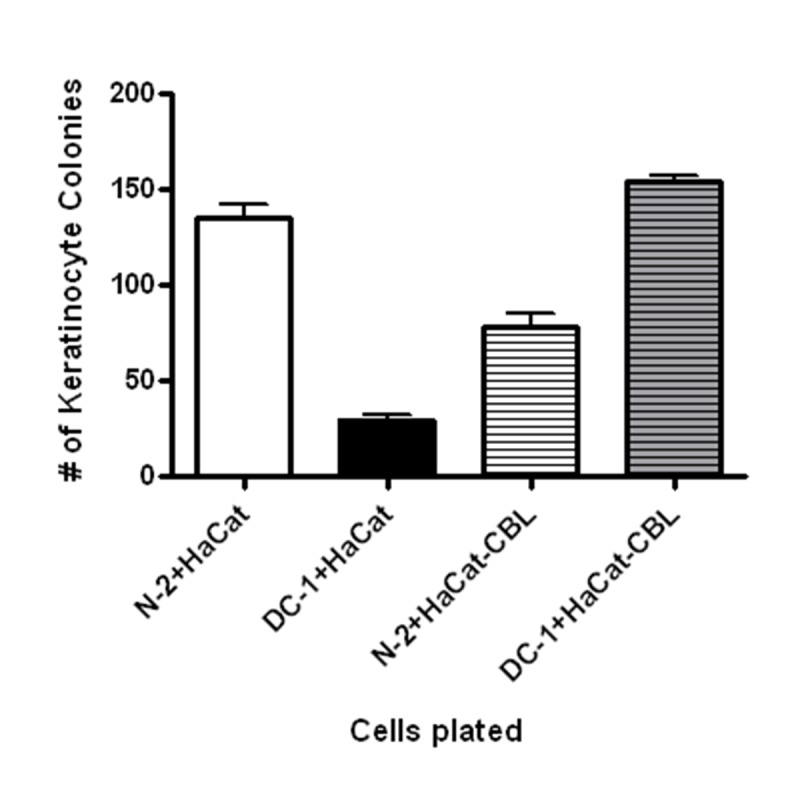
HaCaT cells from different sources exhibit differential growth in co-culture. HaCaT cells used to generate results shown in figures 1 to 4 were obtained from an early passage source (39) whereas HaCaT-CBL (Click Beetle Luciferase) were obtained from Dr. Stewart’s laboratory where they are used to study transformative effects of tumor microenvironment on epithelial cells (48). The different HaCaT cells were plated at 500 cells per 60 mm dish with fibroblasts (DC-1 or N-2). Error bars represent standard error of results from triplicate plates.
While the present studies have focused on skin cells, the phenomenon that we have observed could be applicable to many other tissue types and could be important for organismal aging as a whole. Since cell aging can lead to defective production of cytokines and other growth regulatory factors, it would be expected that telomere shortening in one cell type can have profound effects on other cell types and tissues. Telomere shortening in mice has been shown to lead to defects in support cells of the bone marrow niche that give rise to the proper development of the hematopoietic system (55). Lower IGF1 levels in serum have been implicated as being important for many processes and have been associated with diseases that are related to aging, including diabetes (56). Short telomeres are associated with lower serum IGF1 (57, 58) and with diabetes (59, 60). It is possible that telomere length and concomitant senescence in certain cell types may be important initiating events that lead to lower levels of IGF1 and other growth factors that are needed for maintaining normal tissue regeneration, function, and wound repair. Interestingly, mice that have been engineered to exogenously express TERT in their skin have an increased lifespan and higher serum levels of IGF1 (20). The mechanism for this increased level of IGF1 in TERT transgenic mice is unknown, but the fact that the transgene is expressed through a keratin specific promoter suggests that the IGF1 may be originating from changes in skin associated with exogenous TERT expression.
In summary, our results have uncovered a potential means by which telomere shortening might lead to the aging of skin, and possibly other tissues. Further studies are needed to explore the mechanism by which telomere shortening leads to altered gene expression, how this changes the secretory profile of cells, and how to safely reverse this process.
Acknowledgments
We thank the University of Iowa DNA Facility for assistance with quantitative PCR. This work was supported by NIH R01 AG0227388.
References
- [1].Robert L, Labat-Robert J, Robert AM. Physiology of skin aging. Pathol Biol (Paris) 2009;57:336–341. doi: 10.1016/j.patbio.2008.09.007. [DOI] [PubMed] [Google Scholar]
- [2].Buckingham EM, Klingelhutz AJ. The role of telomeres in the ageing of human skin. Exp Dermatol. 2011;20:297–302. doi: 10.1111/j.1600-0625.2010.01242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–334. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- [4].O’Sullivan RJ, Karlseder J. Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol. 2010;11:171–181. doi: 10.1038/nrm2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Allsopp RC, Chang E, Kashefiaazam M, Rogaev EI, Piatyszek MA, Shay JW, Harley CB. Telomere Shortening Is Associated with Cell-Division in-Vitro and in-Vivo. Exp Cell Res. 1995;220:194–200. doi: 10.1006/excr.1995.1306. [DOI] [PubMed] [Google Scholar]
- [6].von Zglinicki T. Oxidative stress shortens telomeres. Trends Biochem Sci. 2002;27:339–344. doi: 10.1016/s0968-0004(02)02110-2. [DOI] [PubMed] [Google Scholar]
- [7].Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43:405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- [8].Nakamura TM, Morin GB, Chapman KB, Weinrich SL, Andrews WH, Lingner J, Harley CB, Cech TR. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997;277:955–959. doi: 10.1126/science.277.5328.955. [DOI] [PubMed] [Google Scholar]
- [9].Wright WE, Piatyszek MA, Rainey WE, Byrd W, Shay JW. Telomerase activity in human germline and embryonic tissues and cells. Dev Genet. 1996;18:173–179. doi: 10.1002/(SICI)1520-6408(1996)18:2<173::AID-DVG10>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- [10].Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- [11].Buchkovich KJ, Greider CW. Telomerase regulation during entry into the cell cycle in normal human T cells. Mol Biol Cell. 1996;7:1443–1454. doi: 10.1091/mbc.7.9.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Prowse KR, Greider CW. Developmental and tissue-specific regulation of mouse telomerase and telomere length. Proc Natl Acad Sci U S A. 1995;92:4818–4822. doi: 10.1073/pnas.92.11.4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hu BT, Insel RA. Up-regulation of telomerase in human B lymphocytes occurs independently of cellular proliferation and with expression of the telomerase catalytic subunit. Eur J Immunol. 1999;29:3745–3753. doi: 10.1002/(SICI)1521-4141(199911)29:11<3745::AID-IMMU3745>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- [14].Bickenbach JR, Vormwald-Dogan V, Bachor C, Bleuel K, Schnapp G, Boukamp P. Telomerase is not an epidermal stem cell marker and is downregulated by calcium. J Invest Dermatol. 1998;111:1045–1052. doi: 10.1046/j.1523-1747.1998.00420.x. [DOI] [PubMed] [Google Scholar]
- [15].Krunic D, Moshir S, Greulich-Bode KM, Figueroa R, Cerezo A, Stammer H, Stark HJ, Gray SG, Nielsen KV, Hartschuh W, Boukamp P. Tissue context-activated telomerase in human epidermis correlates with little age-dependent telomere loss. Biochim Biophys Acta. 2009;1792:297–308. doi: 10.1016/j.bbadis.2009.02.005. [DOI] [PubMed] [Google Scholar]
- [16].Butler MG, Tilburt J, DeVries A, Muralidhar B, Aue G, Hedges L, Atkinson T, Schwartz H. Comparison of chromosome telomere integrity in multiple tissues from subjects at different ages. Cancer GenetCytogenet. 1998;105:138–144. doi: 10.1016/s0165-4608(98)00029-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997;91:25–34. doi: 10.1016/s0092-8674(01)80006-4. [DOI] [PubMed] [Google Scholar]
- [18].Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, DePinho RA. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell. 1999;96:701–712. doi: 10.1016/s0092-8674(00)80580-2. [DOI] [PubMed] [Google Scholar]
- [19].Gonzalez-Suarez E, Geserick C, Flores JM, Blasco MA. Antagonistic effects of telomerase on cancer and aging in K5-mTert transgenic mice. Oncogene. 2005;24:2256–2270. doi: 10.1038/sj.onc.1208413. [DOI] [PubMed] [Google Scholar]
- [20].Tomas-Loba A, Flores I, Fernandez-Marcos PJ, Cayuela ML, Maraver A, Tejera A, Borras C, Matheu A, Klatt P, Flores JM, Vina J, Serrano M, Blasco MA. Telomerase reverse transcriptase delays aging in cancer-resistant mice. Cell. 2008;135:609–622. doi: 10.1016/j.cell.2008.09.034. [DOI] [PubMed] [Google Scholar]
- [21].Sarin KY, Cheung P, Gilison D, Lee E, Tennen RI, Wang E, Artandi MK, Oro AE, Artandi SE. Conditional telomerase induction causes proliferation of hair follicle stem cells. Nature. 2005;436:1048–1052. doi: 10.1038/nature03836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Siegl-Cachedenier I, Flores I, Klatt P, Blasco MA. Telomerase reverses epidermal hair follicle stem cell defects and loss of long-term survival associated with critically short telomeres. J Cell Biol. 2007;179:277–290. doi: 10.1083/jcb.200704141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tejera AM, Stagno d’Alcontres M, Thanasoula M, Marion RM, Martinez P, Liao C, Flores JM, Tarsounas M, Blasco MA. TPP1 is required for TERT recruitment, telomere elongation during nuclear reprogramming, and normal skin development in mice. Dev Cell. 2010;18:775–789. doi: 10.1016/j.devcel.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Martinez P, Thanasoula M, Munoz P, Liao C, Tejera A, McNees C, Flores JM, Fernandez-Capetillo O, Tarsounas M, Blasco MA. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 2009;23:2060–2075. doi: 10.1101/gad.543509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hockemeyer D, Palm W, Wang RC, Couto SS, de Lange T. Engineered telomere degradation models of dyskeratosis congenita. Genes Dev. 2008;22:1773–1785. doi: 10.1101/gad.1679208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Watt FM, Fujiwara H. Cell-extracellular matrix interactions in normal and diseased skin. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a005124. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Parrinello S, Coppe JP, Krtolica A, Campisi J. Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci. 2005;118:485–496. doi: 10.1242/jcs.01635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pazolli E, Alspach E, Milczarek A, Prior J, Piwnica-Worms D, Stewart SA. Chromatin remodeling underlies the senescence-associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer Res. 2012;72:2251–2261. doi: 10.1158/0008-5472.CAN-11-3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Davalos AR, Coppe JP, Campisi J, Desprez PY. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010;29:273–283. doi: 10.1007/s10555-010-9220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A. 2001;98:12072–12077. doi: 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kirwan M, Dokal I. Dyskeratosis congenita: a genetic disorder of many faces. Clin Genet. 2008;73:103–112. doi: 10.1111/j.1399-0004.2007.00923.x. [DOI] [PubMed] [Google Scholar]
- [33].Vulliamy T, Marrone A, Goldman F, Dearlove A, Bessler M, Mason PJ, Dokal I. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature. 2001;413:432–435. doi: 10.1038/35096585. [DOI] [PubMed] [Google Scholar]
- [34].Knudson M, Kulkarni S, Ballas Z, Bessler M, Goldman F. Association of Immune Abnormalities with Telomere Shortening in Autosomal Dominant Dyskeratosis Congenita. Blood. 2005;105:682–688. doi: 10.1182/blood-2004-04-1673. [DOI] [PubMed] [Google Scholar]
- [35].Goldman FD, Aubert G, Klingelhutz AJ, Hills M, Cooper SR, Hamilton WS, Schlueter AJ, Lambie K, Eaves CJ, Lansdorp PM. Characterization of primitive hematopoietic cells from patients with dyskeratosis congenita. Blood. 2008;111:4523–4531. doi: 10.1182/blood-2007-10-120204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Westin ER, Chavez E, Lee KM, Gourronc FA, Riley S, Lansdorp PM, Goldman FD, Klingelhutz AJ. Telomere restoration and extension of proliferative lifespan in dyskeratosis congenita fibroblasts. Aging Cell. 2007;6:383–394. doi: 10.1111/j.1474-9726.2007.00288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gourronc FA, Robertson M, Herrig AK, Lansdorp PM, Goldman FD, Klingelhutz AJ. Proliferative defects in dyskeratosis congenita skin keratinocytes are corrected by expression of the telomerase reverse transcriptase, TERT, or by activation of endogenous telomerase through expression of papillomavirus E6/E7 or the telomerase RNA component, TERC. Exp Dermatol. 2010;19:279–288. doi: 10.1111/j.1600-0625.2009.00916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Darbro BW, Schneider GB, Klingelhutz AJ. Co-regulation of p16INK4A and migratory genes in culture conditions that lead to premature senescence in human keratinocytes. J Invest Dermatol. 2005;125:499–509. doi: 10.1111/j.0022-202X.2005.23844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988;106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pazolli E, Luo X, Brehm S, Carbery K, Chung JJ, Prior JL, Doherty J, Demehri S, Salavaggione L, Piwnica-Worms D, Stewart SA. Senescent stromal-derived osteopontin promotes preneoplastic cell growth. Cancer Res. 2009;69:1230–1239. doi: 10.1158/0008-5472.CAN-08-2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Rheinwald JG, Green H. Epidermal growth factor and the multiplication of cultured human epidermal keratinocytes. Nature. 1977;265:421–424. doi: 10.1038/265421a0. [DOI] [PubMed] [Google Scholar]
- [42].Mildner M, Mlitz V, Gruber F, Wojta J, Tschachler E. Hepatocyte growth factor establishes autocrine and paracrine feedback loops for the protection of skin cells after UV irradiation. J Invest Dermatol. 2007;127:2637–2644. doi: 10.1038/sj.jid.5700938. [DOI] [PubMed] [Google Scholar]
- [43].Catalan V, Gomez-Ambrosi J, Rodriguez A, Ramirez B, Silva C, Rotellar F, Hernandez-Lizoain JL, Baixauli J, Valenti V, Pardo F, Salvador J, Fruhbeck G. Up-regulation of the novel proinflammatory adipokines lipocalin-2, chitinase-3 like-1 and osteopontin as well as angiogenic-related factors in visceral adipose tissue of patients with colon cancer. J Nutr Biochem. 2011;22:634–641. doi: 10.1016/j.jnutbio.2010.04.015. [DOI] [PubMed] [Google Scholar]
- [44].Wenghoefer M, Pantelis A, Najafi T, Deschner J, Allam JP, Novak N, Reich R, Martini M, Berge S, Fischer HP, Jepsen S, Winter J. Gene expression of oncogenes, antimicrobial peptides, and cytokines in the development of oral leukoplakia. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2010;110:351–356. doi: 10.1016/j.tripleo.2009.08.013. [DOI] [PubMed] [Google Scholar]
- [45].Bialas M, Borczynska A, Rozwadowska N, Fiszer D, Kosicki W, Jedrzejczak P, Kurpisz M. SCF and c-kit expression profiles in male individuals with normal and impaired spermatogenesis. Andrologia. 2010;42:83–91. doi: 10.1111/j.1439-0272.2009.00960.x. [DOI] [PubMed] [Google Scholar]
- [46].Pei K, Yu C, Shi X, Jia M. The effects of mifepristone on the expressions of osteopontin, interleukin-6 and leukemia inhibitory factor in the villi of early pregnant women. Contraception. 2010;82:379–384. doi: 10.1016/j.contraception.2010.04.009. [DOI] [PubMed] [Google Scholar]
- [47].Boukamp P. Skin aging: a role for telomerase and telomere dynamics? Curr Mol Med. 2005;5:171–177. doi: 10.2174/1566524053586644. [DOI] [PubMed] [Google Scholar]
- [48].Kosmadaki MG, Gilchrest BA. The role of telomeres in skin aging/photoaging. Micron. 2004;35:155–159. doi: 10.1016/j.micron.2003.11.002. [DOI] [PubMed] [Google Scholar]
- [49].Lewis DA, Travers JB, Somani AK, Spandau DF. The IGF-1/IGF-1R signaling axis in the skin: a new role for the dermis in aging-associated skin cancer. Oncogene. 2010;29:1475–1485. doi: 10.1038/onc.2009.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Spix JK, Chay EY, Block ER, Klarlund JK. Hepatocyte growth factor induces epithelial cell motility through transactivation of the epidermal growth factor receptor. Exp Cell Res. 2007;313:3319–3325. doi: 10.1016/j.yexcr.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Dunsmore SE, Rubin JS, Kovacs SO, Chedid M, Parks WC, Welgus HG. Mechanisms of hepatocyte growth factor stimulation of keratinocyte metalloproteinase production. J Biol Chem. 1996;271:24576–24582. doi: 10.1074/jbc.271.40.24576. [DOI] [PubMed] [Google Scholar]
- [52].Westin ER, Aykin-Burns N, Buckingham EM, Spitz DR, Goldman FD, Klingelhutz AJ. The p53/p21(WAF/CIP) pathway mediates oxidative stress and senescence in dyskeratosis congenita cells with telomerase insufficiency. Antioxid Redox Signal. 2011;14:985–997. doi: 10.1089/ars.2010.3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Fusenig NE, Boukamp P. Multiple stages and genetic alterations in immortalization, malignant transformation, and tumor progression of human skin keratinocytes. Mol Carcinog. 1998;23:144–158. doi: 10.1002/(sici)1098-2744(199811)23:3<144::aid-mc3>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- [55].Ju Z, Jiang H, Jaworski M, Rathinam C, Gompf A, Klein C, Trumpp A, Rudolph KL. Telomere dysfunction induces environmental alterations limiting hematopoietic stem cell function and engraftment. Nat Med. 2007;13:742–747. doi: 10.1038/nm1578. [DOI] [PubMed] [Google Scholar]
- [56].Ezzat VA, Duncan ER, Wheatcroft SB, Kearney MT. The role of IGF-I and its binding proteins in the development of type 2 diabetes and cardiovascular disease. Diabetes Obes Metab. 2008;10:198–211. doi: 10.1111/j.1463-1326.2007.00709.x. [DOI] [PubMed] [Google Scholar]
- [57].Kaplan RC, Fitzpatrick AL, Pollak MN, Gardner JP, Jenny NS, McGinn AP, Kuller LH, Strickler HD, Kimura M, Psaty BM, Aviv A. Insulin-like growth factors and leukocyte telomere length: the cardiovascular health study. J Gerontol A Biol Sci Med Sci. 2009;64:1103–1106. doi: 10.1093/gerona/glp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Moverare-Skrtic S, Svensson J, Karlsson MK, Orwoll E, Ljunggren O, Mellstrom D, Ohlsson C. Serum insulin-like growth factor-I concentration is associated with leukocyte telomere length in a population-based cohort of elderly men. J Clin Endocrinol Metab. 2009;94:5078–5084. doi: 10.1210/jc.2009-1450. [DOI] [PubMed] [Google Scholar]
- [59].Salpea KD, Humphries SE. Telomere length in atherosclerosis and diabetes. Atherosclerosis. 2010;209:35–38. doi: 10.1016/j.atherosclerosis.2009.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Salpea KD, Talmud PJ, Cooper JA, Maubaret CG, Stephens JW, Abelak K, Humphries SE. Association of telomere length with type 2 diabetes, oxidative stress and UCP2 gene variation. Atherosclerosis. 2010;209:42–50. doi: 10.1016/j.atherosclerosis.2009.09.070. [DOI] [PMC free article] [PubMed] [Google Scholar]